Open Journal of Genetics
Vol.06 No.01(2016), Article ID:65275,11 pages
10.4236/ojgen.2016.61004
Genetic Linkage Map and Comparative Genome Analysis for the Atlantic Killifish (Fundulus heteroclitus)
Eric R. Waits1*, John Martinson1, Brian Rinner1, Stephen Morris1, Dina Proestou2, Denise Champlin3, Diane Nacci3
1Office of Research and Development, US Environmental Protection Agency, Cincinnati, USA
2Agricultural Research Service, US Department of Agriculture, Kingston, USA
3Office of Research and Development, US Environmental Protection Agency, Narragansett, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 November 2015; accepted 28 March 2016; published 31 March 2016
ABSTRACT
Genetic linkage maps are valuable tools in evolutionary biology; however, their availability for wild populations is extremely limited. Fundulus heteroclitus (Atlantic killifish) is a non-migratory estuarine fish that exhibits high allelic and phenotypic diversity partitioned among subpopulations that reside in disparate environmental conditions. An ideal candidate model organism for studying gene-environment interactions, the molecular toolbox for F. heteroclitus is limited. We identified hundreds of novel microsatellites which, when combined with existing microsatellites and single nucleotide polymorphisms (SNPs), were used to construct the first genetic linkage map for this species. By integrating independent linkage maps from three genetic crosses, we developed a consensus map containing 24 linkage groups, consistent with the number of chromosomes reported for this species. These linkage groups span 2300 centimorgans (cM) of recombinant genomic space, intermediate in size relative to the current linkage maps for the teleosts, medaka and zebrafish. Comparisons between fish genomes support a high degree of synteny between the consensus F. heteroclitus linkage map and the medaka and (to a lesser extent) zebrafish physical genome assemblies.
Keywords:
Fundulus heteroclitus, Genetic Linkage Map, Synteny Analysis, Microsatellite, Single Nucleotide Polymorphism (SNP)

1. Introduction
The estuarine fish Fundulus heteroclitus (Atlantic killifish) is an important ecological model for studying regional and local adaptation [1] . Killifish are relatively non-migratory and have large effective population (Ne) [2] sizes which provide evolutionary arenas for natural selection to occur. Their extensive genetic diversity coupled with their occurrence in varying environmental conditions has led to adaptive variation in several physiological and biochemical traits, including susceptibility to toxic xenobiotics [1] [3] - [6] . The extensive phenotypic variation and genetic diversity exhibited by killifish make them an ideal candidate for studying evolutionary biology including questions of quantitative genetic variation.
Since the post-genomic era, the molecular toolbox for F. heteroclitus has expanded from a few genetic loci allowing for population genetic studies [7] [8] to numerous loci distributed across the genome [9] - [11] . As the repertoire of genetic markers increases, it is becoming more feasible to perform genetic association studies using Fundulus as a model. However, a genetic linkage map has not yet been developed for this species, prohibiting localization of chromosomal regions containing the genetic variation responsible for phenotypic traits.
To further advance F. heteroclitus as a functional genetic model organism, we developed enriched microsatellite libraries and identified 330 novel microsatellites. Parental (P) and Second Filial (F2) generations of three genetic crosses were genotyped with the microsatellites reported herein, existing microsatellites [7] - [9] , and single nucleotide polymorphisms (SNPs) [10] [11] . Based on the segregation patterns of polymorphic loci in the recombinant generations, we constructed the first genetic linkage map of the killifish genome. Comparisons between this killifish genetic map and published genomic assemblies for medaka (Oryzias latipes) and zebrafish (Danio rerio) suggest relatively high degrees of synteny among fish species, and provide additional resources to test evolutionary models and hypotheses.
