Open Journal of Ecology
Vol.04 No.07(2014), Article ID:46286,22 pages
10.4236/oje.2014.47034
Assessment of Genetic Variability of 142 Sweet Sorghum Germplasm of Diverse Origin with Molecular and Morphological Markers
A. Lekgari, I. Dweikat
Department of Agronomy and Horticulture, University of Nebraska, Lincoln, USA
Email: Idweikat2@unl.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 January 2014; revised 3 February 2014; accepted 10 February 2014
ABSTRACT
Sorghum bicolor (L.) Moech is the fifth most important crop in the world. Recently, its agronomics and genetics have drawn interest among scientists. Sweet sorghum, a variety of sorghum, may po- tentially become a bioenergy source because of the high sugar content in its juicy stems. Exploring the diversity of sweet sorghum around the world is important to the development and improve- ment of the crop as an energy source. In exploring the diversity of sweet sorghum, three types of markers (simple sequence repeats [SSR], sequence-related amplified polymorphisms [SRAP], and morphological markers) are used on 142 sweet sorghum accessions from around the world. The accessions show a high significance (P < 0.05) for all the morphological traits measured. The morphological markers cluster the accessions into five groups based primarily on plant height (PH), anthesis data (AD), and moisture content (ML), with the principal component analysis (PCA) showing these traits to explain 92.5% of the total variation. The furthest accessions were PI571103 from Sudan, and N99 from the United States. The Nei’s genetic standard distances ranged from 0.024 to 1.135 and 0.078 to 0.866 for SSR and SRAP, respectively. As expected, accessions of the same origin or breeding history had the lowest genetic distance (e.g. Mokula and Marupantse, both from Botswana; NSL83777 and NSL83779 from Cameroon). Neighbor joining clusters the sweet sorghum accessions into five major groups using SSR and four major groups using SRAP, based on their origin, or breeding history. The three marker types complement each other, and the presence of accessions of different origins across clusters indicate similar genetics, and evi- dence of germplasm movement between countries.
Keywords:
SRAPs, SSRs, Energy Crops, Genetic Distance

1. Introduction
Sorghum (Sorghum bicolor (L.) Moench) is one crop species that can survive harsh climatic conditions of arid environments [1] . Sorghum bicolor contains both cultivated and wild races and possess a significant amount of genetic diversity for traits of agronomic importance [2] . Commonly, it is used as a source of grain food, syrup fuel, and feed for livestock. Sweet sorghum, a variety of sorghum with high sucrose accumulation in their stems, has recently received a lot of attention as a source of biofuel [3] . Sweet sorghums are selected to accumulate high levels of sucrose in the parenchyma of the juicy stems [4] [5] . The stems are desired for food grade syrup, fresh chewing, and alcohol production in areas like Brazil [5] , India, and Africa [6] . In the United States, sweet sorghum has been researched for biofuel for over 30 years [7] . The primary research regarding sweet sorghum, its development, and breeding began in the late 1970s [5] because of the high oil costs and the need for alternative energy sources. Under favorable conditions, a sweet sorghum crop is capable of producing up to 13.2 metric tons per hectare of total sugars, which is equivalent to 7682 liters of ethanol per hectare [5] . Sweet sorghum has a compelling advantage for cellulosic biofuel production over seed-based ethanol production, and its adaptation to marginal lands makes the per unit value of biomass production economical [8] .
Sweet sorghum gene pool creation has not received much attention, primarily because it is not considered to be among the important crops in the US, and the pedigree information is scarce and incomplete. Most sweet sorg- hums released in the US are developed by public breeding programs in the 1900s and are mainly open pollinated [9] . The crop improvement is done mainly on the sweet sorghums’ syrup, sugar concentration, and biomass, with lines primarily selected for improved disease resistance [5] . Genetic diversity or knowledge of patterns of diver- sity of genetic resources is of great importance (Warburton et al., 2001) and is a key component in crop im- provement and plant breeding.
Murray et al. [5] stated that the majority of the sweet sorghum cultivars released in the United States have a narrow genetic base that can be traced to six African landraces. There are currently no criteria (molecular or morphological markers) to differentiate sweet sorghums from grain sorghums [5] , and most of the accessions lack the proper information to help distinguish them. Therefore, when requesting sweet sorghum germplasm, one is limited to a few characters that are common in sweet sorghum, such as tall leafy plants (high biomass), and the brix degree where available, which is also subjective as there is no definite value for distinguishing grain sorg- hums from sweet ones. The Meridian, Mississippi Station attempted to curate what may be the world sweet sorghum collection, and when it closed, materials were transferred to the USDA sorghum collection in Griffin, Georgia [10] . Thus, many diversity studies have concentrated on cultivars/lines that are common and known, leaving the vast majority of the collection (genetic sources) unexploited. In this study, we tried to incorporate the commonly used lines together with some of the uncommon lines, and accessions from other sorghum collections.
The use of morphological traits in plants as markers for determining the genetic relationship dates back many years. Mendel followed visible phenotypic traits in progeny of sexual crosses, and the use of morphological markers has continued to the present day [11] . Phenotypic variables include continuous variables such as height, maturity, and yield, as well as discrete variables such as grain color, texture, and insect and disease resistance [12] . Franco et al. [12] stated that the truth underlying homogeneous groups or sub-populations of genotypes and their shape and structure is unknown. This is due to the fact that the association between the traits affects the shape of the groups and their structure is dependent on the composition of the group.
