Open Journal of Immunology
Vol.2 No.4(2012), Article ID:26258,7 pages DOI:10.4236/oji.2012.24020
Cell-type specific regulation of MARCH1 E3 ubiquitin ligase by the anti-inflammatory cytokine IL-10*
![]()
Department of Microbiology and Immunology, University of Montreal, Montreal, Canada; #Corresponding Author: Jacques.thibodeau@umontreal.ca
Received 18 August 2012; revised 15 September 2012; accepted 5 October 2012
Keywords: MARCH1; Interleukin-10; MHC-II; Ubiquitin; Antigen-Presenting Cells
ABSTRACT
Membrane-associated RING-CH-1 (MARCH1) is an E3 ubiquitin ligase which targets MHC-II, CD86 and various other molecules for degradation. It is one of the most efficient post-translational regulators of antigen presentation. MARCH1 is expressed in resting immature dendritic cells and B cells but can be induced in other cell types. While activation of most immune cells results in a reduction in MARCH1 gene expression, its anti-inflammatory properties are highlighted by its induction in response to IL-10. Here, we review what is known about the regulation of MARCH1 gene expression in response to IL-10 by various immune cells. We speculate on the role of MARCH1 in infection, its differential expression pattern and the promise that this E3 ubiquitin ligase holds to influence immune responses and mitigate immune pathologies.
1. INTRODUCTION
The membrane-associated RING-CH (MARCH) family of proteins is an eleven-member group characterized by the presence of a conserved RING-CH domain conferring ubiquitin ligase activity. They were first identified as human homologues of the K3 family of ubiquitin ligases which specifically target surface MHC molecules [1-3] . Phylogenic analyses have revealed that three pairs of closely related members could be identified: MARCH1 and MARCH8, MARCH2 and MARCH3 and MARCH4 and MARCH9 [4]. MARCH8 was found to be identical to the previously characterized c-MIR [5] . Functionally, the MARCH proteins represent the last step leading to the ubiquitination of target proteins. Briefly, the ubiquitin-conjugating system is supported by three distinctive steps. Ubiquitins are activated, in an ATPdependent manner, by an E1-enzyme which then transfers the ubiquitin moiety to E2-enzymes. Ubiquitin ligases (E3) are the final enzymes in the ubiquitination process, in which they serve as a bridge between the E2- enzymes and the substrate. As such, the MARCH proteins confer specificity to this reaction and each have a different set of targets.
2. INTERLEUKIN-10
IL-10 is a pleiotropic cytokine with a wide spectrum of biological effects on lymphoid and myeloid cells [6] . The IL-10 receptor complex is coupled to the JAK1- STAT3 signal transducers [7] . By this pathway, IL-10 inhibits the production of pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6, IL-10 itself, IL-12, GM-CSF and TNFα by macrophages after Toll-like receptor (TLR) activation [8-10] . Nowadays, it is widely accepted that IL-10 limits and down-regulates inflammatory responses in general. IL-10 is predominantly produced by Tcells, macrophages and dendritic cells. Most types of Tcells produce IL-10 including Th1- or Th2-polarised Tcells and regulatory T-cells [11] . IL-10 also inhibits the production of chemokines, e.g. MCP-1, MIP-1α and IP- 10 [12,13], as well as the expression of MHC-II on the surface of APCs.
3. MARCH PROTEINS IN IMMUNITY
While the different MARCHs share sequence homology and a functional similarity, they however target very different substrates. Of these, multiple were identified for particular MARCHs. For instance, MARCH3 down-regulates the transferrin receptor (TfR) and B7.2 (CD86), MARCH4 and MARCH9 down-regulate MHC-I and CD4 while the closely related MARCH1 and MARCH8 down-regulate TfR, CD86, Fas (CD95) and MHC-II [4, 5,14]. New targets were recently identified for MARCH 8 in primary human fibroblasts, namely CD44 and CD81 [15] . These were particularly interesting findings as they implied a regulatory role of the adaptive arm of immunity by the MARCH proteins. Moreover, even though MARCH1 and MARCH8 are functionally closely related, they present a differential expression pattern. Whereas MARCH8 is more ubiquitously expressed and primarily found in the lungs and pancreas, MARCH1 was found to be highly expressed in secondary lymphoid organs, mainly in lymph nodes and splenic tissues, highlighting, again, a potential role in adaptive immunity [4] . Herein, we will concentrate on the recent evidence that MARCH1 is transcriptionally regulated in professional APCs by the anti-inflammatory cytokine IL-10 [16-18].