2. Methods and Materials
2.1. Killifish Collection and Breeding Design
Parental (P) fish for genetic crosses were collected from New Bedford Harbor (NBH), Massachusetts, USA, and Block Island (BI), Rhode Island, USA, and were maintained as previously described [4] . Following a classic F2-intercross design, BI males were crossed with NBH females to generate the first filial generation (F1). Once sexually mature, F1 siblings were crossed (F1 × F1) to generate the recombinant F2 generation. F2 larvae from each of three independent pedigrees were collected and used to construct an integrated consensus map of the killifish genome. All research including fish collection, culture, and breeding have been approved and adhere to the guidelines set forth by USEPAs Institutional Animal Care and Use Committee (IACUC).
2.2. Microsatellites
Genomic libraries enriched for microsatellites were developed following the methods of Hamilton et al. [12] and Glenn et al. [13] . Briefly, genomic DNA extracted from eight larval fish (DNeasy, Qiagen) was pooled and digested with Hae III and Rsa I (NEB) and ligated to double-stranded linkers (SNX-f-5’-CTAAGGCCTT- GCTAGCAGAAGC-3’and SNX-r-5’-pGCTTCTGCTAGCAAGGCCTTAGAAAA-3’). Microsatellite probes (5’-biotinylated) were used to capture fragments containing microsatellite repeats on streptavidin-coated beads. Microsatellite-enriched single stranded DNA was amplified using the SNX-f primer to recover double-stranded DNA. PCR amplification products of clones (TOPO 2.1, Invitrogen) were sequenced (ABI 3730 Genetic Analyser) and screened for microsatellites. Primers for candidate microsatellites were designed using Primer 3 [14] .
2.3. Genotyping
Parental fish (n = 6) and 95 F2 embryos from each of three genetic crosses (total n = 291) were genotyped with the microsatellites developed herein, as well as with previously published markers including microsatellites [7] - [9] , and SNPs [10] [11] . Polymerase chain reactions and genotyping were performed according to Waits and Nebert, 2011 [15] , for microsatellite markers, and according to Williams and Oleksiak, 2011 [10] , for SNPs.
2.4. Linkage Mapping
Linkage groups were identified based on estimated two-point logarithm of odds (LOD) scores and Haldane distances. For each genetic cross, a minimum LOD score of 3.0 and a maximum recombination rate (rf) of 0.5 were used as threshold values to determine linkage groups [16] . Within each linkage group, marker order was determined by examining all pairs of markers, starting with the pair having the highest maximum likelihood and incrementally adding markers (most strongly linked first) always choosing the highest log likelihood and the best insertion point . Linkage maps were then improved and validated by perturbation of the initial maps in a systematic manner [16] . First, the “flips” algorithm was used to evaluate all possible permutations of marker order in a sliding window of fixed size (window = 5) on the initial map. Initial maps were replaced with improved maps as identified [16] . These maps were then interrogated by “polishing,” which displaces each marker in all possible intervals in search of a map having a better log-likelihood [16] . For the consensus map, markers in common among the three families were used to anchor an integrated map. Linkage groups were identified and an initial framework map was built using stringent criteria (LOD > 10, rf < 0.2), which were then incrementally relaxed (to LOD ≥ 3.0, rf ≤ 0.5). Marker order within each consensus linkage group was determined as described above.
2.5. Marker Identity
To identify microsatellite and SNP marker identities, an annotated draft of the F. heteroclitus genome (supported by National Science Foundation grant, DEB-1120512) was queried. Specifically, marker sequences were assigned to annotated killifish gene models by performing nucleotide BLAST searches against the killifish genome sequence. Markers were assigned a gene name when the marker sequence fell within an annotated gene model and the match between the marker sequence and the gene model sequence spanned at least 150 bp and exceeded 96% identity.
