Open Journal of Ecology
Vol.4 No.8(2014), Article ID:46468,4 pages DOI:10.4236/oje.2014.48040
Evaluation of Molecular Techniques in Characterization of Deep Terrestrial Biosphere
Malin Bomberg1*, Mari Nyyssönen1, Aura Nousiainen1, Jenni Hultman2, Lars Paulin3, Petri Auvinen3, Merja Itävaara1
1VTT Technical Research Centre of Finland, Espoo, Finland
2Faculty of Veterinary Medicine, University of Helsinki, Helsinki, Finland
3Institute of Biotechnology, University of Helsinki, Helsinki, Finland
Email: *malin.bomberg@vtt.fi
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 7 March 2014; revised 7 April 2014; accepted 15 April 2014
ABSTRACT
A suite of molecular methods targeting 16S rRNA genes (i.e., DGGE, clone and high-throughput [HTP] amplicon library sequencing) was used to profile the microbial communities in deep Fennoscandian crystalline bedrock fracture fluids. Variation among bacterial 16S rRNA genes was examined with two commonly used primer pairs: P1/P2 and U968f/U1401r. DGGE using U968f/ U1401r mostly detected β-, γ-proteobacteria and Firmicutes, while P1/P2 primers additionally detected other proteobacterial clades and candidate divisions. However, in combination with clone libraries the U968f/U1401r primers detected a higher bacterial diversity than DGGE alone. HTP amplicon sequencing with P1/P2 revealed an abundance of the DGGE bacterial groups as well as many other bacterial taxa likely representing minor components of these communities. Archaeal diversity was investigated via DGGE or HTP amplicon sequencingusing primers A344F/ 519RP. The majority of archaea detected with HTP amplicon sequencing belonged to uncultured Thermoplasmatales and Pendant 33/DHVE3, 4, 6 groups. DGGE of the same samples detected mostly SAGMEG and Methanosarcinales archaea, but almost none of those were revealed by HTP amplicon sequencing. Overall, our results show that the inferred diversity and composition of microbial communities in deep fracture fluids is highly dependent on analytical technique and that the method should be carefully selected with this in mind.
Keywords:DGGE, 16S rRNA Gene, Amplicon Library, Sequencing, Deep Subsurface

1. Introduction
Due to the difficulty of obtaining samples, microbial communities in deep terrestrial subsurface environments are some of the least studied in the world. In deep fracture fluids of Fennoscandian crystalline bedrock, microbes typically occur at densities of 103 to 106 cells∙ml−1 [1] -[5] . Cell numbers are the highest in groundwater layers near the surface but rapidly decrease and experience a tenfold drop at 100 m and up to a 100-fold decrease at 500 m [1] [2] [4] [6] . Despite the relatively low concentration of cells, taxonomic and functional diversity of microbial communities in the deep terrestrial subsurface is believed to be high [1] -[6] .
According to culture dependent techniques such as Most Probable Number (MPN), microbial communities in the anaerobic deep subsurface of the Fennoscandian Shield reduce iron, sulphate, manganese, nitrate and oxygen and produce acetate and methane [7] [8] . While this information is important for estimating the functional potential of these communities, identification of the microbes detected by MPN remains challenging due to the difficulty of in vitro culture. It has been estimated that only 1% - 5% of the microbial cells in an environmental sample can be successfully cultured on laboratory media. Thus, in vitro cultivation techniques offer a very incomplete picture of microbial communities [9] [10] and it is unclear if and how artificial conditions affect the metabolism or functional activity of isolated microbes [11] . Given that microbial communities in the deep subsurface may comprise thousands of species, in vitro cultivation is an impractical option [12] .
Analysis of phylogenetic marker genes using molecular methods offers a way to detect and identify the microbial taxa present in fracture fluids and can thereby be used to generate estimates of community diversity and composition [4] [13] . Gene for ribosomal 16S subunit (16S rRNA) is commonly used as a taxonomic marker because it is highly conserved, found in all bacteria and archaea, and a rich 16S rRNA database has been generated by systematists and evolutionary biologists against which sequences from fracture fluids can be compared and identified [14] [15] .
Molecular techniques such as denaturing gradient gel electrophoresis (DGGE) and clone library construction [2] -[5] [13] most frequently detect Actinobacteria, Firmicutes and various Proteobacteria in deep subsurface fracture fluids of the Fennoscandian Shield. Furthermore, Bacteroidetes, Green non-sulphur bacteria, Candidate Divisions JS1, TM7, OP3, OP8, OP10, OP11, Deinococcus/Thermus, Verrucomicrobia, Bacilli and even Cyanobacteria are often detected in terrestrial subsurface, although in lower abundance [8] [16] -[18] . To date, archaeal groups identified in Fennoscandian deep subsurface belong to uncultured Thermoplasmatales and methanogenic archaea [2] -[5] [13] [19] , and novel archaeal phylotypes recently detected in deep marine and terrestrial subsurface environments have also been found [20] .
Recent advances in high-throughput (HTP) sequencing technologies have further enhanced the study of microbial communities in deep subsurface environments. HTP sequencing is becoming increasingly popular due to improved cost-efficiency, robust techniques, and the possibility for greater coverage and resolution of the microbial community. With higher detection sensitivity, it has become possible to obtain more complete estimates of community composition and infer relationships between microbes and their geochemical surroundings [5] . HTP amplicon library sequencing techniques can also detect microbes that occur infrequently or at low abundances in environmental samples much better than other methods [5] [21] [22] .
However, molecular techniques, like all methods, are limited or biased according to their design and sophistication. For example, selection of PCR primers used to amplify 16S rRNA genes has a tremendous impact on the microbial diversity detected [23] -[25] . In addition, the method used to separate and identify PCR amplicons is important. DGGE is widely used in molecular ecology to rapidly screen microbial communities in different habitats (e.g., [3] [26] [27] ). However, PCR-DGGE is believed to be a rather coarse technique that can over-emphasize abundant groups while failing to detect taxa comprising <1% of microbial cells in a sample [27] -[30] . Furthermore, different techniques can be biased with respect to the detection of certain taxa, e.g., the SAR11 cluster of aquatic α-proteobacteria has been shown by clone library screening and fluorescent in situ hybridization (FISH) to be the dominant group of bacteria in most marine ecosystems, but they are only rarely detected by DGGE [31] [32] . DGGE can also underestimate microbial diversity in that different amplicons with similar migration properties will not be resolved on the gel [33] -[35] . On the other hand, while sequences obtained from clone libraries are generally of better quality than those amplified from DGGE bands, the technique is biased towards gene fragments that are better compatible with the cloning system and leaves “difficult” fragments undetected [36] [37] . The HTP sequencing of amplicon libraries yield a much higher number of sequences per sample than what is possible for DGGE or practical for clone libraries. However, although the read length for amplicon library sequencing is increasing, it remains relatively short and can consequently yield imprecise or uncertain phylogenetic resolution. In addition, the higher rate of sequencing error in comparison to Sanger sequencing can mislead estimates of species richness and community diversity.
In this study, we used DGGE, clone libraries and HTP amplicon sequencing to investigate microbial (i.e., bacteria and archaea) community composition in terrestrial groundwater 78 - 546 m deep collected from Olkiluoto Island, Finland. We aimed to determine the influence of classical (DGGE and clone libraries) and novel molecular tools (amplicon library sequencing) on the characterization of microbial communities and population diversity. We also investigated the effect of primer selection on the detection of bacterial groups in groundwater samples.
2. Materials and Methods
Sample preparation. Groundwater samples were obtained from several drillholes on Olkiluoto Island, Finland (61˚14'13"N, 21˚26'27"E). The site is described in detail in [4] . Samples were drawn at depths from 78 - 546 m below ground surface (mbgs) (Table 1). All samples except ONK-PVA3/83m were obtained with devices that
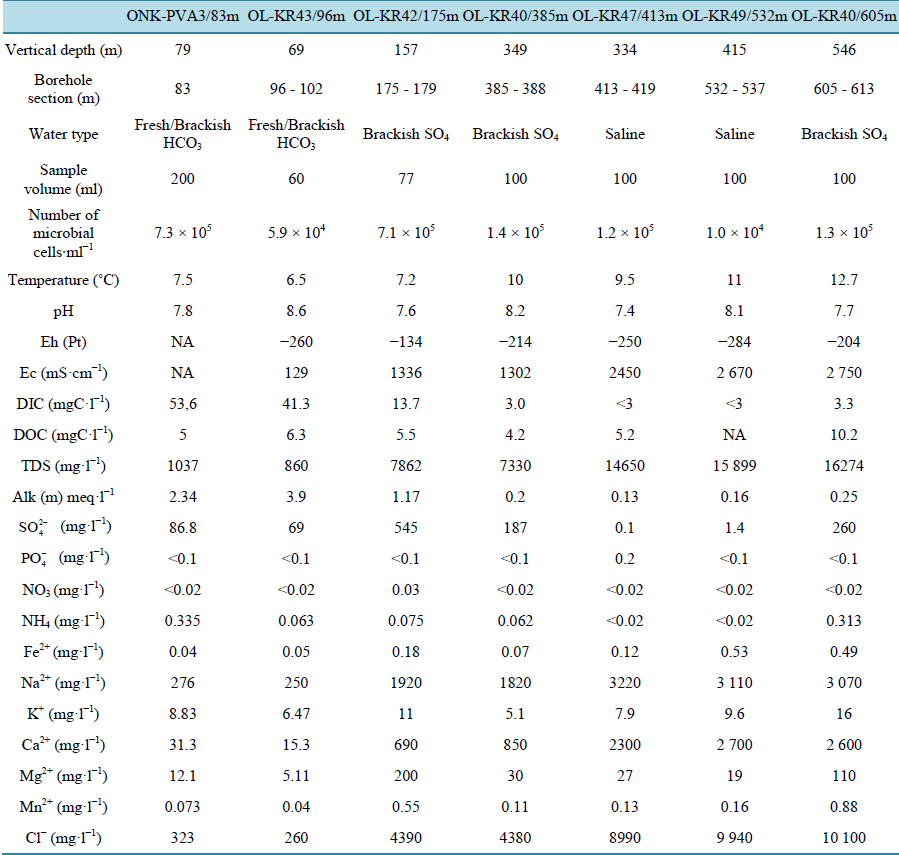
Table 1. Geochemical and physical characteristics of the samples (courtesy of Posiva Oy).