4. REGULATION OF MARCH1
Multiple functional studies have been conducted in recent years for the MARCH proteins. Still, little is currently known about the regulation of their expression. Existing MARCH1 antibodies fail to detect the endogenous molecule, which is expressed at very low levels. Thus, current analyses of MARCH1 expression patterns are principally looking at the mRNA levels. Quantitative real-time PCR and microarrays have shown that MARCH1 was constitutively expressed in resting immature dendritic cells and B cells and could be induced in monocytes and macrophages [16-18] . Activation of most dendritic cells and B cells results in a complete loss of expression, implying a deleterious role of MARCH1 for optimal effector functions. As MARCH1 targets MHC-II trafficking in APCs, redirecting MHC-II molecules from recycling endosomes to degradative compartments, it is of no surprise that maturation/activation signals would lead to a decrease in MARCH1 expression [14] (Figure 1). Moreover, by targeting the co-stimulatory molecule CD86 in APCs, MARCH1 not only reduces the amount of MHC-II-peptide complex to be presented to CD4+ T cells but also the activation stimuli required for proper T cell activation [19] . Overall, regulating MARCH1 expression in APCs might provide an optimal and easily malleable way to control antigen presentation without affecting transcription of the implicated genes. While proper antigen presentation machinery induction can take up to 60 hours for ultimate effectiveness, regulating the expression of MARCH1 can have a rapid and reversible effect allowing APCs to fine-tune T cell stimulation. Indeed, the half-life of MARCH1 has been estimated to be around 30 minutes, providing an efficient way to modulate not only MHC-II but also CD86 output [20] . In addition, CD83 has recently been shown to block the interaction between MARCH1 and some of its targets leading to an impaired MARCH1-mediated ubiquitination of MHC-II and CD86 [21] . While the activation of dendritic cells leads to a decrease in MARCH1 mRNA expression, it also induces an up-regulation of CD83, providing a second mechanism to counter remaining MARCH1-mediated ubiquitination. This process could be especially useful in the context of a previously activated APC responding to the presence of IL-10. Pathogen-inducedIL-10-mediated MARCH1 expression could lead to a decrease in antigen presentation in infected APCs, where CD83 could provide the only mechanism to block MARCH1 activity. Ultimately, this might allow an APC to rapidly adapt its antigen presentation machinery in response to persistent infections.
5. IL-10-INDUCED MARCH1
As up to now, only the anti-inflammatory cytokine IL-10 has been shown to induce MARCH1. While IL-10 diminishes the production of inflammatory mediators by monocytes/macrophages and antigen presentation, it increases their uptake of antigens. IL-10 signals through a trans-membrane receptor complex composed of IL-10R1 and IL-10R2. The former being mainly expressed in immune cells where its expression is normally low but can be induced by an LPS-driven activation. In contrast, IL-10R2 is widely and strongly expressed in most cells [22,23]. A nice parallel can be drawn between the pattern of expression of both subunits of the IL-10 receptor complex and the pattern of expression of closely related MARCH1 and MARCH8 which possess overlapping functions. While most immune cells express either constitutive or inducible amounts of MARCH1 (like IL- 10R1), MARCH-8 is found in most cells and tissues like (IL-10R2). Thus, the IL-10-MARCH1 axis might be a master regulating pathway for a wide range of the IL-10 functions on APCs.