2.6. Comparative Genome Analysis
Syntenic relationships were explored for the consensus linkage map against two other teleost species for which an annotated genome assembly is available: medaka (Oryzias latipes) [17] and zebrafish (Danio rerio) [18] . Synteny with Tilapia (Oreochromis niloticus) was also explored, however, the structure of tilapia genome is less well defined precluding the comparison. Orthologs were assigned by reciprocal BLAST and predicted proteins from the annotated killifish gene models were compared to the zebrafish and medaka non-redundant protein databases (NCBI). Orthology was confirmed if the BLAST search of zebrafish/medaka genes against the killifish genome returned the original killifish gene model as the primary hit and exhibited an e-value < 1e−5. To validate and augment the list of orthologs, an additional query was performed by blasting the individual marker sequences against sequence data for D. rerio and O. latipes (Ensembl).
3. Results
3.1. Novel Microsatellite Markers
Approximately 1200 clones were sequenced from the microsatellite-enriched libraries and surveyed for tandem repeats with sufficient flanking sequence for oligonucleotide design. One-third of the sequences contained di-, tri-, or tetra-nucleotide repeats that met these criteria. Of these, 330 candidate loci were selected for primer design and evaluated for amplification success and polymorphism in the parental generation used to establish genetic crosses. In the parental generation of the three genetic crosses, 59, 65, and 94 loci, respectively, proved polymorphic and Informative. A X2 (chi-square) goodness-of-fit test for deviation from expected 1:2:1 Mendelian ratios identified a small proportion of markers showing segregation distortion (Supplementary Data, Table 1(a)) in the 285 F2 larvae genotyped (following Bonferonni correction, α = 0.00015). Moderate departures from these frequencies are not unusual and may indicate the presence of partially lethal alleles. Gross departures from these frequencies may indicate problematic markers or null alleles [19] .
3.2. Linkage Mapping
Linkage maps were constructed for each of the three mapping families. A total of 75, 88, and 105 loci clustered into 16, 21, and 23 linkage groups (Lg), respectively (Supplementary Data, Table 1(b)). Genetic loci incommon among the three families were used to anchor and join the individual maps into one integrated consensus map (Figure 1). At a threshold of LOD ≥ 3.0 and a recombination frequency ≤ 0.5, twenty-four linkage groups were identified. Together, the 24 linkage groups contain 240 loci covering 2300 cM total. Individual linkage groups range from 35 to 195 cM with an average marker resolution of 9.5 cM.