enable samples to be retrieved at in situ pressure (as described in [4] [6] ). Sample ONK-PVA3/83m originated from a free-flowing drillhole in the ONKALO tunnel (mbsl) (http://www.posiva.fi/en/final_disposal/onkalo#.U329rC-D2Go) and was collected anaerobically and aseptically into a sterile and anaerobic headspace vial (vol. 120 ml) sealed with a butyl rubber septum and aluminum crimp cap (Bellco Glass, NJ, USA). Groundwater was drawn into the glass vial via airtight poly-acetate tubing attached to a sterile hypodermic needle.
The concentration of biomass and protocol for DNA extraction was as described in [4] . In short, the microbial cells from each water sample were collected on 0.2 µm pore size nitrocellulose acetate filters (Corning Inc., NY, USA) by vacuum suction. Prior to DNA extraction the filters were cut to approximately 2 × 2 mm pieces in a laminar fume hood with sterile scalpels. The DNA was extracted with the Power Soil DNA extraction kit (MoBio Laboratories Inc., Solana Beach, CA, USA). The microbial biomass collected on the filter pieces was lysed by bead beating with a Ribolyser (Hybaid Ltd., Ashford, UK) device for 30 s at 6 m·s−1 after which the DNA extraction proceeded according to the manufacturer’s instructions.
The samples were subsequently submitted to different molecular biological methods (Table 2).
PCR. Two different primer pairs were used for amplification of bacterial 16S rRNA gene fragments. Primer pair P1 and P2 [26] was used to amplify a 193bp long fragment covering the V3 variable region of the bacterial 16S rRNA gene, while primers U968f and U1401r [38] were used to amplify a 473 bp long fragment covering the V6-8 regions. For DGGE analysis, the forward primers were modified with a GC-clamp at their 5’ end [26] [38] .
PCR amplification was carried out in 50 µl reaction volumes containing 1 × Dynazyme® II buffer (10 m MTris-HCl, pH 8.8, 1.5 mM MgCl2, 50 mM KCl and 1% Triton-X-100), 0.2 mM each deoxynucleoside triphosphate, 20 or 50 pmol of each primer for primer pairs P1/P2 and U968f/U1401r, respectively, and 1U Dynazyme® II DNA polymerase (Finnzymes, Espoo, Finland). For primer pair P1/P2, 0.5 µl formamide was added to each PCR, and 10 µg BSA was added for PCRs with U968f/U1401r. 2 µl undiluted DNA extract (approximately 1 - 3 ng DNA µl−1) was used as template. The PCR program for P1/P2 consisted of an initial denaturation step of 5 min at 94˚C followed by 35 cycles of 1 min at 94˚C, 1 min at 55˚C and 1 min at 72˚C, followed by a final elongation at 72˚C for 5 min. PCR program for U968f/U1401r consisted of 5 min initial denaturation at 94˚C followed by 30 cycles of 30 s at 94˚C, 20 s at 55˚C and 40 s at 72˚C and final elongation at 72˚C for 7 min.
The archaeal 16S rRNA gene fragments were produced by a nested PCR [4] . An 806 bp fragment of archaeal 16S rRNA gene was first amplified with A109f and Arch915R [39] [40] . The reaction mixture was as described above. The PCR program consisted of an initial denaturation of 5 min at 95˚C followed by 30 cycles of 1 min at 95˚C, 1 min at 54˚C and 2 min at 72˚C. In the second PCR, a 227 bp fragment was produced with A344F-GC (GC-clamp at the 5’ end) and 519RP [40] [41] flanking the V3 hypervariable region of 16S rRNA. The reaction mixture was as in the first PCR except that 2 µl of purified amplification product was used as template. The

Table 2. Assays used for each sample.
*Not sequenced.
PCR program was 5 min at 95˚C, 30 cycles of 1 min at 95˚C, 1 min at 50˚C, 1 min at 72˚C followed by 10 min at 72˚C.
PCR products were visualized via electrophoresis in a 1.2% agarose gel stained with EtBr (0.2 µg·ml−1). The gel was run in 1× TAE buffer for 1 h at 150 V and subsequently photographed in UV light with the Bio-Rad GelDoc imager (Bio-Rad, CA, USA).
DGGE. Bacterial and archaeal 16S rRNA amplicons were resolved by denaturing gradient gel electrophoresis (DGGE) in an 8% acrylamide/bis (37.5:1) gel. The denaturing gradient for amplicons obtained with primer pair P1/P2 and the archaeal primers A344FGC/519RP was 20% - 65%, and 38% - 60% for amplicons obtained with U968f/U1401r. Electrophoresis was performed at 60˚C for 19 h and 65 V for P1/P2 products, 16 h and 85 V for U968f/U1401r products, and 18 h and 65 V for archaeal products. Following electrophoresis, DGGE gels were stained with Sybr Green II (Lonza, Switzerland) according to the manufacturer’s recommendations and visual-ized under UV light with a Bio-Rad GelDoc imager. Bands were cut from gels with sterile disposable plastic Pasteur pipettes. Each excised band was incubated in 20 µl ultra clean water overnight at 4˚C in order to elute the DNA. Eluted products were reamplified with the same primers and conditions as in the original PCRs and resulting amplicons were purified with the Qiaquick PCR Purification kit (Qiagen, Germany). Sequencing was performed from both ends of the amplicon with the BigDye® Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, California, USA) in the ABI Prism 310 Genetic Analyzer (Applied Biosystems, California, USA).
Clone libraries. Bacterial U968f/U1401r amplicons were used to construct clone libraries. Amplicons were purified with the QIAquick PCR purification kit (Qiagen, Germany) according to the manufacturer’s protocol. The TOPO-TA cloning kit (Life Technologies, UK) was used for ligation and chemically competent DH5α Escherichia coli cells were used for transformation. Cloning was performed according to the manufacturer’s instructions. Two parallel LB plates containing 50 µg·ml−1 kanamycin were prepared for each cloning reaction and 40 µl X-gal (40 mg·ml−1) was spread on the surface. Plates were pre-incubated for 1 h at 37˚C before 10 and 50 µl aliquots were taken from each reaction and plated. Plates were incubated overnight at 37˚C. Between 128 and 229 white clones from each reaction were checked for insertion by colony PCR. Bacterial cells from individual colonies were suspended in 50 µl sterile molecular grade water. Of this suspension, 2.5 µl was used as template in 10 µl PCR reactions. PCR reactions consisted of 1 × reaction buffer, 0.25 m MdNTP, 10 pmol each of vector specific primers M13F and M13R and 0.25 U Dynazyme II. The amplification program consisted of an initial cell lysis and denaturation step of 5 min at 94˚C, followed by 35 cycles of 1 min at 94˚C, 1 min at 54˚C and 3 min at 72˚C, and a final extension step of 10 min at 72˚C. The colony PCR products were checked on a 1.2% agarose gel as described above. Ninety-five positive clones were randomly selected for growth in Luria broth containing 50 µg∙ml−1 kanamycin and plasmids were extracted using the QIAprep Spin Miniprep Kit (Qiagen). The clones were sequenced using the M13 primers and BigDye® Terminator v3.1 cycle sequencing kit in an ABI Prism 310 Genetic Analyzer.
Amplicon libraries. Amplicon libraries for HTP sequencing were prepared by PCR from OL-KR40/385m, OL-KR49/532m and OL-KR40/609m. For bacterial 16S rRNA gene libraries, P1/P2 with adapter and barcode sequence modifications at their 5’ end were used in PCR (Table 2). For archaeal 16S rRNA gene libraries, a nested PCR approach was used with A109f and Arch915R [39] [40] in the first round after which A344FGC/ 519RP modified with 5’ adapter and barcode sequences were used for the production of the amplicon library.
The amplicon libraries for bacterial 16S rRNA were produced using the Dynazyme II polymerase (Finnzymes, Finland) in 50 µl reaction volumes as described above. For the archaeal 16S rRNA gene amplicon libraries were generated with 50 µl reactions consisting of 1 × GC reaction buffer, 0.2 m MdNTP, 2.5 pmol of each primer and 1 unit of Phusion polymerase (Finnzymes, Finland). As template, 2 µl first round PCR product was used. The PCR program for amplicon libraries consisted of an initial denaturation at 98˚C for 30 s followed by 35 cycles of 98˚C for 10 s, 55˚C for 15 s and 72˚C for 15 s and a final extension of 5 min at 72˚C. Amplicon libraries were sequenced at the Institute of Biotechnology using Roche 454 FLX technology.