6. MONOCYTES/DENDRITIC CELLS
Antigen presentation can be modulated on many levels, both transcriptionally and post-transcriptionally. The master regulator of transcription, CIITA, can be induced by IFN-gamma to increase MHC-II gene expression but also accessory molecules HLA-DO, HLA-DM and the invariant chain. In the endocytic pathway, HLA-DO and HLA-DM control peptide loading directly and modulate the intrinsic characteristics of classical MHC-II molecules. Antigen presentation can also be regulated indirectly through the action of cytokines, such as IL-6 and IL-10. IL-6 induces the STAT3 pathway, leading to an increase in cathepsin S activity and thus a reduction in MHC-II, the invariant chain and HLA-DM [24]. Paradoxically, Koppelman et al. showed that IL-10, despite signaling also mainly through STAT3, could reduce the amount of surface MHC class II molecules in monocytes without affecting transcription, polypeptide synthesis or subunit assembly, but the exact mechanism was unknown [25]. Later, it was shown that MARCH1 was induced by IL-10 in monocytes and immature dendritic cells leading
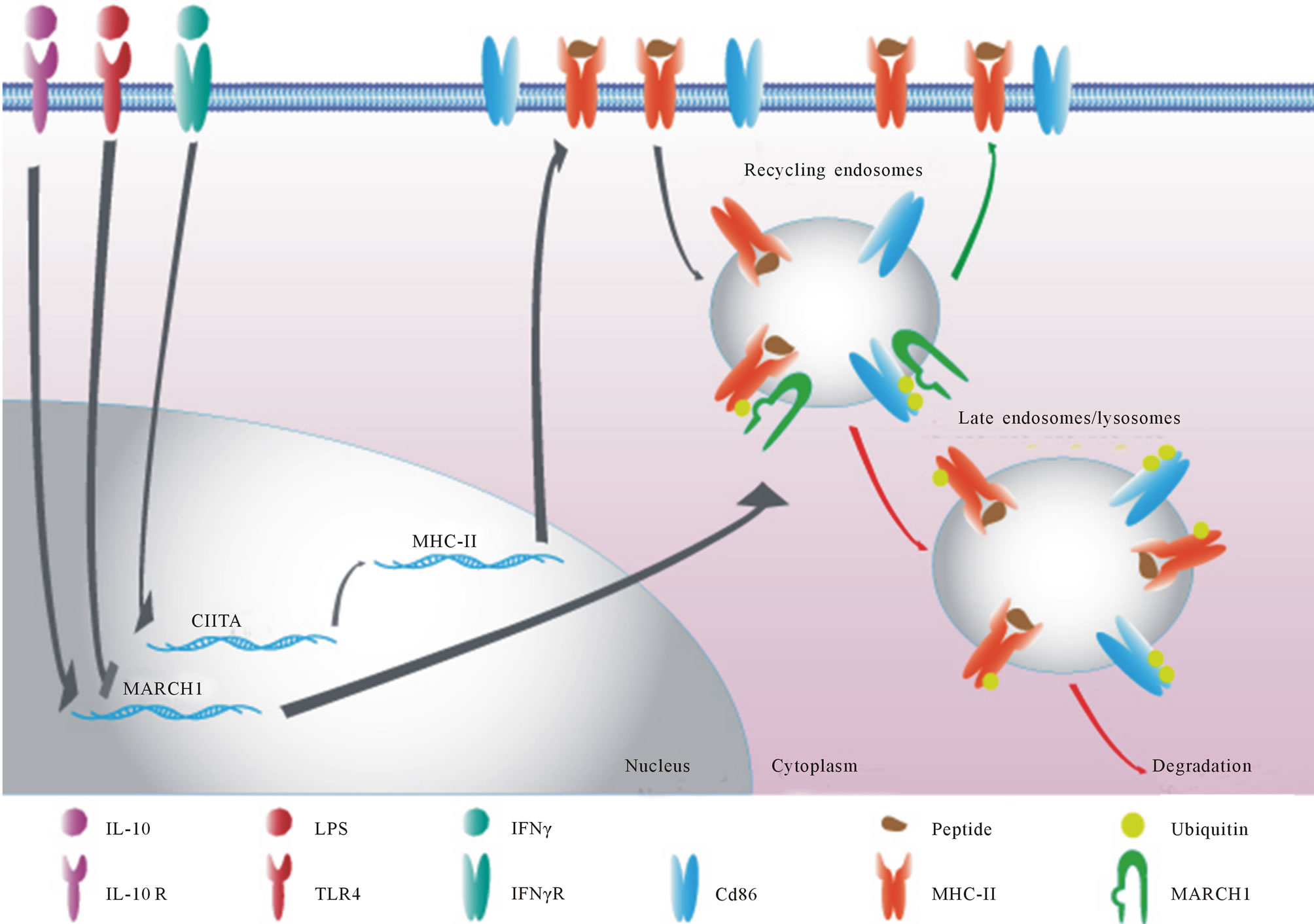
Figure 1. Schematic representation of the regulation of MARCH1 in APCs. Monocytes/Dendritic cells and macrophages only express MARCH1 after stimulation with IL-10. On the other hand, B cells express constitutive amounts of MARCH1 and its expression is repressed upon activation with IL-10. MARCH1 redirects MHC-II from recycling endosomes to degradative compartments by ubiquitinating the MHC-II beta chain. It is thought that MARCH1 uses a similar mechanism to regulate CD86 expression [16,17].
to the intracellular sequestration of MHC-II molecules [16] . In this fashion, through MARCH1, IL-10 is able to act as an immunosuppressive cytokine by impairing antigen presentation, leading to a decreased CD4+ T cells activation. Moreover, it was shown that upon LPS-driven maturation, moDCs decreased their MARCH1 mRNA expression leading to an increase in surface MHC-II molecules stability and antigen presentation [26]. Young et