However, clustering methods attempt to recover the true shape and structure of the sub-population. When using both the morphological and molecular marker data, two types of hierarchical classification are carried out inde- pendently. The morphological marker data first utilizes the computation of standard distances (e.g. Euclidean distances) and clustering strategies such as UPGMA or neighbor joining are applied. On the other hand, when applying the molecular marker data, genetic similarities or dissimilarities using each band fragment as an attribute (0 for absence and 1 for presence) are determined, then a clustering strategy applied [12] . This enables genotypes to be clustered into as homogenous groups as possible. Phenotypic and genetic diversity are important in genetic conservation, evaluation, and utilization of genetic resources, and the study of breeding germplasm for deter- mining uniqueness and genetic constitution for the purpose of breeder’s property rights [12] . The morphological markers are highly influenced by environmental conditions. Therefore, there is a need to supplement or complement their clustering with molecular marker data.
Polymerase chain reaction (PCR) is widely used in genomic DNA analysis, and one of its main applications has been in the development of DNA-based markers for map construction, breeding taxonomy, evolution, and gene cloning [11] [14] . Molecular markers are basically nucleotide sequence corresponding to a physical position in the genome, and their polymorphisms between accessions allow the pattern of inheritance to be easily traced [11] . The availability of molecular markers to assess diversity is a quicker way to help breeders select suitable lines/ge- notypes for crossing. The use of molecular markers as a tool to assess relatedness in and between cultivated and wild sorghum have been successfully used [1] [15] - [19] . PCR-based markers are widely used in fingerprinting crops because of their high level of polymorphisms [20] and their ease of detection [21] . Several PCR-based markers vary in their complexity, reliability, and information generating capacity.
Simple sequence repeats (SSR), also known as microsatellites, are based on tandem repeats of one to six core nucleotide elements. These codominant markers are dispersed throughout the genome, and have multiple alleles that often have conserved loci between related species [11] [22] . Powell et al. [23] stated that SSRs are able to discriminate among closely related individuals and have advantage over other markers in their ability to trace pedigrees in plants. Therefore, SSRs have been used in a variety of genetic studies such as diversity analysis, quantitative trait locus mapping, gene tagging, and cultivar identification.
Several studies involving either SSR markers alone or in combination with other marker types have been conducted on sorghum varieties [5] [24] - [27] . Polymerase chain reaction made possible the development of many other marker methods. Schulman [11] indicated that some marker methods detect specific, cloned, and sequenced targets in the genome, while others use conserved or general primers that amplify from many anonymous sites throughout the genome.
Sequence-related amplified polymorphism (SRAP) markers are based on two primer amplification, which preferentially amplifies open reading frames (ORFs) or coding regions that result in a number of dominant and codominant markers [14] [28] - [30] . Li and Quiros [14] and Zhao et al. [30] explained that the forward primer amplifies the exon regions while reverse primer amplifies the intron and promoter regions. They also stated that the polymorphisms resulted from the variation in length of these exons, introns, promoters, and spacers among both individuals and species. Sequence-related amplified polymorphism markers are more reproducible, stable, and less complex [30] [31] , in addition to being more powerful in revealing the genetic diversity among closely related individuals than other marker types [32] . Sequence-related amplified polymorphism markers have also been used in a wide range of plant species such asalfalfa [28] , Brassica [14] , buffalo grass ( [28] , 2004b), cotton [33] , Cucubita [34] , tree peony [31] and wheat [35] [36] . Ferriol et al. [34] also reported that the information obtained from SRAP markers agreed with the morphological variations and evolutionary history of morphotypes more than that found with AFLP.
Several diversity studies of sorghum and/or its wild relatives [1] [15] [16] [26] are limited to either grain sorghum or to germplasm from or within an individual country. In this era, germplasm sharing is an important factor in breeding as breeders try to develop modern cultivars with improved agronomic performance. The use of molecular markers has proven to be an effective tool in assessing the genetic relatedness of different species [1] . Regarding this, many types of markers have been used in sorghum. These studies have revealed both a wide and narrow genetic variation between agroecological zones. Folkertsma et al. [37] indicated that there is a wide va- riability within accessions in the semi-arid regions of Africa. However, the south Asian accessions have narrower diversity compared to those in Africa. Therefore, it is important to establish the genetic similarity among some of the world germplasm collection of sweet sorghum, especially since its potential as an agro-industrial crop con- tinues to draw more attention. Therefore the objectives of this study are to:
Examine the genetic variability within sweet sorghum germplasm from different regions of the world for traits associated with biofuel production.
Classify/group the sweet sorghum germplasm based on SSRs, SRAPs, and several morphological data.
2. Materials and Methods
2.1. Plant Material
One hundred and forty-two 142 sweet sorghum accessions were used in this study (Appendix 1). These acces- sions were obtained from the USDA-ARS, University of Nebraska-Lincoln, NE; National Center for Genetic Resources Preservation (NCGRP), Fort Collins, CO; National Plant Germplasm System, Griffin, GA; Texas Agricultural System Station, College station, TX; University of Kentucky, KY; and the Department of Agricul- tural Research (Ministry of Agriculture), Botswana. These 142 sweet sorghum accessions consisted of landraces, released improved cultivars, and breeding lines. The available pedigree information were obtained from GRIN website (http://www.ars-grin.gov/cgi-bin/npgs/html/index.pl), ICRISAT website (http://www.icrisat.org), and other resource publications [5] [37] , or accompanied in the seed information.