Community analyses and phylogeny. Sequences obtained from clone and amplicon libraries were analyzed with Mothur [42] . First, the adapter and barcode sequences from the amplicon library sequences as well as the primer sequences from both clone and amplicon library sequences were removed. Sequences were then aligned to reference alignments composed of bacterial (14,956) or archaeal (2297) 16S rRNA gene sequences obtained from the Silva database [43] . Bacterial amplicon library sequences were at least 95 bp long or 320 bp long for the clone library sequences. Archaeal amplicon library sequences were at least 90 bp long. All sequences that aligned to the reference were included in subsequent analyses. Sequences that did not align well (e.g., due to poor sequence quality) were discarded at this point.
Sequences at least 90% identical were combined and treated as Operational Taxonomic Units (OTUs). Representativeness of sequencing was tested by rarefaction analysis of the resulting OTUs. The estimated Chao [44] and ACE [45] species richness, rarefaction analyses as well as the Shannon [46] and Simpson [47] diversity indices were analyzed for archaea and bacteria separately for each sample with Mothur [43] .
Representative sequences of each OTU as well as the sequences obtained from DGGE were subjected to phylogenetic analysis. Sequence sets obtained with different primer pairs were analyzed separately. Sequences were aligned with appropriate references and the most similar sequences were identified from the NCBI database using the blastn tool as applied in Geneious Pro (Biomatters Ltd, New Zealand). Sequences were aligned in Geneious Pro using ClustalW and then checked and edited by eye before being trimmed to a region represented by all OTUs. The trimmed alignment was then realigned using Muscle with default settings included in Geneious Pro. Maximum likelihood trees were calculated using the PhyML algorithm [48] included in Geneious Pro. Support values for inferred branches were calculated with 1000 bootstrap pseudoreplicates.
In silico testing of primer coverage. The taxonomic coverage of the primers used in this study was tested against the 16S rRNA gene sequence database of the Ribosomal Database Project (RDP) release 11 [49] using the Probe Match tool. The search was restricted to the area covered by the primers, i.e. positions 341-534 for primers P1/P2, 960-1450 for U986f/U1401r, 109-934 for A109f/A934r and 344-354 for A344f/A519r, E. coli numbering. In addition, the primers were compared to the NCBI genome database using blastn.
Statistical analyses. Pearson’s linear r correlation between the presence or absence of bacterial taxa in correlation to community profiling technique (DGGE, clone libraries, HTP sequencing), primer pair, number of identified sequences and the geochemical parameters were calculated using PAST v. 3.0 [50] in order to determine which of these parameters had the greatest effect on the detected taxa.
Accession numbers. All new sequences have been submitted to the European Nucleotide Archive (ENA) Sequence Read Archive (SRA) (http://www.ebi.ac.uk/ena) under accession numbers HG976872-HG976887 for bacterial 16S rRNA gene DGGE fragments with primers P1/P2, HG976626-HG976871 bacterial 16S rRNA gene clone libraries and PRJEB6086 bacterial and archaeal 16S rRNA gene amplicon libraries (http://www.ebi.ac.uk/ena/data/view/PRJEB6086). Bacterial 16S rRNA gene sequences obtained with DGGE with primers U968f/U1401r and archaeal DGGE band sequences have been published before in [4] .
3. Results
DGGE. Bacteria. DGGE analysis of bacterial communities was performed with two different primer sets described above. U968f/U1401r [38] amplified 5 - 18 DGGE bands in each sample (Figure 1, Table 3(a), Table 4(a)), 2 - 7 of which were successfully sequenced per sample. P1/P2 [26] amplified 11 - 19 products per sample (Figure 1, Table 3(a), Table 4(a)), 3 - 10 of which were sequenced but only few of these sequences were of sufficient quality for subsequent analysis. Richness estimates were not calculated for DGGE results because of relatively low number of bands and the difficulty of determining the abundance of a given DGGE band sequence.
Most of the bacterial 16S rRNA gene sequences obtained from OL-KR40/385m and OL-KR47/413 m with U968f/U1401r belonged to γ-proteobacterial lineages (Figure 1, Figure S1). γ-proteobacteria sequences were detected with P1/P2, in addition to α- and β-proteobacteria and Firmicutes (Figure 1). Sequences obtained from OL-KR49/532m with P1/P2 were diverse and belonged to α-, β-, γ-proteobacteria, Clostridia, and low G+C Firmicutes (Figure 1, Figure S2(a), Figure S2 (b), Table 3(a)), while primers U968f/U1401r again produced only γ-proteobacterial sequences. In sample ONK-PVA3/83m, 16S rRNA fragments amplified with P1/P2 belonged to Firmicutes and δ-proteobacteria while U968f/U1401r detected Candidate Division OD1 and γ-proteobacteria. Sample OL-KR43/96m contained γ- and ε-proteobacteria and Firmicutes similar to Clostridia when amplified with U968f/U1401r. OL-KR40/609m was found to contain β-proteobacteria, γ-proteobacteria and Clostridia with U968f/U1401r, while primers P1/P2 detected α- and β-proteobacteria, Clostridia and Spirochaetes.
Archaea. The archaeal community in samples OL-KR40/385m, OL-KR49/532m and OL-KR40/609m was characterized using nested PCR with A109f and Arch915R as first-round primers and A344FGC and 519RP in the second round. Between 8 - 23 DGGE bands were detected per sample (Table 3(b), Table 4(b)) and good quality sequence was obtained from 2 - 5 bands per sample. Archaeal sequences belonged to uncultured Thermoplasmatales and SAGMEG/SAGMA1 (OL-KR40/385m), Methanosarcinales and SAGMEG/SAGMA1
Table 3. The (a) bacterial and (b) archaeal diversity obtained by HTP amplicon sequencing or clone library squencing in relation to DGGE. The number indicate relative abundance of sequences for HTP amplicon sequencing and clone libraries, and number of identified DGGE bands for the DGGE.
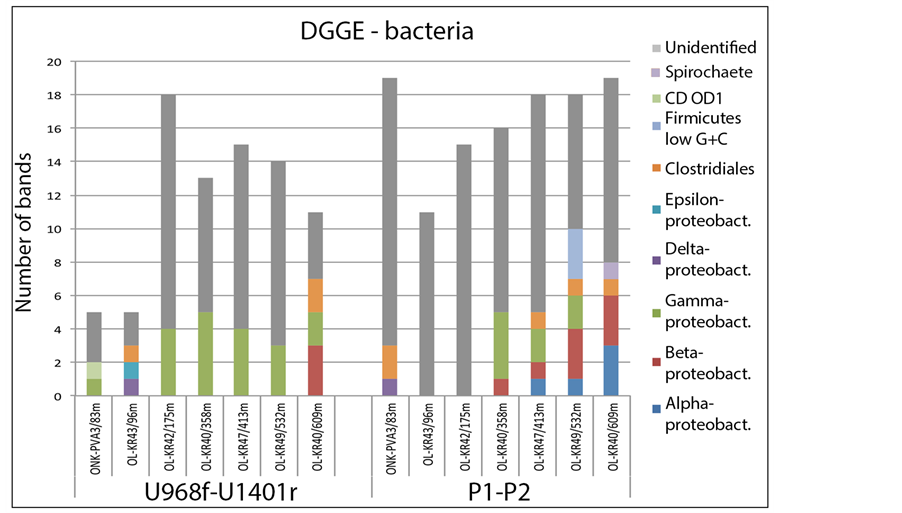
Figure 1. Number and classification of the different DGGE bands obtained with the bacterial 16S rRNA gene specific primers U968f/U1401r and P1/P2.
(OL-KR49/532m) and Pendant 33/DHVE3, 4, 6, Methanosarcinales, uncultured Thermoplasmatales and SAGMEG/SAGMA1 (OL-KR40/609m) (Figure S3, Table 3(b)).
Clone libraries. Clone libraries of the bacterial 16S rRNA genes were constructed from three samples, ONK-PVA3/83m, OL-KR43/96m and OL-KR42/175m with U968f/U1401r. Out of 96 clones chosen for sequencing from each clone library, good quality sequences were obtained from 88, 90 and 66 clones, respectively (Table 3(a), Table 4(a)). These clones belonged to 17 different classes of bacteria and comprised 75 operational taxonomic units (OTUs). The number of different bacterial classes in ONK-PVA3/83m, OL-KR43/96m and OL-KR42/175m were 15, 14 and 11, respectively. According to the Chao estimator, the total estimated number of OTUs was 78, 115 and 79, respectively (Table 3(a)). Together with rarefaction analysis this demonstrated that clone libraries only partially captured the bacterial diversity of the samples (Figure 2). A majority of sequences belonged to the Phylum Proteobacteria (33%, 64% and 45% of clone libraries, respectively) with many sequences affiliating with γ- and δ-proteobacteria. The second largest group was the Firmicutes (23%, 7.3% and 7.7% of sequences, respectively). Less common bacterial groups, such as the Actinomycetes, Chlamydiae, Bacteroidetes, Verrucomicrobia, Chloroflexi, Chlorobi, Nitrospirae, as well as the Candidate Divisions OP3, OD1, OP11 and TM7 were also present in the clone libraries (Figure 1(b)) The total number of OTUs was 45, 36 and 23 in samples ONK-PVA3/83m, OL-KR43/96m and OL-KR42/175m, respectively.