al. have shown that as opposed to cDCs, pDCs failed to down-regulate MARCH1 mRNA after maturation, promoting the turnover of MHC-II-peptide complexes on their surface and the presentation of endogenous viral peptides [27] . These results demonstrate that MARCH1 is used by some APCs to fine tune their antigen presenting functions to promote a constantly “updated’’ peptide repertoire on their surface, maximizing T cell priming in rapidly changing infection profiles. On the other hand, ten Broeke et al. suggested that increased turnover of MHC-II-peptide complexes on the surface of MARCH1- expressing iDCs would prevent presentation of peptides loaded before the danger stimulus has been received [28] . In another set of mind, higher MARCH1 expression would lower both the MHC-II and co-stimulatory molecule CD86 on the surface of DCs, leading to a weak stimulatory potential and a tolerogenic phenotype. Thus the fate of MARCH1 following exposure of different APCs to tolerogenic cytokines will have to be assessed.
7. MACROPHAGES
Macrophages play a central role in infections as a tart for multiple pathogens and a producer of a wide range of cytokines. A recent publication by the group of Drake [18] demonstrated that PGE2 could induce IL-10, through a yet unidentified factor, which in turn induces MARCH1 in Francisella tularensis-infected macroages. These results suggest that MARCH1 could be used by pathogens, through the production of IL-10, to evade the immune system by down-modulating pathogenic antigen presentation. Many human pathogens, such as Salmolla typhi [29], Mycobacteriumum tuberculosis [30] and Legionella pneumophila [31] , and tumors elicit the production of significant levels of PGE2. Another mechanism by which MARCH1 could contribute to immune evasion is by targeting surface Fas (CD95) molecules. IL-10 has been reported to block TNF-induced apoptosis, but could also impair apoptotic signals by inducing MARCH1 and reducing the amount of Fas molecules. Again, providing a useful mechanism by which pathogens could evade destruction of their host cell.