2.2. Agronomic Traits
The 142 sorghum lines and two check cultivars (M81-E and sugar drip) were planted under rain-fed conditions at Mead, NE during the 2009 growing season. The experiment was laid in an incomplete block design with 12 in- complete blocks of 12 entries each (12 × 12 alpha lattice) and two replications. Single row plots measuring five meters long with between row spacing of 0.75 m were over sown at the rate of 160,000 seeds per hectare. The seeding rate was assumed to compensate for situation where there might be low seed viability, and the final population density was on average of 140,000 plants per hectare.
Four agronomic traits were measured and included anthesis date measured as the duration in days from planting to 50% of the plants within a plot were 1) shedding pollen; 2) plant height measured as the distance from the base of the plant to the tip of the panicle; 3) dry matter yield in Mg∙ha−1 when plants had reached their physiological maturity; and 4) moisture content as the percentage difference between wet and dry biomass weight. Dry matter yield was calculated from a sample taken at harvest as follows: DM = (Dry weight of total 0.50 m row)/(plot area in m2) then calculated as Mg∙ha−1. Plants were weighed immediately after cutting the 0.5 m samples, bagged, and placed into an oven at 120˚C - 160˚C for ten days to completely dry the samples. Samples were reweighed to obtain the dry weight.
2.3. DNA Extraction and Marker Analysis
Genomic DNA of each accession was extracted from fresh leaf tissues from plants planted in the greenhouse using cetyltrimethyl ammonium bromide (CTAB) protocol [39] . The ground tissue was incubated in an extraction buffer (50 mM Tris-HCl, 25 mM EDTA, 1 M NaCl, 1% CTAB, 1 mM 1,10-phenathroline and 0.15% 2-mercaptoethanol) at 65˚C for one hour; then equal volume of chloroform:isoamyl alcohol (24:1) was added to the tissue mixture. After centrifugation at 3000 rpm, the supernatant was transferred to a new clean tube and DNA was precipitated with equal volume of cold isopropanol. DNA was air died at room temperature for one hour and then re-suspended in TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) with 20 ng RNase and incubated at 37˚C overnight. Equal volume of 24:1 chloroform:isoamyl alcohol was added to the DNA-RNase mix and centrifuged at 3000 for five minutes, and the resulting supernatant was transferred to new tube. Two volumes of cold absolute ethanol and 5 μl of 8 M ammonium acetate were added to the supernatant in order to precipitate the DNA. After centrifugation, DNA pellets were air dried at room temperature, and later re-suspended with 200 to 400 μl TE buffer, depending on the size of the pellet. DNA concentration was determined using a spectrophotometer (TKO 100 Fluorometer, Hoefer Scientific Instruments, San Francisco, California).
A collection of 82 oligonucleotide primer pairs that included 33 sorghum SSRs ( [40] ; Lubbock, TX [unpub- lished]) and 49 SRAP combinations [14] [41] were synthesized, and marker assays were conducted following the procedure of Kuleung et al. [42] . A 25 μl total/reaction was used, which consisted of 75 ng genomic DNA, 100 ng primer pair, 125 μMdNTP, 50 mM KCl and 10 mM Tris-HCl, 2 mM MgCl2, and one unit Taq polymerase. The amplification procedure consisted of one cycle at 94˚C for three minutes, followed by 35 cycles of one minute at 94˚C, one minute at 55˚C to 58˚C for SSRs depending on the primer pair, and 47˚C for SRAPs, one minute at 72˚C, and final extension step at 72˚C for five minutes. The reaction was then cooled to a resting temperature of 4˚C and resolved by electrophoresis in 12% non-denatured polyacrylamide gels (37:1 of acrylamide: bis-acrylamide). The gels were stained in 1 µg/ml ethidium bromide for 10 minutes, distained in deionized water for 15 minutes, then photographed using the Gel Doc2000 (Bio-Rad, Hercules, Califorlia).
2.4. Data Analysis
Analysis of variance was performed on agronomic data using PROC MIXED, where incomplete blocks were treated as random effects. In the next step, principal component analysis using a correlation matrix from least square means (LSMEAN) was done using PROC PRINCOMP to determine the traits that account for most varia- tion between lines. A simple Pearson correlation was done on the means of the four agronomic traits measured. Due to the large difference in the unit of each trait, agronomic data were standardized using the standard deviation of mean by PROC STANDARD. Afterwards, we used PLOC CLUSTER using “Average Linkage Cluster Analysis” based on Euclidean distance [43] for the clustering. Average Linkage algorithms were used for cluster analysis and then dendrogram was constructed using PROC TREE [44] .
Two genetic distance and clustering methods were used for the marker data to determine how the sorghum accessions grouped, using a band scoring of “1” to indicate the presence of an allele and “0” when absent. Po- lymorphism information content (PIC) values were calculated as per formula developed by Anderson et al. [45] , which assumes homologous alleles. Polymorphic information content is calculated as:
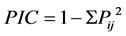
where Pij is the frequency of jth allele of ith locus, summed across all the alleles for the locus over all lines. A marker with a PIC value of more than 0.5 is considered highly informative, between 0.25 and 0.5 is considered informative, and less than 0.25 is considered slightly informative [46] . The genetic diversity was estimated by similarity index calculation from band sharing data of each pair of DNA fingerprints. Genetic similarity (GS) between cultivars i and j was calculated using all loci for both the SSR and SRAP markers according to Nei and Li’s formula for estimating coefficient of similarity [47] [48] based on shared allele frequency. The formula is as follows:
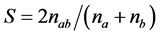
where S is the similarity coefficient, nab is the number of bands common to A and B cultivars, na and nb are number of bands in A and B cultivar, respectively. A similarity matrix was used to construct a similar dendogram by cluster analysis using the neighbor joining method to determine how sorghum accessions were related. The ge- netic distances were calculated based on Nei’s [49] standard genetic distance, as follows:
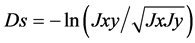
where Jx = ΣΣ Xij2/r, Jy = ΣΣ Yij2/r, and Jxy = ΣΣ XijYij/r with Xij and Yij being the frequencies of allele i at j locus of populations X and Y, respectively [50] . Population genetics software (Version 1.2.30) was used for genetic distance calculation (http://bioinformatics.org/~tryphon/populations). The dendogram construction from the POPULATIONS program used the TreeView program (version 1.6.6) (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html; [51] ).