Amplicon Library Sequencing
Bacteria. Bacterial 16S rRNA gene fragments for amplicon library sequencing were amplified with P1/P2 from samples OL-KR40/385m, OL-KR49/532m and OL-KR40/609m (Table 3(b)). The 2781, 228, and 8449 sequences obtained from the respective libraries comprised 60, 12, and 216 OTUs (90% internal sequence homology), respectively (Table 4(b)). Rarefaction analysis indicated that the number of OTUs detected were a good representation of the total species richness in samples OL-KR40/385m and OL-KR49/532m, but not in sample OL-KR40/609m (Table 3(a)) where an order of magnitude fewer sequences were obtained. The total number of OTUs determined by the Chao estimator [44] was 90 (OL-KR40/385m) and 18 (OL-KR49/532m) indicating that at least 66% of species richness was captured in the sequence data (Table 4(b)). A total of 407 OTUs was observed for OL-KR40/609m, indicating that 53% of species richness was detected. The ACE estimator [45] indicated even higher species richness for OL-KR40/385m (104 OTUs) and OL-KR40/609m (567 OTUs), but only 16 OTUs for OL-KR49/532m, from which only few sequences were obtained.
(a) (b)
Table 4. The number of identified sequences, diversity and richness indices of the samples characterized by HTP amplicon library sequencing and clone library sequencing in relation to DGGE of (a) bacteria and (b) archaea.
*% based on sequenced DGGE bands vs. observed DGGE bands.
According to phylogenetic analysis, OTUs belonged to at least 12 classes of bacteria and one archaeal group (Figure S2(a) Figure S2(b), Table 3(a)). 83% of bacterial sequences in OL-KR40/385m belonged to γ-proteobacteria (Table 3(a)). In OL-KR49/532m and OL-KR40/609m the proportion of γ-proteobacteria was only 11% and 24%, and the most abundant bacterial taxa belonged to β-proteobacteria (68% and 41%, respectively) (Figure S2(a)). In OL-KR40/385m, 1.7% of sequences belonged to β-proteobacteria. α-, δ- and ε-proteobacteria sequences comprised 0.3% - 2.7% of the samples obtained from OL-KR40/385m and OL-KR40/609m, and were
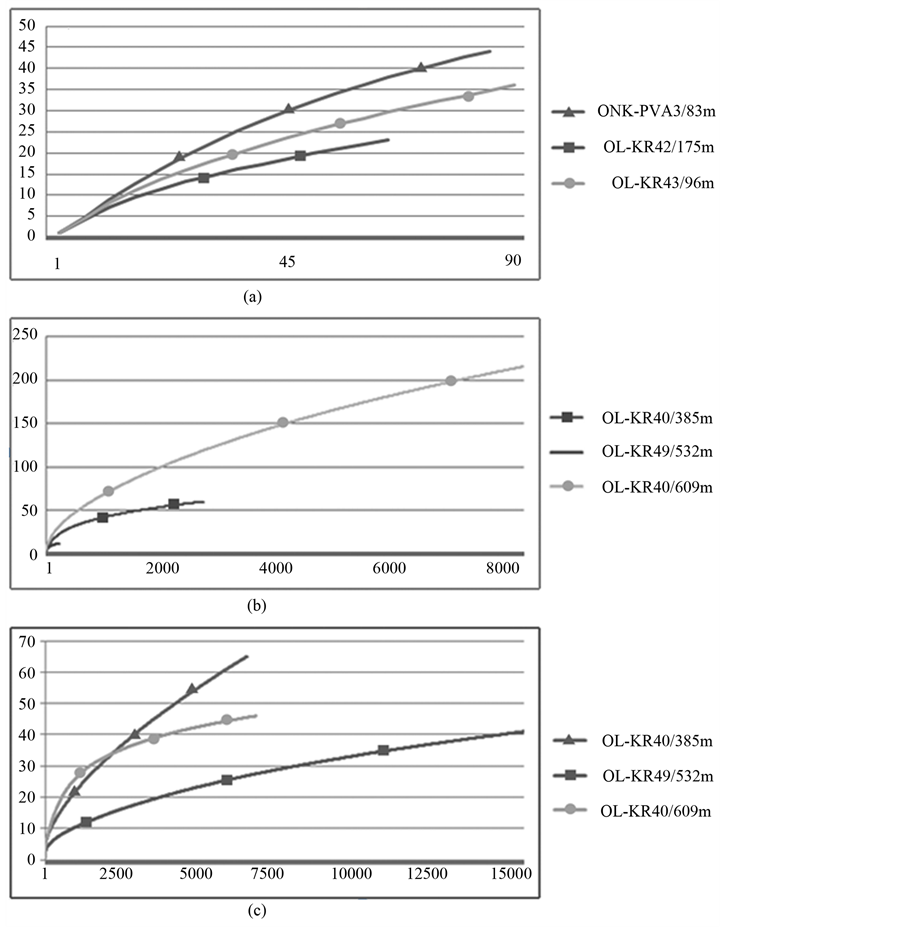
Figure 2. Rarefaction curves on the sequences obtained from (a) the clone libraries of bacterial 16S rRNA genes with primers U968f/U1401r, (b) the amplicon library sequencing of bacterial 16S rRNA genes with primers P1/P2, and (c) the amplicon library sequencing of archaeal 16S rRNA gene fragments. The distance 0.1 was used.
not detected in sample OL-KR49/532m (Table 3(a)). Firmicutes belonging to the Clostridia constituted one of the major groups in OL-KR49/532m (20.2%), but were inferred to represent <1% in OL-KR40/385m and OL-KR40/609m. Between 8% - 26% of sequences in OL-KR40/385m and OL-KR40/609m grouped with Bacteroidetes (Flavobacteria).
Archaea. Amplicon libraries of archaeal 16S rRNA genes were produced from samples OL-KR40/385m, OL-KR49/532m and OL-KR40/609m (Table 3(b)). Between 6577 and 15615 sequences were obtained from each amplicon library (Table 4(b)). The number of archaeal OTUs (90% internal sequence homology) was 65, 41 and 46 in OL-KR40/385m, OL-KR49/532m and OL-KR40/609m, respectively. Rarefaction analysis indicated that the number of detected OTUs only partially represented the total species richness in OL-KR40/385m where the estimated number of OTUs was 147 (Chao) or 304 (ACE) (Table 3(b), Table 4(b)). Better coverage was implied for OL-KR49/532m and OL-KR40/609m, where rarefaction analysis and richness estimators indicated that 56% - 84% of the OTUs present were successfully detected and sequenced (Table 4(b)).
The 127 archaeal OTUs clustered within eight major clades (Table 3(b), Figure S3). The greatest part of the archaeal 16S rRNA gene sequences (52%, 81% and 58% in OL-KR40/385m, OL-KR49/532m and OL-KR40/ 609m, respectively) belonged to uncultured Thermoplasmatales (Table 3(b)). 13.7% of the archaeal community in OL-KR40/385m clustered with cultured Thermoplasma species. Pendant 33/DHVE3, 4, 6 archaea was a major group in OL-KR40/609m (47.3%), but was only minor or absent in the other samples. Methanosarcinales comprised a significant part of the archaeal community in OL-KR49/532m, but was a minor component or missing in other samples. The South African Gold Mine Euryarchaeotal Group (SAGMEG/SAGMA1) was a significant part of archaeal communities in OL-KR40/609m and OL-KR49/532m but absent in OL-KR40/385m. Rare or low abundance archaeal groups belonged to Methanobacterium sp., the Miscellaneous Euryarchaeotic Group (MEG) and the C3 group of uncultured Crenarchaeota.
Coverage of primers. The P1/P2 primer pair covered 81% of the bacterial 16S rRNA gene sequences of the RDP database spanning the predetermined positions of the primer pair when allowing for no mismatches between the primer and target sequences. The primers U968f/U1401r, on the other hand covered less than 10% of the total number of bacterial 16S rRNA gene sequences spanning the predetermined position of the 16S rRNA genes. While P1/P2 matched between 79% - 99% of the 16S rRNA gene sequences of the different proteobacterial classes U968f/U1401r matched only 0.1% of the β-proteobacteria, and none of the other proteobacterial groups. In general, the primers U968f/U1401r matched between 0% - 0.5% of the sequences in the bacterial groups detected by P1/P2. However, without restricting the search to only positions between nucleotides 960-1450 the detected diversity was much greater and included representatives of all proteobacterial classes and representatives of all bacterial classes and phyla detected by P1/P2. By allowing a mismatch in primer U968f in position 9 and restricting the search to positions 960-1450 the detection increased to 15.2% of the bacterial 16S rRNA genes in the RDP, increasing especially the detectability of β-proteobacteia, Clostridia, Chlamydiae and Verrucomicrobia.
The archaeal primers matched 38.7% (A109f/A934r) and 19% (A344f/A519r) of the archaea 16SrRNA gene sequences of the RDP, covering mainly Euryarchaeota and Crenarchaeota.