8. B CELLS
Matsuki et al. have shown that MARCH1 plays a pival role in the post-translational regulation of MHC-II in B cells [14] . Their group and ours have demonstrated that MARCH1 was constitutively expressed in all B cell subtypes but highly enriched in mature follicular B cells [14,17]. Constitutive MARCH1 expression in B cells could prevent a too strong MHC-II expression and anergy induction through B cell maturation. On the other hand, a high MARCH1 expression in follicular B cells could prevent these cells from presenting self-antigens prior to meeting with exogenous peptides. Follicular B cells patrol secondary lymphoid organs in search of signs of infection; upon antigen encounter they seek T cell help for activation. At this point, the activation stimuli would drive down the expression of MARCH1 and allow for full antigen presentation. While IL-10 decreases MHC-II amounts in dendritic cells, its effects on B lymphocytes are anything but immunosuppressive. By increasing their survival and MHC-II expression, IL-10 has a stimulary/ activating role on B cells. We have previously demonstrated that IL-10 suppresses the expression of MARCH1 in B cells leading to the well-characterized increase in surface MHC-II molecules levels [17]. Other activating stimuli such as LPS or CD40-CD40L interaction also results in a complete loss of MARCH1 expression in B cells. These phenotypes match accordingly to the idea that MARCH1 is deleterious for optimal effector functions in most APCs. The group of Lesage [32] has recently highlighted the role of IL-10 on the CD4-CD8- double-negative (DN) T cell and auto-reactive B cells interaction. Indeed, IL-10 has been shown to induce apoptosis of DN T cells which normally specifically lyse auto-reactive B cells loaded with self-antigens. As this effector function is dependent on MHC-II recognition, MARCH1 could prevent B cells from being targeted by DN T cells. However, in the presence of IL-10, apoptosis of DN T cells would prevent the elimination of activated B cells that express more MHC-II molecules due to MARCH1 down-regulation. If the activity of IL-10 on the deletion of auto-immune B cells and/or the development of Tregs is mediated, at least in part, through the down-regulation of MARCH1 remains to be determined.
The interaction between B and T cells is required for entry into germinal centers, thus a constant low amount of antigenic presentation is required for GC formation. The effectiveness of GC creation by individual B cells is dependent upon help provided by T follicular helper (TFH) cells and antigen presentation by these B cells. As shown by Draghi et al. [33] , germinal center entry by B cells can be modulated by antigen presentation players. They elegantly demonstrated that H2-O, a biochemical inhibitor of peptide loading onto MHC-II, acted as a repressor of GC entry, thus, highlighting the importance of antigen presentation for this selective process. Moreover they showed that upon entry into germinal centers, B cells significantly decrease their amount of H2-O proteins. We hypothesize that MARCH1, because of its ability to modulate antigen presentation, and making a parallel with H2-O, could act as a germinal center guardian. MARCH1-expressing follicular B cells have reduced antigen presenting abilities and thus are restrained from GC entry. H2-O indirectly selects for higher affinity peptides to be presented and thus creates a selective pressure for specific antigens to be mounted on MHC-II in order for B cells to enter GCs. While H2-O modulates the quality of presented peptides, MARCH1 is a non-qualitative antigen presentation repressor. MARCH1 relocates MHC-II molecules, disregarding its antigen load, leading to the accumulation and degradation of the MHC-IIpeptide complex in lysosomes. Thus, only highly responsive follicular B cells that previously increased their MHC-II pool may effectively prime TFH cells and eventually take place in the germinal center reaction. These results suggest that MARCH1 may set a threshold for germinal center entry in the spleen by selecting B cell clones that have optimal antigen presenting abilities.