3. Results and Discussion
3.1. Agronomic Traits
Harvest was done at the same time for all lines. However, due to the wide range of maturity existing among them, some lines were past the physiological maturity stage at the time of harvest. The analysis of variance showed a highly significant differences (P < 0.01) for all the traits measured among sorghum accessions (Table 1). The anthesis data showed a wide range of maturity among the lines (70 to 147 days to anthesis) (Appendix 2). Plant height ranged from 76.0 cm to 423.8 cm, moisture content ranged from 45.4% to 80.6%, and dry matter yield weight ranged from 3.81 Mg∙ha−1 for PI 276804 to 59.19 Mg∙ha−1 for N99 (Appendix 2).

Table 1. Mean squares of anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) measured at Mead in 2009 season.
†The degree of freedom for lines was less than expected because of some missing data; **, *** indicate significance at probability values of 0.05 and 0.01 respectively.
PI 276804 is an Ethiopian landrace with moderate tillering and medium height (ICRISAT website), while N99 is an F7 selection from a cross between a Fremont forage sorghum and Theis sweet sorghum [38] . Theis is a high biomass producer that may contribute to N99 high yield. There were highly significant correlations between an- thesis date and plant height (r = 0.53***), anthesis date and dry matter yield (r = 0.57***), and plant height with moisture content and plant height with dry matter yield (r = 0.285*** and 0.712*** respectively) (see Table 2).
Cluster analysis of agronomic traits grouped the lines into five main groups (Figure 1, Appendix 3). Although the agronomic traits did not distinctly group lines according to their geographic origin/area, materials from the same area tended to cluster together within each group, indicating that their origin played a role in the selection or development of germplasm used (Figure 1). Apart from the germplasm origin, the lines tended to group together according to plant height and percent moisture content. For example, Group 1 consisted of materials that were 248.0 cm tall (176.3 - 288.3 cm) on average Group 2 averaged 328.5 cm (287.3 - 390.5 cm); Group 3 averaged 399.5 cm (379.2 - 423.8 cm); Group 4 averaged 170.7 cm (104.3 - 200.8 cm) and Group 5 was 123.8 cm tall (76.0 - 281.8 cm) on average.
Data obtained from the principal component analysis in which anthesis date, plant height, and moisture loss accounted for the 79.1% of the variation (i.e. principal component 1 and 2) supported the above reasoning (Table 3). Principal component 3 was mainly associated with anthesis date, together with the first two principal com- ponents, which accounted for 92.5% of the variation. The two furthest genetic distances exist between accession PI 571103 (a landrace from Sudan) and N99 with distance of 7.818. On the other hand, the closest accessions were PI 569520 (a breeding line from Sudan) and ICSR90017 (a restorer line from ICRISAT), with a genetic distance coefficient of 0.189.
3.2. Molecular Marker Data
From the 33 SSR marker pairs screened, 29 produced 84 polymorphic alleles with a mean of 2.90. This was lower

Figure 1. The dendrogram using average distances of 142 accessions based on anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) measured at Mead in 2009. Five major groups at threshold distance of 0.40.
Table 2. Correlation coefficients of anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) measured at Mead in 2009 season (n = 142).
***Significance at probability value of 0.01 respectively; nsNon-significance at P < 0.05.
Table 3. The eigenvalues and principal components for anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) measured at Mead in 2009 showing the pro- portion explaining variation.
than the 3.22 that Ali et al. [26] observed, or the 3.4 that Schloss et al. [40] observed. The polymorphic informa- tion content of SSR markers ranged from 0.22 to 0.75 with a mean value of 0.52 (Table 4). These values were higher than those of 0.40 and 0.44 observed by Ali et al. [26] and Folkertsma et al. [37] , respectively. The dif- ferences may be attributed to the number of bands scored and the type of SSR markers used. Botstein et al. [46] suggested that markers with PIC > 0.5 be considered highly informative. Thus, we could conclude that the mean PIC value of 0.52 indicated that the markers used were highly informative. By using 72 US sorghums in their experiment, Ali et al. [26] reported PIC value range of 0.03 to 0.87.
For each pairwise similarity estimate, a dendrogram was constructed using Nei’s standard genetic distance [49] . The accessions were grouped mainly according to their origin or breeding history (Figure 2). Nei’s standard genetic distance ranged from 0.024 to 1.135, with Marupantse and Mokula having the smallest genetic distance, while PI 154844 and NSL 55404 had the largest genetic distance, followed by NSL 55429 and NSL 87920, and PI 602982 and PI 571103 with a value of 1.099. Marupantse and Mokula are both from Botswana. Marupantse is an advanced/improved cultivar while Mokula is of unknown parentage. However, the two do not belong to the same sorghum race (kafir vs. durra-caudatum). PI 154844 is a landrace from Uganda and NSL 55404 is from India. However, both belong to the Durra race. NSL 53429 is a landrace from India, while NSL 87920 is from Cameroon; PI 602982 is a line developed in Mali with pedigree (SPV 35/E35-1)/CS 3541, and PI 571103 is a landrace from Sudan.