Statistics. The detection of the different bacterial groups did not correlate with the pair of bacterial primers used. Instead, the detection of Actinomycetes, Chloroflexi, Chlorobi, Candidate Divisions OP3, OP11 and TM7 correlated positively (>0.6, p < 0.01) with the method of detection. These groups were detected from the clone libraries, but not by the corresponding DGGE. The detection of α-proteobacteria, Clostridia, Tenericutes, Erysipelotrichi, Clamydiae and Spitochaete on the other hand corresponded strongly (0.5 - 0.9, p < 0.05) with the number of sequences identified per sample. These bacterial groups were only detected by HTP or clone library sequencing. Of the geochemical parameters measured, Chloroflexi, Chlorobi, Candidate Divisions OD1 and OP11 correlated with DIC (>0.5, p < 0.03), Verrucomicrobia, Chloroflexi, Chlorobi and Candidate Division OP11 corresponded with alkalinity (>0.5, p < 0.05) and Candidate Divisions OD1 correlated with ammonia (>0.5, p < 0.03), however with clearly lower correspondences than shown for the method of detection or number of identified sequences.
4. Discussion
Primers used during PCR amplification of target gene regions and the techniques used to analyze their products can greatly influence the results of microbial community studies (e.g. [25] [51] [52] ). In this study, we evaluated two commonly used primer sets for the amplification of bacterial 16S rRNA genes from deep groundwater samples in three alternative analytical approaches: DGGE, clone libraries and recently developed amplicon libraries for HTP sequencing. Primers P1/P2 gave a much better resolution of bacterial communities than primers U968f/U1401r. While U968f/U1401r in most samples mainly detected γ-proteobacteria in the DGGE analysis, P1/P2 also detected a variety of more rare bacterial lineages, such as Bacteroidetes and α-proteobacteria. But in general, both primer sets amplified 16S rRNA genes from a range of distantly related bacteria when used for clone libraries or HTP amplicon sequencing (Figure 1, Table 3(a)). Although neither primer set had statistically significant effect on the diversity of detected bacterial groups, sequences obtained with U968f/U1401r were generally of higher quality and longer than those obtained with P1/P2. Longer sequences are desired as they provide greater phylogenetic resolution promoting the use of U968f/U1401r for molecular screening of microbial diversity.
DGGE enabled a rapid visualization of differences in community composition among samples. Earlier research of the Fennoscandian deep subsurface has demonstrated its potential for monitoring changes in microbial community composition at different depths over time and for screening samples prior to the construction of clone libraries or metagenomic work [2] [5] [13] . However, despite use of two universal bacteria primer pairs, DGGE appeared to be biased towards Proteobacteria, especially the γ- and β-groups. In fact, most of the bacterial community was not detected by DGGE, regardless of which primer set was used. Groups such as Nitrospirae, Candidate Divisions OD1, OP3, OP11 and TM7, Verrucomicrobia and the Chlorobi-Bacteroidetes group detected in the clone libraries with primers U968f/U1401r were not obtained in the DGGE analysis (Table 3). Similar results have been shown in aquatic environments, e.g., for the SAR11 α-proteobacteria [31] [32] . These microorganisms were not detected by DGGE but were inferred to be the dominant bacteria in the water column by FISH and clone libraries. According to [31] , low primer affinity was the main reason for the low detection of SAR11. The authors also speculated that SAR11 is not readily detected by DGGE due to high microdiversity among similar species or strains that may cause a multitude of faint bands in DGGE that elude detection. This is in accordance with our DGGE results where numerous bands were detected but only a few of which were strong enough to be isolated and reamplified. In addition, many DGGE bands were not successfully sequenced which may bias the DGGE results towards the abundant and readily amplified taxa. Kisand and Vikner [30] also showed that using a broad denaturing gradient (20% - 70%) resulted in so-called multi-domain melting profiles (i.e., smears) for 11 flavobacterial isolates but a defined DGGE band for γ-proteobacteria.
In comparison to DGGE, clone libraries revealed significantly greater bacterial diversity, sequences were of higher quality than those re-amplified from DGGE bands, and a higher number of sequences were obtained per sample. While only two sequences were obtained from each sample by DGGE, 23 - 45 different OTUs were observed from the clone libraries of the same samples. As such, clone libraries appear to be better able to characterize uncultured microbial communities in the deep terrestrial subsurface but can fail to detect unknown parts of the community due to host-vector-insert incompatibility.
As expected, amplicon library sequencing surpassed both DGGE and clone library analyses in terms of sequence coverage and microbial diversity detected. By obtaining tento hundred-fold higher number of sequence reads from each sample, both dominant and rare microbial groups were detected. According to DGGE performed with the same primers, γ- and β-proteobacteria dominated the amplicon libraries. However, other groups (e.g., Bacteroidetes) were detected by amplicon library sequencing at high abundances in samples OL-KR40/ 385m and OL-KR40/609m (7.9% - 26%) yet were not detected by DGGE. Similarly, α-, β-, δ- and ε-proteobacteria, and Firmicutes were also detected in amplicon libraries but not in DGGE (Table 3(a)).
In this study, clone libraries and amplicon libraries were produced from different samples, which limits a direct comparison of the two methods. Nevertheless, the two methods statistically significantly increased the detection of specific bacterial groups. This agrees with two related studies by Itävaara et al. [2] [3] , where the deep biosphere of Outokumpu was examined using the P1/P2 primer set for DGGE and amplicon library sequencing and U968f/U1401r for clone libraries. Despite diverse fingerprints of the deep subsurface bacterial community, only a few bands were successfully sequenced from DGGE. Most of these sequences belonged to Clostridia with only one sequence affiliating with β-proteobacteria [2] . In contrast, clone libraries showed that β-proteobacteria dominated the community close to the surface while Clostridia dominated at greater depths [3] . Results obtained with amplicon library sequencing corresponded well with those obtained with clone libraries, although the relative abundance of β-proteobacteria and Clostridia differed between methods.
In case of archaeal communities, results of both amplicon sequencing and DGGE of the Olkiluoto samples investigated in this study agreed but were not identical. For example, Thermoplasmatales archaea was detected as a dominant group in all amplicon libraries, but was not detected by DGGE from sample OL-KR49/532m. Likewise, SAGMEG/SAGMA1 archaea were detected in all samples by DGGE but only by the amplicon library of OL-KR40/385m. This is in accordance with Nyyssönen et al. [5] , where archaeal communities in the Outokumpu deep biosphere were studied using the same primers for DGGE and amplicon library sequencing as in this study.
5. Conclusion
We compared different PCR primers and molecular methods (i.e., DGGE, clone libraries and HTP amplicon library sequencing) used to survey the community of uncultured microorganisms in deep terrestrial fracture fluids. Our results show that primer selection is of critical importance and, as yet, no single primer pair is able to produce a comprehensive picture of the microbial community in fracture fluids. In addition, different methods provide diverse results depending on their sensitivity and resolving power and should be used judiciously. While DGGE analysis can be used for rapid sample screening, it is often stated that taxa comprising <1% of the community fail to be detected. Our results, however, indicate that the detection limit may be even higher and other factors such as PCR primers play a role in the detection rates or probabilities of certain groups. Clone libraries can provide longer sequences than DGGE or amplicon libraries, resulting in more precise classification of uncultured microorganisms, but this approach is laborious and expensive compared to amplicon sequencing. Amplicon library sequencing covers the microbial diversity most efficiently because a great number of sequences are obtained and may provide the most reliable picture of microbial community composition in deep subsurface environments.
Acknowledgements
The authors would like to thank Posiva Oy for providing samples for this study and for financial support. This work was also funded by VTT Technical Research Centre of Finland, KYT Finnish Research Program on Nuclear Waste Management (projects Geomol 2006-2010, Geomicro 2011-2014), the Academy of Finland (projects Deep life, Methano) and the Finnish Funding Agency for Technology and Innovation (Tekes) project Metageno. Marjo Öster is thanked for excellent technical assistance in the laboratory and Dr Michael Hardman for language editing.
References
- Haveman, S.H., Pedersen, K. and Routsalainen, P. (1999) Distribution and Metabolic Diversity of Microorganisms in Deep Igneous Rock Aquifers of Finland. Geomicrobiology Journal, 16, 277-294. http://dx.doi.org/10.1080/014904599270541
- Itävaara, M., Nyyssönen, M., Bomberg, M., Nousiainen, A., Kapanen, A., Ahonen, A., Hultman, J., Paulin, L., Auvinen, P. and Kukkonen, I. (2011) Microbiological Sampling and Analysis of Outokumpu Deep Borehole Biosphere in 2007-2008. Outokumpu Deep Drilling Project 2003-2010, Special Paper Vol. 51, Kukkonen, I., Ed., Geological Survey of Finland, Espoo, 119-206.