9. CONCLUSION
There can be little doubt that MARCH1 is a master regulator of MHC class II antigen presentation at the post-translational level. It is expressed in APCs and has been found to be repressed upon immune activation. The biological relevance of MARCH1 is better understandable if a parallel with the anti-inflammatory activity of IL-10, its only known inducer, is kept in mind. IL-10 is a rather late cytokine, usually being produced after the pro-inflammatory mediators. Thus MARCH1 would probably have a special significance in limiting and preventing an excessive immune response. Its first function is probably highlighted by its ability to specifically target MHC-II and CD86 molecules, leading to an impaired antigen presentation, and T cell activation, one of the main modes of action of IL-10. What strikes us is that some cells, mainly immature dendritic cells, plasmacytoid dendritic cells and B cells, maintain a constitutive MARCH1 expression. We can hypothesize that MARCH1 could be expressed to maintain a constant turnover of various MHC-II-peptide pools for immune sampling. But also, MARCH1 could have a role as to limit the amount of MHC-II on the surface of APCs for specific purposes, such as tolerization. For instance, throughout B cell maturation, the amount of MHC-II molecules could dictate the fate of B cells, whether they would go on to full maturation or plain anergy. Also, through its action on Fas and the transferrin receptor, MARCH1 could alter the fate of infected macrophages. By preventing their apoptosis, MARCH1 could increase survival of infected cell and the formation of a persistent pathogen pool. This possibility is especially striking considering that many intracellular pathogens such as mycobacteria and viruses either lead to a robust production of IL-10 or even encode their own IL-10 homologue. In this respect, it will be interesting to assess the effect of the different members of the IL-10 cytokine family on MARCH1 expression considering that they don’t use the same receptors but they signal through the same cascade of mediators, namely STAT1 and STAT3 [34]. The knowledge regarding MARCH1 effects forces us to think about modulation of MARCH1 activity as a potential therapy for chronic infections (by decreasing MARCH1 activity) or chronic inflammatory disease (by increasing MARCH1 activity). Moreover, MARCH1 has been recently shown to dimerize and auto-ubiquitinate in order to regulate its own activity [35] . Future experiments should look at the potential implication of MARCH1 auto-regulation on its role in immune suppression. We could hope to dissociate the effect of MARCH1 from the bulk of IL-10 effects for ultimate therapy designs. The IL-10 knock-out (IL-10-/-) mice develop a chronic enterocolitis similar to human inflammatory bowel disease. Clinical symptoms are loss of bodyweight, hunchback and piloerection, diarrhea, blood stools, and consequently anaemia [36] . It would be interesting to compare the MARCH1-KO mice to see if these effects are MARCH1-mediated. Finally, it is fascinating to ponder on the opposing effect of IL-10 on MARCH1 expression between different cell types. As such, a systemic IL-10 treatment would increase MARCH1 in macrophages, monocytes and dendritic cells but decrease it in B cells. This would in turn lead to a massive B cell activation skew and should be assessed when considering the use of systemic IL-10 instead of localised IL-10 treatment.
10. ACKNOWLEDGEMENTS
We thank Sylvie Lesage for helpful discussions.
REFERENCES
- Coscoy, L. and Ganem, D. (2000) Kaposi’s sarcomaassociated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proceedings of the National Academy of Sciences of USA, 97, 8051-8056. doi:10.1073/pnas.140129797
- Ishido, S., Wang, C., Lee, B.S., Cohen, G.B. and Jung, J.U. (2000) Down-regulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. Journal of Virology, 74, 5300-5309. doi:10.1128/JVI.74.11.5300-5309.2000
- Stevenson, P.G,. Efstathiou, S., Doherty, P.C. and Lehner, P.J. (2000) Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proceedings of the National Academy of Sciences of USA, 97, 8455-8460. doi:10.1073/pnas.150240097
- Bartee, E., Mansouri, M., Hovey Nerenberg, Gouveia, B.T.K. and Fruh K. (2004) Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. Journal of Virology, 78, 1109-1120. doi:10.1128/JVI.78.3.1109-1120.2004
- Goto, E., Ishido, S., Sato, Y., Ohgimoto, S., Ohgimoto, K., Nagano-Fujii, M., et al. (2003) c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. Journal of Biological Chemistry, 278, 14657-14658. doi:10.1074/jbc.M211285200
- Moore, K.W., de Waal, M.R., Coffman, R.L. and O’Garra, A. (2001) Interleukin-10 and the interleukin-10 receptor. Annual Review of Immunology, 19, 683-765. doi:10.1146/annurev.immunol.19.1.683
- Pestka, S., Krause, C.D., Sarkar, D., Walter, M.R., Shi, Y. and Fisher, P.B. (2004) Interleukin-10 and related cytokines and receptors. Annual Review of Immunology, 22, 929-979. doi:10.1146/annurev.immunol.22.012703.104622
- de Waal, M.R., Abrams, J., Bennett, B., Figdor, C.G. and De Vries, J.E. (1991) Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. Journal of Experimental Medicine, 174, 1209-1220. doi:10.1084/jem.174.5.1209
- Fiorentino, D.F., Zlotnik, A., Mosmann, T.R., Howard, M. and O’Garra, A. (1991) IL-10 inhibits cytokine production by activated macrophages. Journal of Immunology, 147, 3815-3822.