By using Nei’s standard genetic distances [49] through neighbor joining (Figure 2), cluster analysis grouped the accessions into five major groups. Group 1 consisted of germplasm mainly from East Africa (Sudan, Kenya, and Ethiopia), while Group 2 consisted of germplasm from different regions. Group 3 is occupied by Nebraska lines (released and breeding lines) and some Ethiopian germplasm. The largest group was Group 4, which con- sisted of 47 accessions, and occupied mainly by germplasm from Botswana and the US (particularly Nebraska). The SC accessions from Botswana might have been part of the late 1960s USDA sorghum conversion program, thus creating a link between Botswana sorghums and the US sweet sorghums, e.g. 65D, which is an introduction to Botswana from the United States, with unknown parentage. The last group was the smallest (nine accessions) and also had germplasm from different regions.
Within each major group, accessions from the same country/region grouped together to form smaller clusters. This is in agreement with Wang et al. [52] and Murray et al. [5] , who observed that both sweet and grain sorghums germplasm corresponded well with the geographic locations where the accessions originated. Since most of the accessions used were landraces with unknown parentage, it can only be assumed that accessions with the same origin may be highly related. However, those with known parentage such as the Nebraska breeding lines, the ones with similar pedigree, tend to cluster together. For example, the lines that have wheatland in their parentage (05C09882 [5] tan, 05C09881 [4] ppbmr, and 05C09892 [6] ppbmrsw, etc.) were closer to wheatland, while lines like 05C09889 [1] vtallsw grouped with N99. Ali et al. [26] reported that Dale, N108, Theis, Cowley, and Norkan clustered in the same major group but different subgroups. In this study, they were also in the same major group.


Figure 2. Dendrogram constructed by neighbor joining analysis using Nei’s (1972) genetic standard distances of 142 sorghum accessions based on SSRs data.
Table 4. Polymorphic information content (PIC) of markers used to analyze 142 sorghum accessions.
From the 49 SRAP marker pairs screened, 40 polymorphic pairs produced 109 alleles, with a mean of 2.73 alleles. This value was lower than that of SSR markers. However, the PIC for SRAP was higher than that of the SSR because SRAP markers had lower allele frequency, and it ranged from 0.15 to 0.94 with a mean of 0.56 (Table 4). The pairwise similarity estimate using Nei’s standard genetic distance ranged from 0.078 (NSL 83777 and NSL 83779) to 0.866 (PI 286245 and Orange). Both NSL 83777 and NSL 83779 are sorghum landraces from Cameroon, PI 286245 is from Sudan, and orange has no clear origin, as there are various versions of orange from different origins. The SRAP markers also grouped the accessions according to their origin or breeding history, although the groups were different from the SSR groups. The differences between markers in clustering the ac- cessions may be due to differences in genomic regions amplified by each marker type.
Cluster analysis based on neighbor joining using Nei’s standard distances produced four major groups (Figure 3). Group 1 was occupied by accessions from East and West Africa and consisted of 59 accessions. Group 2 was the smallest with 9 accessions mainly from East Africa. Group 3 consisted of germplasm from both Botswana and North and South America. Unlike the SSRs grouping, the Nebraska lines featured in this group were very few and were mainly the released ones. Finally, Group 4 consisted mainly of the Nebraska breeding lines with some ICRISAT and India accessions. The SRAP markers seem to have separated the accessions well based on their breeding and origin compared to SSRs. Budak et al. [32] reported that SRAP markers are suitable in showing true variation within and among buffalo grass cultivars. Zhao et al. [30] also observed SRAPs clustering seemed to agree with morphological classification, although that was not the case with this study.
This study focused more and was limited by the number of morphological traits measured as well as the number of field experiments conducted. When looking at other types of molecular markers, Ritter et al. [1] observed that clusters developed based on agronomic data could not approximate groupings produced by molecular markers. When looking at within group clustering, one could observe smaller subgroups aligned to each country or breeding program .The main difference between the marker clusters could be due to the differences in marker type. Simple sequence repeats amplified randomly in the genome, whereas the SRAP amplified from the open reading frames or promoters of genes. Therefore, based on the study, SRAP was more informative in grouping accessions based on their breeding history.


Figure 3. Dendrogram constructed by neighbor joining based on Nei’s (1972) standard distances of 142 sorghum accessions based on SRAP data.
Comparing the three clustering procedures, it has been shown that each data type has its own strength but all seem to reflect the breeding history of the germplasms. The morphological data grouped accessions were based primarily on plant height, which is one of the characters that breeders base their selection on. Therefore, what the molecular markers (particularly SRAP) showed was what potential genes were selected for in these accessions. Wang et al. [52] reported that most of agronomic traits are affected by different levels of population structure and may therefore contribute to the differences observed between clustering conducted with different marker data.
4. Conclusions
The agronomic and molecular marker data produced distinct cluster groups for the sorghum accessions evaluated. Although the groups were not identical, they complemented each other. The agronomic data clusters provided clues to which important characters separated individual accessions, while SSR clusters further narrowed the groups based on their origin. The SRAP markers then even refined the groups as they showed the breeding pat- tern/history of the accessions. Perumal et al. [25] made similar observations, indicating that a more comprehensive and composite index based on pedigree, morphological, biochemical, and molecular data is expected to improve accuracy of grouping individuals.