- Itävaara, M., Nyyssönen, M., Kapanen, A., Nousiainen, A., Ahonen, L. and Kukkonen, I. (2011) Characterization of Bacterial Diversity Down to a Depth of 1500 m of the Outokumpu Deep Borehole. FEMS Microbiology Ecology, 77, 295-309. http://dx.doi.org/10.1111/j.1574-6941.2011.01111.x
- Nyyssönen, M., Bomberg, M., Kapanen, A., Nousiainen, A., Pitkänen, P. and Itävaara, M. (2012) Methanogenic and Sulphate-Reducing Microbial Communities in Deep Groundwater of Crystalline Rock Fractures in Olkiluoto, Finland. Geomicrobiology Journal, 29, 863-878. http://dx.doi.org/10.1080/01490451.2011.635759
- Nyyssönen, M., Hultman, J., Ahonen, L., Kukkonen, I., Paulin, L., Laine, P., Itävaara, M. and Auvinen, P. (2014) Taxonomically and Functionally Diverse Microbial Communities in Deep Crystalline Rocks of the Fennoscandian Shield. The ISME Journal, 8, 126-138. http://dx.doi.org/10.1038/ismej.2013.125
- Haveman, S.A. and Pedersen, K. (2002) Distribution of Culturable Anaerobic Microorganisms in Fennoscandian Shield Groundwater. FEMS Microbiology Ecology, 39, 129-137. http://dx.doi.org/10.1111/j.1574-6941.2002.tb00914.x
- Hallbeck, L. and Pedersen, K. (2008) Characterization of Microbial Processes in Deep Aquifers of the Fennoscandian Shield. Applied Geochemistry, 23, 1796-1819. http://dx.doi.org/10.1016/j.apgeochem.2008.02.012
- Pedersen, K., Arlinger, J., Eriksson, S., Hallbeck, A., Hallbeck, L. and Johansson, J. (2008) Numbers, Biomass and Cultivablediversity of Microbialpopulationsrelate to Depth and Borehole-Specific Conditions in Groundwater from Depths of 4 - 450 m in Olkiluoto, Finland. The ISME Journal, 2, 760-775. http://dx.doi.org/10.1038/ismej.2008.43
- Torsvik, V. and Øvreås, L. (2002) Microbial Diversity and Function in Soil: From Genes to Ecosystems. Current Opinions in Microbiology, 5, 240-245. http://dx.doi.org/10.1016/S1369-5274(02)00324-7
- Amann, R.I., Ludwig, W. and Schleifer, K.H. (1995) Phylogenetic Identification and in Situ Detection of Individual Microbial Cells without Cultivation. Microbiology Reviews, 59, 143-169.
- Eydallin, G., Ryall, B., Maharjan, R. and Ferenci, T. (2014) The Nature of Laboratory Domestication Changes of Freshly Isolated Escherichia coli Strains. Environmental Microbiology, 16, 813-828. http://dx.doi.org/10.1111/1462-2920.12208
- Teske, A. and Biddle, J.F. (2008) Analysis of Deep Subsurface Microbial Communities by Functional Genes and Genomics. In: Dilek, Y., Furnes, H. and Muehlenbachs, K., Eds., Links between Geological Processes, Microbial Activities and Evolution of Life, Springer Science+Business Media B.V., Berlin, Heidelberg, 159-176.
- Purkamo, L., Bomberg, M., Nyyssönen, M., Kukkonen, I., Ahonen, L., Kietäväinen, R. and Itävaara, M. (2013) Dissecting the Deep Biosphere: Retrieval and Analysis of Authentic Microbial Communities from Packer-Isolated Deep Crystalline Bedrock Fractures Zones. FEMS Microbiology Ecology, 85, 324-337. http://dx.doi.org/10.1111/1574-6941.12126
- Fox, G.E., Stackebrandt, E., Hespell, R.B., Gibson, J., Maniloff, J., Dyer, T.A., Wolfe, R.S., Balch, W.E., Tanner, R.S., Magrum, L.J., Zablen, L.B., Blakemore, R., Gupta, R., Bonen, L., Lewis, B.J., Stahl, D.A., Luehrsen, K.R., Chen, K.N. and Woese, C.R. (1980) The Phylogeny of Prokaryotes. Science, 209, 457-463. http://dx.doi.org/10.1126/science.6771870
- Yarza, P., Richter, M., Peplies, J., Euzeby, J., Amann, R., Schleifer, K.H., Ludwig, W., Glöckner, F.O. and Rosselló- Móra, R. (2008) The All-Species Living Tree Project: A 16S rRNA-Based Phylogenetic Tree of All Sequenced Type Strains. Systematic and Applied Microbiology, 31, 241-250. http://dx.doi.org/10.1016/j.syapm.2008.07.001
- Akob, D.M., Mills, H.J. and Kostka, J.E. (2007) Metabolically Active Microbial Communities in Uranium-Contaminated Subsurface Sediments. FEMS Microbiology Ecology, 59, 95-107. http://dx.doi.org/10.1111/j.1574-6941.2006.00203.x
- Fry, J.C., Horsefield, B., Sykes, R., Cragg, B.A., Heywood, C., Kim, G.T., Mangelsdorf, K., Mildenhall, D.C., Rinna, J., Vieth, A., Zink, K.G., Sass, H., Weightman, A.J. and Parkes, R.J. (2009) Prokaryotic Populations and Activities in an Interbedded Coal Deposit, Including a Previously Buried Section (1.6 - 2.3 km) above ~150 Ma Basement Rock. Geomicrobiology Journal, 26, 163-178. http://dx.doi.org/10.1080/01490450902724832
- Gihring, T.M., Moser, P.D., Lin, L.H., Davidson, M., Onstott, T.C., Morgan, L., Milleson, M., Kieft, T.L., Trimarco, E., Balkwill, D.L. and Dollhopf, M.E. (2006) The Distribution of Microbial Taxa in the Subsurface Water of the Kalahari Shield, South Africa. Geomicrobiology Journal, 23, 415-430. http://dx.doi.org/10.1080/01490450600875696
- Kotelnikova, S. and Pedersen, K. (1997) Evidence for Methanogenic Archaea and Homoacetogenic Bacteria in Deep Granitic Rock Aquifers. FEMS Microbiology Reviews, 20, 339-349. http://dx.doi.org/10.1111/j.1574-6976.1997.tb00319.x
- Bomberg, M., Nyyssönen, M. and Itävaara, M. (2011) Characterization of Olkiluoto Bacterial and Archaeal Communities by RNA Based High-Throughput 454 Pyrosequencing. Posiva Working Report 2011-31, Posivaoy, Eurajoki.
- Huse, S.M., Dethlefsen, L., Huber, J.A., Welch, D.M., Relman, D.A. and Sogin, M.L. (2008) Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. PLoSGenetics, 4, e1000255. http://dx.doi.org/10.1371/journal.pgen.1000255
- Sogin, M.L., Morrison, H.G., Huber, J.A., Welch, D.M., Huse, S.M., Neal, P.R, Arrieta, J.M. and Herndl, G.J. (2006) Microbial Diversity in the Deep Sea and the Underexplored “Rare Biosphere”. Proceedings of the National Academy of Science of the United States of America, 103, 12115-12120. http://dx.doi.org/10.1073/pnas.0605127103
- Teske, A. and Sørensen, K.B. (2008) Uncultured Archaea in Deep Marine Subsurface Sediments: Have We Caught Them All? The ISME Journal, 2, 3-18. http://dx.doi.org/10.1038/ismej.2007.90
- Baker, B.J., Moser, D.P., MacGregor, B.J., Fishbain, S., Wagner, M., Fry, N.K., Jackson, B., Speolstra, N., Loos, S., Takai, K., Sherwood Lollar, B., Fredrickson, J., Balkwill, D., Onstott, T.C., Wimpee, C.F. and Stahl, D.A. (2003) Related Assemblages of Sulphate-Reducing Bacteria Associated with Ultradeep Gold Mines of South Africa and Deep Basalt Aquifers of Washington State. Environmental Microbiology, 5, 267-277. http://dx.doi.org/10.1046/j.1462-2920.2003.00408.x
- Banning, N., Brock, F., Fry, J.C., Parkes, R.J., Hornibrook, E.R.C. and Weightman, A.J. (2005) Investigation of the Methanogen Population Structure and Activity in a Brackish Lake Sediment. Environmental Microbiology, 7, 947-960. http://dx.doi.org/10.1111/j.1462-2920.2004.00766.x
- Muyzer, G., de Waal, E.C. and Uitterlinden, A.G. (1993) Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Applied and Environmental Microbiology, 59, 695-700.