- Gruber, M.F., Williams, C.C. and Gerrard, T.L. (1994) Macrophage-colony-stimulating factor expression by antiCD45 stimulated human monocytes is transcriptionally up-regulated by IL-1 beta and inhibited by IL-4 and IL-10. Journal of Immunology, 152, 1354-1361.
- Murray, P.J. (2006) Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Current Opinion in Pharmacology, 6, 379-386. doi:10.1016/j.coph.2006.01.010
- Berkman, N., John, M., Roesems, G., Jose, P.J., Barnes, P.J. and Chung, K.F. (1995) Inhibition of macrophage inflammatory protein-1 alpha expression by IL-10. Differential sensitivities in human blood monocytes and alveolar macrophages. Journal of Immunology, 155, 4412- 4418.
- Kopydlowski, K.M., Salkowski, C.A., Cody, M.J., van R.N., Major, J., Hamilton, T.A., et al. (1999) Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. Journal of Immunology, 163, 1537-1544.
- Matsuki, Y., Ohmura-Hoshino, M., Goto, E., Aoki, M., Mito-Yoshida, M., Uematsu, M., et al. (2007) Novel regulation of MHC class II function in B cells. EMBO Journal, 26, 846-854. doi:10.1038/sj.emboj.7601556
- Bartee, E., Eyster, C.A., Viswanathan, K., Mansouri, M., Donaldson, J.G. and Fruh, K. (2010) Membrane-Associated RING-CH proteins associate with Bap31 and target CD81 and CD44 to lysosomes. PLoS One, 5, e15132. doi:10.1371/journal.pone.0015132
- Thibodeau, J., Bourgeois-Daigneault, M.C., Huppe, G., Tremblay, J., Aumont, A., Houde, M., et al. (2008) Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. European Journal of Immunology, 38, 1225-1230. doi:10.1002/eji.200737902
- Galbas, T., Steimle, V., Lapointe, R., Ishido, S. and Thibodeau, J. (2012) MARCH1 down-regulation in IL-10- activated B cells increases MHC class II expression. Cytokine, 59, 27-30. doi:10.1016/j.cyto.2012.03.015
- Hunt, D., Wilson, J.E., Weih, K.A., Ishido, S., Harton, J.A., Roche, P.A., et al. (2012) Francisella tularensis elicits IL-10 via a PGE(2)-inducible factor, to drive macrophage MARCH1 expression and class II downregulation. PLoS ONE, 7, e37330. doi:10.1371/journal.pone.0037330
- Corcoran, K., Jabbour, M., Bhagwandin, C., Deymier, M.J., Theisen, D.L. and Lybarger, L. (2011) Ubiquitinmediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1). Journal of Biological Chemistry, 286, 37168- 37180. doi:10.1074/jbc.M110.204040
- Jabbour, M., Campbell, E.M., Fares, H. and Lybarger, L. (2009) Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. Journal of Immunology, 183, 6500-6512. doi:10.4049/jimmunol.0901521
- Tze, L.E., Horikawa, K., Domaschenz, H., Howard, D.R., Roots, C.M., Rigby, R.J., et al. (2011) CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10- driven MARCH1-mediated ubiquitination and degradation. Journal of Experimental Medicine, 208, 149-165. doi:10.1084/jem.20092203
- Wolk, K., Witte, E., Reineke, U., Witte, K., Friedrich, M., Sterry, W., et al. (2005) Is there an interaction between interleukin-10 and interleukin-22? Genes & Immunity, 6, 8-18.