This study also showed that there was a considerable amount of germplasm movement across different regions of the world, and there is still a large genetic diversity even within some regions. For example, some of the lines that were far from each other came from the same region. Previous reports have also indicated that the diversity of sorghum is limited in certain regions compared to others. Therefore, sorghum improvement will benefit from this wide range of diversity, and germplasm exchange will be the key to the success of improving sweet sorghum cultivars as a source for biofuel. This study has also strengthened the point that the use of molecular markers is essential and beneficial to plant breeders. The molecular markers are used to compliment the agronomic data when pedigree information is limited or unavailable.
References
- Ritter K.B., McIntyre, C.L. Godwin, I.D. Jordan, D.R. and Chapman, S.C. (2007) An Assessment of the Genetic Relationship between Sweet and Grain Sorghums, within Sorghum bicolor ssp. bicolor (L.) Moench, Using AFLP Markers. Euphytica, 157, 161-176. http://dx.doi.org/10.1007/s10681-007-9408-4
- Hart, G., Schertz, K., Peng, Y. and Syed, N. (2001) Genetic Mapping of Sorghum bicolor (L.) Moench QTLs That Control Variation in Tillering and Other Morphological Character. Theoretical and Applied Genetics, 103, 1232-1242. http://dx.doi.org/10.1007/s001220100582
- Ferraris, R. (1981) Early Assessment of Sweet Sorghum as an Agro-Industrial Crop. I. Varietal Evaluation. Australian Journal of Experimental Agriculture and Animal Husbandry, 21, 75-82. http://dx.doi.org/10.1071/EA9810075
- Vietor, D.M. and Miller, F.R. (1990) Assimilation, Partitioning, and Nonstructural Carbohydrate in Sweet Compared with Grain Sorghum. Crop Science, 30, 1109-1115. http://dx.doi.org/10.2135/cropsci1990.0011183X003000050030x
- Murray, S.C., Rooney, W.L., Hamblin, M.T., Mitchell, S.E. and Kresovich, S. (2009) Sweet Sorghum Genetic Diversi- ty and Association Mapping for Brix and Height. Plant Genome, 2, 48-62. http://dx.doi.org/10.3835/plantgenome2008.10.0011
- Vaidyanathan, S., Rao, K.E.P., Mengesha, M.H. and Jambunathan, R. (1987) Total Sugar Content in Sorghum Stalks and Grains of Selected Cultivars from the World Germplasm Collection. Journal of the Science of Food and Agricul- ture, 39, 289-295. http://dx.doi.org/10.1002/jsfa.2740390403
- Lipinsky, E.S., Kresovich, S., McClure, T.A. and Lawhon, W.T. (1977) Fuels from Sugar Crops. First Quarterly Re- port, Battelle Columbus Labs, Columbus.
- Paterson, A.H. (2008) Genomics of Sorghum: A Review. International Journal of Plant Genomics, 2008, Article ID: 362451, 6 p. http://dx.doi.org/10.1155/2008/362451
- Swanson, A. and Laude, H. (1934) Varieties of Sorghum in Kansas. Kansas Bulletin, 266, 2-50.
- Freeman, K.C. (1979) Germplasm Release of Sweet Sorghum Lines with Resistance to Downy Mildew, Leaf Anthracnose and Rust with Adequate Combining Ability to Produce Progeny with Agronomic Characters Acceptable for Commercial Sirup and/or Sugar Production. Mississippi Agricultural and Forestry Experiment Station Research Report, Vol. 4, No. 2.
- Schulman, A. (2007) Molecular Markers to Assess Genetic Diversity. Euphytica, 158, 313-321. http://dx.doi.org/10.1007/s10681-006-9282-5
- Franco, J., Crossa, J., Ribaout, J.M., Betran, J., Warburton, M.L. and Khairallah, M. (2001) A Method for Combining Molecular Markers and Phenotype Attributes for Classifying Plant Genotypes. Theoretical and Applied Genetics, 103, 944-952. http://dx.doi.org/10.1007/s001220100641
- Ramakrishnan, A.P., Meyer, S.E., Walters, J., Stevens, M.R., Coleman, C.E. and Fairbanks, D.J. (2004) Correlation between Molecular Markers and Adaptively Significant Genetic Variation in Bromus tectorum (Poaceae), an Inbreedingannual Grass. American Journal of Botany, 91, 797-803. http://dx.doi.org/10.3732/ajb.91.6.797
- Li, G. and Quiros, C.F. (2001) Sequence-Related Amplified Polymorphism (SRAP), a New Marker System Based on a Simple PCR Reaction: Its Application to Mapping and Gene Tagging in Brassica. Theoretical and Applied Genetics, 103, 455-461. http://dx.doi.org/10.1007/s001220100570
- Tao, Y., Manners, J., Ludlow, M. and Henzell, R. (1993) DNA Polymorphisms in Grain Sorghum (Sorghum bicolor (L.) Moench). Theoretical and Applied Genetics, 86, 679-688. http://dx.doi.org/10.1007/BF00222656
- Ahnert, D., Lee, M., Austin, D., Livini, C., Woodman, W., Openshaw, S., Smith, J., Porter, K. and Dalon, G. (1996) Genetic Diversity among Elite Sorghum Inbred Lines Assessed with DNA Markers and Pedigree Information. Crop Science, 36, 1385-1392. http://dx.doi.org/10.2135/cropsci1996.0011183X003600050049x
- Uptmoor, R., Wenzel, W., Friedt, W., Donaldson, G., Ayisi, K. and Ordon, F. (2003) Comparative Analysis on the Ge- netic Relatedness of Sorghum bicolor Accessions from Southern Africa by RAPDs, AFLPs and SSRs. Theoretical and Applied Genetics, 106, 1316-1325.