- Casamayor, E.O., Schäfer, H., Bañeras, L., Pedrós-Alió, C. and Muyzer, G. (2000) Identification of and Spatio-Temporal Differences between Microbial Assemblages from Two Neighboring Sulfurous Lakes: Comparison by Microscopy and Denaturing Gradient Gel Electrophoresis. Applied and Environmental Microbiology, 66, 499-508. http://dx.doi.org/10.1128/AEM.66.2.499-508.2000
- vonWintzingerode, F., Göbel, U.B. and Stackebrandt, E. (1997) Determination of Microbial Diversity in Environmental Samples: Pitfalls of PCR-Based rRNA Analysis. FEMS Microbiology Reviews, 21, 213-229. http://dx.doi.org/10.1111/j.1574-6976.1997.tb00351.x
- Kan, J., Wang, K. and Chen, F. (2006) Temporal Variation and Detection Limit of an Estuarine Bacterioplankton Community Analyzed by Denaturing Gradient Gel Electrophoresis (DGGE). Aquatic Microbial Ecology, 42, 7-18. http://dx.doi.org/10.3354/ame042007
- Kisand, V. and Wikner, J. (2003) Limited Resolution of 16S rDNA DGGE Caused by Melting Properties and Closely Related DNA Sequences. Journal of Microbiological Methods, 54, 183-191. http://dx.doi.org/10.1016/S0167-7012(03)00038-1
- Sánchez, O., Gasol, J.M., Balagué, V., Massana, R., Mas, J. and Pedrós-Alió, C. (2009) Influence of Primer Mismatch and Microdiversity on DGGE Results: A Case Study with SAR11. Aquatic Microbial Ecology, 54, 211-216. http://dx.doi.org/10.3354/ame01267
- Alonso-Sáez, L., Balagué, V., Sà, E.L., Sánchez, O., González, J.M., Pinhassi, J., Massana, R., Pernthaler, J., Pedrós- Alió, C. and Gasol, J.M. (2007) Seasonality in Bacterial Diversity in North-West Mediterranean Coastal Waters: Assessment through Clone Libraries, Fingerprinting and FISH. FEMS Microbiology Ecology, 60, 98-112. http://dx.doi.org/10.1111/j.1574-6941.2006.00276.x
- Costa, R., Götz, M., Mrotzek, N., Lottmann, J., Berg, G. and Smalla, K. (2006) Effects of Site and Plant Species on Rhizosphere Structure Revealed by Molecular Analysis of Microbial Guilds. FEMS Microbiology Ecology, 56, 236249. http://dx.doi.org/10.1111/j.1574-6941.2005.00026.x
- Bano, N., Ruffin, S., Ransom, B. and Hollibaugh, J.T. (2004) Phylogenetic Composition of Arctic Ocean Archaeal Assemblages and Comparison with Antarctic Assemblages. Applied and Environmental Microbiology, 70, 781-789. http://dx.doi.org/10.1128/AEM.70.2.781-789.2004
- Vetriani, C., Jannasch, H.W., MacGregor, B.J., Stahl, D.A. and Reysenbach, A.L. (1999) Population Structure and Phylogenetic Characterization of Marine Benthic Archaea in Deep-Sea Sediments. Applied and Environmental Microbiology, 65, 4375-4384.
- Huber, J.A, Morrison, H.G., Huse, S.M., Neal, P.R., Sogin, M.L. and Welsh, D.B.M. (2009) Effect on PCR Amplicon Size on Assessments of Clone Library Microbial Diversity and Community Structure. Environmental Microbiology, 11, 1292-1302. http://dx.doi.org/10.1111/j.1462-2920.2008.01857.x
- Cotrelli, M.T. and Kirchman, D.L. (2000) Community Composition of Marine Bacterioplankton Determined by 16S rRNA Gene Clone Libraries and Fluorescence in Situ Hybridization. Applied and Environmental Microbiology, 66, 5116-5122. http://dx.doi.org/10.1128/AEM.66.12.5116-5122.2000
- Nübel, U., Engelen, B., Felske, A., Snaidr, J., Wieshuber, A., Amann, R.I., Ludwig, W. and Backhaus, H. (1996) Sequence Heterogeneities of Genes Encoding 16S rRNAs in Paenibacillus polymyxa Detected by Temperature Gradient Gel Electrophoresis. Journal of Bacteriology, 178, 5636-5643.
- Großkopf, R., Janssen, P.H. and Liesack, W. (1998) Diversity and Structure of the Methanogenic Community in Anoxic Rice Paddy Soil Microcosms as Examined by Cultivation and Direct 16S rRNA Gene Sequence Retrieval. Applied and Environmental Microbiology, 64, 960-969.
- Stahl, D.A. and Amann, R. (1991) Development and Application of Nucleic Acid Probes in Bacterial Systematics. In: Stackebrandt, E. and Goodfellow, M., Eds., Nucleic Acid Techniques in Bacterial Systematics, John Wiley & Sons Ltd., Chichester, 205-248.
- Lane, D.J. (1991) 16S/23S rRNA Sequencing. In: Stackebrandt, E. and Goodfellow, M., Eds., Nucleic Acid Techniques in Bacterial Systematics, John Wiley & Sons Ltd., New York, 115-175.
- Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A., Oakley, B.B., Parks, D.H., Robinson, C.J., Sahl, J.W., Stres, B., Thallinger, G.G., Van Horn, D.J. and Weber, C.F. (2009) Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and Environmental Microbiology, 75, 7537-7541. http://dx.doi.org/10.1128/AEM.01541-09
- Pruesse, E., Quast, C., Knittel, K., Fuchs, B.M., Ludwig, W., Peplies, J. and Glöckner, F.O. (2007) SILVA: A Comprehensive Online Resource for Quality Checked and Aligned Ribosomal RNA Sequence Data Compatible with ARB. Nucleic Acids Research, 35, 7188-7196. http://dx.doi.org/10.1093/nar/gkm864
- Chao, A. (1984) Nonparametric Estimation of the Number of Classes in a Population. Scandinavian Journal of Statistics, 11, 265-270.
- Chao, A. and Lee, S.M. (1992) Estimating the Number of Classes via Sample Coverage. Journal of the American Statistical Association, 87, 210-217. http://dx.doi.org/10.1080/01621459.1992.10475194
- Shannon, C.E. (1948) A Mathematical Theory of Communication. Bell System Technical Journal, 27, 379-423.
- Simpson, E.H. (1949) Measurement of Diversity. Nature, 163, 688. http://dx.doi.org/10.1038/163688a0
- Guidon, S. and Gascuel, O. (2003) A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Systems Biology, 52, 696-704. http://dx.doi.org/10.1080/10635150390235520
- Cole, J.R., Wang, Q., Fish, J.A., Chai, B., McGarrell, D.M., Sun, Y., Brown, C.T., Porras-Alfaro, A., Kuske, C.R. and Tiedje, J.M. (2014) Ribosomal Database Project: Data and Tools for High Throughput rRNA Analysis. Nucleic Acids Research, 42, D633-D642. http://dx.doi.org/10.1093/nar/gkt1244
- Hammer, Ø., Harper, D.A.T. and Ryan, P.D. (2001) PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontologia Electronica, 4, Published Online. http://palaeo-electronica.org/2001_1/past/issue1_01.htm
- Klindworth, A., Preusse, E., Schweer, T., Peplies, J., Quast, C., Horn, M. and Glöckner, F.O. (2012) Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Research, 41, e1. http://dx.doi.org/10.1093/nar/gks808
- Marchesi, J.R., Sato, T., Weightman, A.J., Martin, T.A., Fry, J.C., Hiom, S.J. and Wade, W.G. (1998) Design and Evaluation of Useful Bacterium-Specific PCRE Primers That Amplify Genes Coding for Bacterial 16S rRNA. Applied and Environmental Microbiology, 64, 795-799.
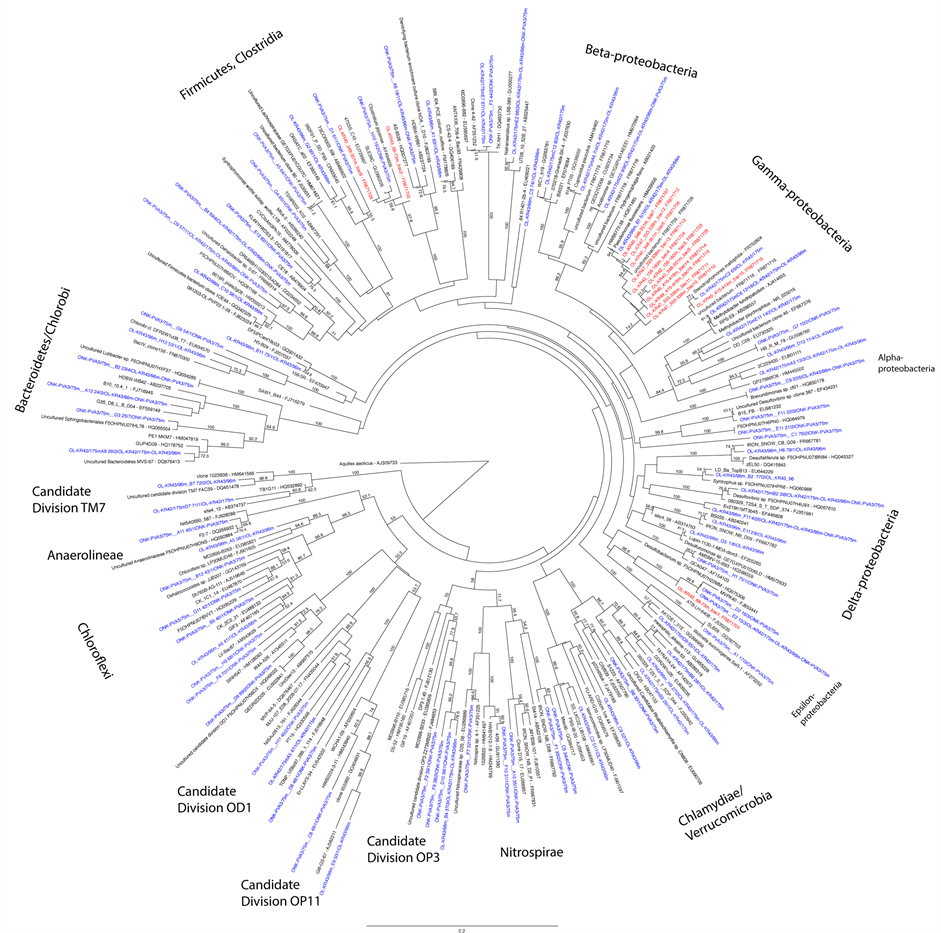
Figure S1. Maximum likelihood tree on the bacterial 16S rRNA gene sequences obtained from DGGE and clone libraries produced with primers U968f/U1401r. The DGGE bands are shown in red and the clone library sequences in blue. Bootstrap support values were calculated with 500 pseudoreplicates. Nodes with >50% bootstrap support are indicated.