- Kunz, S., Wolk, K., Witte, E., Witte, K., Doecke. W.D., Volk, H.D., et al. (2006) Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Experimental Dermatology, 15, 991-1004. doi:10.1111/j.1600-0625.2006.00516.x
- Kitamura, H., Kamon, H., Sawa, S., Park, S.J., Katunuma, N., Ishihara, K., et al. (2005) IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity, 23, 491- 502. doi:10.1016/j.immuni.2005.09.010
- Koppelman, B., Neefjes, J.J., De Vries, J.E. and Malefyt, R.D. (1997) Interleukin-10 down-regulates MHC class II peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity, 7, 861-871. doi:10.1016/S1074-7613(00)80404-5
- De Gassart, A., Camosseto, V., Thibodeau, J., Ceppi, M., Catalan, N., Pierre, P., et al. (2008) MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proceedings of the National Academy of Sciences of USA, 105, 3491-3496. doi:10.1073/pnas.0708874105
- Young, L.J., Wilson, N.S., Schnorrer, P., Proietto, A., Ten, B.T., Matsuki, Y., et al. (2008) Differential MHC class II synthesis and ubiquitination confers distinct antigenpresenting properties on conventional and plasmacytoid dendritic cells. Nature Immunology, 9, 1244-1252. doi:10.1038/ni.1665
- Ten, B.T., de Graaff, A., van’t Veld, E.M., Wauben, M.H., Stoorvogel, W. and Wubbolts, R. (2010) Trafficking of MHC class II in dendritic cells is dependent on but not regulated by degradation of its associated invariant chain. Traffic, 11, 324-331. doi:10.1111/j.1600-0854.2009.01024.x
- Yusof, W.N., Nagaratnam, M., Koh, C.L., Puthucheary, S. and Pang, T. (1993) Release of prostaglandin E2 by human mononuclear cells exposed to heat-killed Salmonella typhi. Microbiology and Immunology, 37, 667-670.
- Rangel, M.J., Estrada, G.I,, De La Luz, G.H., Aguilar, L.D., Marquez, R. and Hernandez, P.R. (2002) The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology, 106, 257- 266.
- N’Guessan, P.D., Etouem, M.O., Schmeck, B., Hocke, A.C., Scharf, S., Vardarova, K., et al. (2007) Legionella pneumophila-induced PKCalpha-, MAPK-, and NF-kappaB-dependent COX-2 expression in human lung epithetlium. American Journal of Physiology: Lung Cellular and Molecular, 292, L267-L277. doi:10.1152/ajplung.00100.2006
- Hillhouse, E.E., Beauchamp, C., Chabot-Roy, G., Dugas, V. and Lesage, S. (2010) Interleukin-10 limits the expansion of immunoregulatory CD4-CD8-T cells in autoimmune-prone non-obese diabetic mice. Immunology & Cell Biology, 88, 771-780. doi:10.1038/icb.2010.84
- Draghi, N.A. and Denzin, L.K.. (2010) H2O, a MHC class II-like protein, sets a threshold for B-cell entry into germinal centers. Proceedings of the National Academy of Sciences of USA, 107, 16607-16612. doi:10.1073/pnas.1004664107
- Hofmann, S.R., Rosen-Wolff, A., Tsokos, G.C. and Hedrich, C.M. (2012) Biological properties and regulation of IL-10 related cytokines and their contribution to autoimmune disease and tissue injury. Clinical Immunology, 143, 116-127. doi:10.1016/j.clim.2012.02.005
- Bourgeois-Daigneault, M.C. and Thibodeau, J. (2012) Autoregulation of MARCH1 expression by dimerization and autoubiquitination. Journal of Immunology, 188, 4959- 4970. doi:10.4049/jimmunol.1102708
- Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. and Muller, W. (1993) Interleukin-10-deficient mice develop chronic enterocolitis. Cell, 75, 263-274. doi:10.1016/0092-8674(93)80068-P
NOTES
*Supported by a grant from the Canadian Institutes for Health Research (MOP 36355).