- Anas and Yoshida, T. (2004) Genetic Diversity among Japanese Cultivated Sorghum Assessed with Simple Sequence Repeats Markers. Plant Production Science, 7, 217-223.
- Menz, M.A., Klein, R.R., Unruh, N.C., Rooney, W.L., Klein, P.E. and Mullet, J.E. (2004) Genetic Diversity of Public Inbreds of Sorghum Determined by Mapped AFLP and SSR Markers. Crop Science, 44, 1236-1244. http://dx.doi.org/10.2135/cropsci2004.1236
- Warburton, M.L., Reif, J.C., Frisch, M., Bohn, M., Bedoya, C., Xia, X.C., Crossa, J., Franco, J., Hoisington, D., Pixley, K., Taba, S. and Melchinger, A.E. (2008) Genetic Diversity in CIMMYT Nontemperate Maize Germplasm: Landraces, Open Pollinated Varieties, and Inbred Lines. Crop Science, 48, 617-624. http://dx.doi.org/10.2135/cropsci2007.02.0103
- Sharon, D., Cregan, P.B., Mhameed, S., Kusharska, M., Hillel, J., Lahav, E. and Lavi, U. (1997) An Integrated Genetic Linkage Map of Avocado. Theoretical and Applied Genetics, 95, 911-921. http://dx.doi.org/10.1007/s001220050642
- Brown, S.M., Hopkins, M.S., Mitchell, S.E., Senior, M.L., Wang, T.Y., Duncan, R.R., Gonzalez-Candelas, F. and Kre- sovich, S. (1996) Multiple Methods for the Identification of Polymorphic Simple Sequence Repeats (SSRs) in Sorghum [Sorghum bicolor (L.) Moench]. Theoretical and Applied Genetics, 93, 190-198. http://dx.doi.org/10.1007/BF00225745
- Powell, W., Morgante, M., Andre, C., Hanafey, M., Vogel, J., Tingey, S. and Rafalski, A. (1996) The Comparison of RFLP, RAPD, AFLP and SSR (Microsatellite) Markers for Germplasm Analysis. Molecular Breeding, 3, 225-238. http://dx.doi.org/10.1007/BF00564200
- Casa, A.M., Mitchell, S.E., Hamblin, M.T., Sun, H., Bowers J.E., Paterson, A.H., Aquadro, C.F. and Kresovich, S. (2005) Diversity and Selection in Sorhum: Simultaneous Analyses Using Simple Sequence Repeats. Theoretical and Applied Genetics, 111, 23-30. http://dx.doi.org/10.1007/s00122-005-1952-5
- Perumal, R., Krishnaramanujan, R., Menz, M.A., Katilé, S., Dahlberg, J., Magill, C.W. and Rooney, W.L. (2007) Genetic Diversity among Sorghum Races and Working Groups Based on AFLPs and SSRs. Crop Science, 47, 1375-1383. http://dx.doi.org/10.2135/cropsci2006.08.0532
- Ali, M.L., Rajewski, J.F., Baenziger, P.S., Gill, K.S., Eskridge, K.M. and Dweikat, I. (2008) Assessment of Genetic Diversity and Relationship among a Collection of US Sweet Sorghum Germplasm by SSR Markers. Molecular Breeding, 21, 497-509. http://dx.doi.org/10.1007/s11032-007-9149-z
- Klein, R.R., Mullet, J.E., Jordan, D.R., Miller, F.R., Rooney, W.L., Menz, M.A., Franks, C.D. and Klein, P.E. (2008) The Effect of Tropical Sorghum Conversion and Inbred Development on Genome Diversity as Revealed by High-Re- solution Genotyping. Crop Science, 48, 512-526.
- Budak, H., Shearman, R.C., Parmaksiz, I., Gaussoin, R.E., Riordan, T.P. and Dweikat, I. (2004) Molecular Characterization of Buffalograss Germplasm Using Sequence-Related Amplified Polymorphism Markers. Theoretical and Applied Genetics, 108, 328-334. http://dx.doi.org/10.1007/s00122-003-1428-4
- Ariss, J.J. and Vandemark, G.J. (2007) Assessment of Genetic Diversity among Non-Dormant and Semi-Dormant Alfalfa Populations Using Sequence-Related Amplified Polymorphism. Crop Science, 47, 2274-2284. http://dx.doi.org/10.2135/cropsci2006.12.0782
- Zhao, W., Fang, R., Pan, Y., Yang, Y., Chung, J.W., Chung, I.M. and Park, Y.J. (2009) Analysis of Genetic Relationships of Mulberry (Morus L.) Germplasm Using Sequence-Related Amplified Polymorphism (SRAP) Markers. African Journal of Biotechnology, 8, 2604-2610.
- Han, X., Wang, L., Liu, Z.A., Jan, D.R. and Shu, Q. (2008) Characterization of Sequence-Related Amplified Polymor- phism Markers Analysis of Tree Peony Bud Sports. Scientia Horticulturae, 115, 261-267. http://dx.doi.org/10.1016/j.scienta.2007.09.003
- Budak, H., Shearman, R.C., Parmaksiz, I. and Dweikat, I. (2004) Comparative Analysis of Seeded and Vegetative Bi- otype Buffalograss Based on Phylogenetic Relationship Using ISSRs, SSRs, RAPDs, and SRAPs. Theoretical and Applied Genetics, 109, 280-288. http://dx.doi.org/10.1007/s00122-004-1630-z
- Lin, Z.X., Zhang, X.L. and Nie, Y.C. (2004) Evaluation of Application of a New Molecular Marker SRAP on Analysis of F2 Segregation Population and Genetic Diversity in Cotton. Acta Genetica Sinica, 31, 622-626.