 (a)
(a) 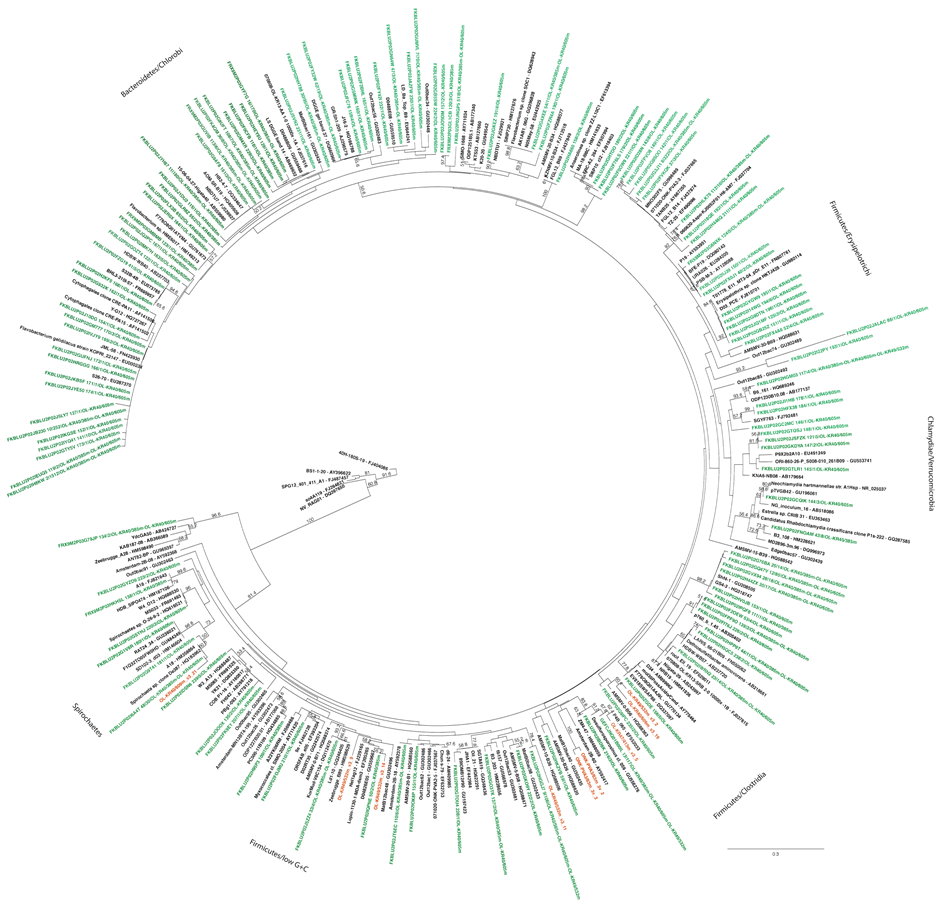 (b)
(b)
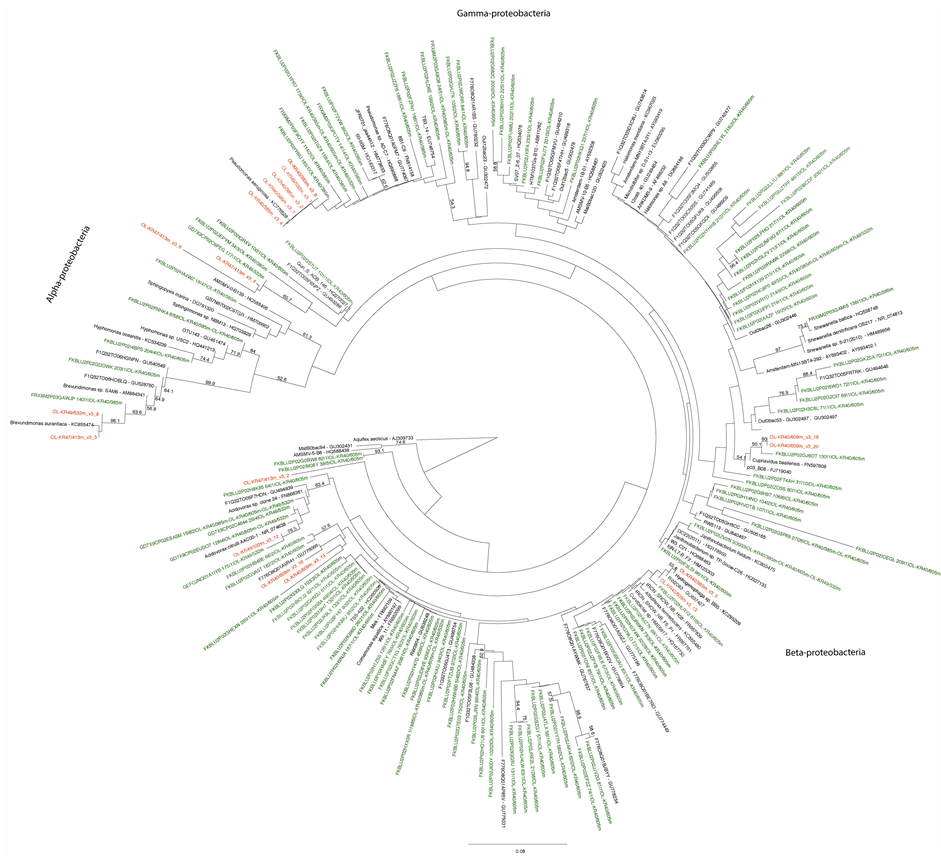
Figure S2. Maximum likelihood tree on the (a) proteobacterial and (b) Bacteroidetes and Firmicutes 16S rRNA gene sequences obtained from DGGE and amplicon library sequencing produced with primers P1/P2. The DGGE bands are shown in red and the clone library sequences in green. Bootstrap support values were calculated with 500 pseudoreplicates. Nodes with >50% bootstrap support are indicated.
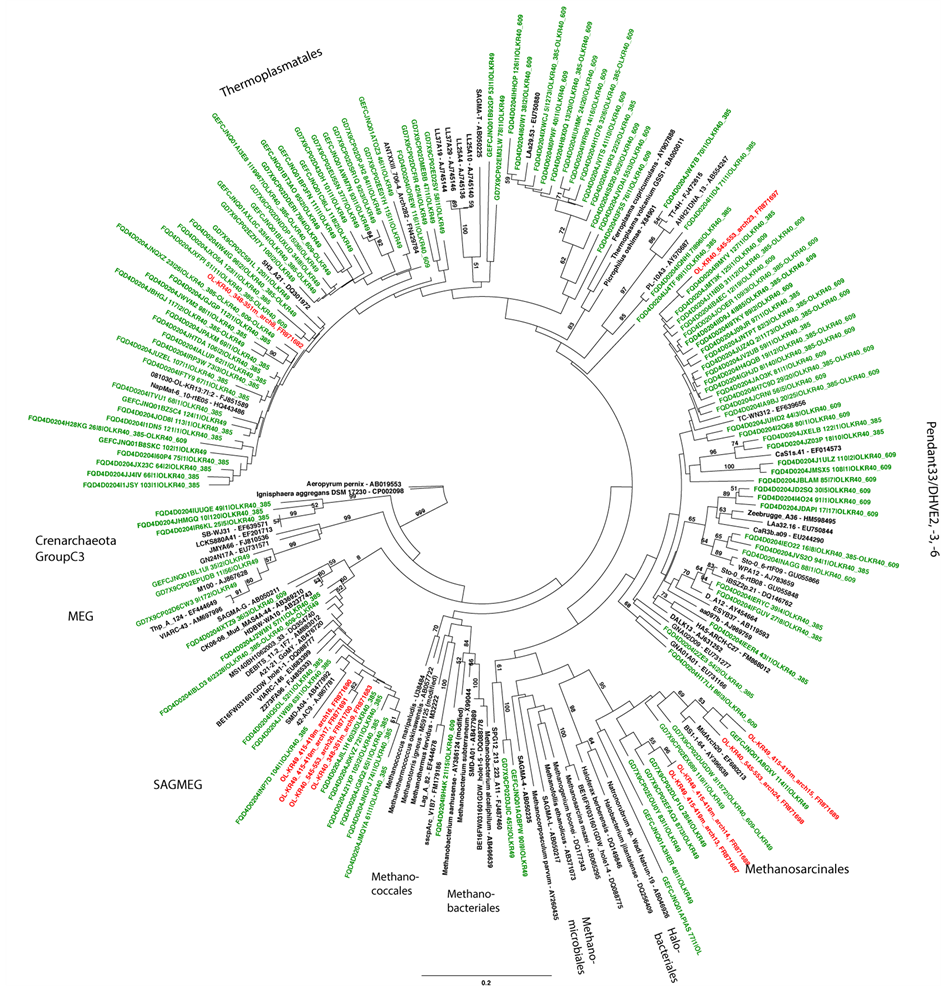
Figure S3. Maximum likelihood tree on the archaeal 16S rRNA gene composition studied by DGGE and amplicon library sequencing. The DGGE bands are shown in red and the clone library sequences in green. Bootstrap support values were calculated with 100 pseudoreplicates. Nodes with >50% bootstrap support are indicated.
NOTES

*Corresponding author.