- Ferriol, M., Pico, B. and Nuze, F. (2003) Genetic Diversity of a Germplasm Collection of Cucurbita pepo Using SRAP and AFLP Markers. Theoretical and Applied Genetics, 107, 271-282. http://dx.doi.org/10.1007/s00122-003-1242-z
- Fufa, H., Baenziger, P.S., Beecher, B.S., Dweikat, I., Graybosch, R.A. and Eskridge, K.M. (2005) Comparison of Phe- notypic and Molecular Marker-Based Classifications of Hard Red Winter Wheat Cultivars. Euphytica, 145, 133-146. http://dx.doi.org/10.1007/s10681-005-0626-3
- Zaefizadeh, M. and Goliev, R. (2009) Diversity and Relationships among Durum Wheat Landraces (Subconvars) by SRAP and Phenotypic Marker Polymorphism. Research Journal of Biological Sciences, 4, 960-966.
- Folkertsma R., Frederick, H., Rattunde, W., Chandra, S., Raju, G. and Hash, C. (2005) The Pattern of Genetic Diversity of Guinea-Race Sorghum bicolor (L.) Moench Landraces as Revealed with SSR Markers. Theoretical and Applied Genetics, 111, 399-409. http://dx.doi.org/10.1007/s00122-005-1949-0
- Gorz, H.J., Haskins, F.A. and Johnson, B.E. (1990) Registration of 15 Germplasm Lines of Grain Sorghum and Sweet Sorghum. Crop Science, 30, 762-763. http://dx.doi.org/10.2135/cropsci1990.0011183X003000030089x
- Dweikat, I. (2005) A Diploid, Interspecific, Fertile Hybrid from Cultivated Sorghum, Sorghum bicolor, and the Common Johnsongrass Weed Sorghum halepense. Molecular Breeding, 16, 93-101. http://dx.doi.org/10.1007/s11032-005-5021-1
- Schloss, S.J., Mitchell, S.E., White, G.M., Kukatla, R., Bowers, J.E., Paterson, A.H. and Kresovich, S. (2002) Characterization of RFLP Probe Sequences for Gene Discovery and SSR Development in Sorghum bicolor (L.) Moench. Theoretical and Applied Genetics, 105, 912-920. http://dx.doi.org/10.1007/s00122-002-0991-4
- Riaz, A., Li, G., Quresh, Z., Swati, M.S. and Quiros, C.F. (2001) Genetic Diversity of Oilseed Brassica napus Inbred Lines Based on Sequence-Related Amplified Polymorphism and Its Relation to Hybrid Performance. Plant Breeding, 120, 411-415. http://dx.doi.org/10.1046/j.1439-0523.2001.00636.x
- Kuleung, C., Baenziger, P. S. and Dweikat, I. (2004) Transferability of SSR Markers among Wheat, Rye and Triticale. Theoretical and Applied Genetics, 108, 1147-1150. http://dx.doi.org/10.1007/s00122-003-1532-5
- Flury, B., and Riedwyl, H. (1986) Standard Distance in Univariate and Multivariate Analysis. The American Statistician, 40, 249-251.
- SAS Institute Inc. (2008) User’s Guide, Version 9.2. SAS Institute Inc., Cary.
- Anderson, J.A., Churchill, G.A., Autrique, J.E., Sorells, M.E. and Tanksley, S.D. (1993) Optimizing Parental Selection for Genetic Linkage Maps. Genome, 36, 181-186. http://dx.doi.org/10.1139/g93-024
- Botstein, D., White, R.L. Skolnick, M. and Davis, R.W. (1990) Construction of a Genetic Linkage Map in Man Using Restriction Fragment Length Polymorphism. The American Journal of Human Genetics, 32, 314-331.
- Nei, M. and Li, W.H. (1979) Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonuclea- ses. Proceedings of the National Academy of Sciences of the United States of America, 76, 5269-5273. http://dx.doi.org/10.1073/pnas.76.10.5269
- Weising, K., Nyborn, H., Wolff, K. and Kahl, G. (2005) DNA Fingerprinting in Plants: Principles, Methods, and Applications. 2nd Edition, CRC Press, New York. http://dx.doi.org/10.1201/9781420040043
- Nei, M. (1972) Genetic Distance between Populations. The American Naturalist, 106, 283-292. http://dx.doi.org/10.1086/282771
- Takezaki, N. and Nei, M. (1996) Genetic Distances and Reconstruction of Phylogenetic Tree from Microsatellite DNA. Genetics, 144, 389-399.
- Page, R.D.M. (1996) TreeView: An Application to Display Phylogenetic Trees on Personal Computers. Computer Applications in the Biosciences, 12, 357-358.
- Wang, M.L., Zhu, C., Barkley, N.A., Chen, Z., Erpelding, J.E., Murray, S.C., Tuinstra, M.R., Tesso, T., Paterson, G.A. and Yu, J. (2009) Genetic Diversity and Population Structure Analysis of Accessions in the US Historic Sweet Sorghum Collection. Theoretical and Applied Genetics, 120, 13-23. http://dx.doi.org/10.1007/s00122-009-1155-6
Appendix 1
List of germplasm accessions used in diversity study, their origin, year of registration and parentage.
Appendix 2
Least square means of anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) for the 142 accessions evaluated at Mead in 2009.
Appendix 3
The cluster groups based on average distances of anthesis date (AD), plant height (PH), moisture content (ML) and total biomass (DM) planted at Mead in 2009.