Overall, the integrated consensus map is in good agreement with the maps based on individual genetic crosses. Marker order is conserved between the consensus map and individual maps with few exceptions. Exceptions include the relative locations of Fhe_2179 and Fhe_495 in cross A, which are reversed relative to cross B, C, and the consensus map. This discrepancy is likely due to heterogeneity in recombination rates between source popula- tions and the extreme segregation distortion of Fhe_2179 (p = 2.27E−10). Other loci with inconsistent positioning due to segregation distortion in at least one of the three genetic crosses include: Fhe_2254 (p = 5.88E−19), Fhe_2277 (p = 7.31E−06), and Fhe_2027 (p = 2.50E−06). Inter-marker distances in the consensus map are

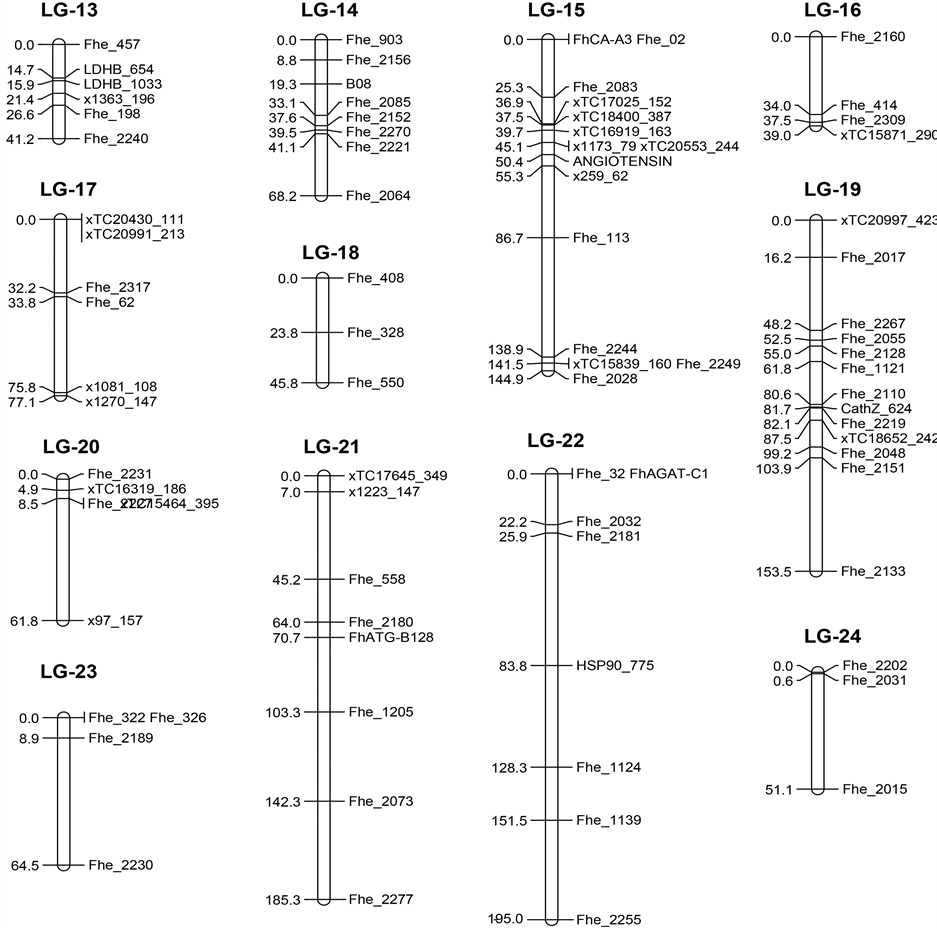
Figure 1. Consensus genetic linkage map for Fundulus heteroclitus. Detailed information for the microsatellite and SNP markers (gene, DNA sequence, map position, source publication) used to construct linkage maps can be found in Supplementary Data, Table 1. All novel microsatellite loci reported herein can be accessed at GenBank at the NCBI (GenBank: KT001557-KT001910).
also consistent (±5 cM) with individual based maps barring three exceptions: xTC15606_316 and xTC19271_848 (Lg-9 consensus, Lg-7 genetic cross B) are separated by 35.6 cM on the consensus map and 15.5 cM on the individual map; Fhe_2151 and Fhe_2133 (Lg-19 consensus, Lg-13 genetic cross C) are separated by 84 cM on the individual map and 49.6 cM on the consensus map; and xTC21107_340 and Fhe_2069 (Lg-5 consensus, Lg-9 genetic cross C) are separated by 20.5 cM on the consensus map and 13.8 cM on the individual map.
3.3. Comparative Genome Analysis
Of the 240 loci comprising the consensus linkage map, 181 loci fell within a gene model in the draft Fundulus genome (Supplementary Data, Table 1(a)) and were assigned identities. Four additional loci were named based on significant hits to the zebrafish and medaka genome assemblies. In order to perform a comparative genome analysis, zebrafish and medaka orthologs and their genomic coordinates were identified (NCBI; non- redundant protein database) based on the predicted protein sequences of the Fundulus gene models. The identified homologous genes were used to determine homologous chromosomes in medaka and zebrafish (Figure 2). Comparing killifish and medaka, 10 killifish linkage groups (2, 20, 10, 9, 6, 11, 21, 3, 16, 1) exhibit 1:1 relationships with medaka chromosomes and for 7 other linkage groups (5, 19, 7, 4, 17, 15, 8, 22), >75% of killifish loci fall on a single medaka chromosome with only a single locus on a second medaka chromosome. For LG4, 7 loci fall on medaka CHR10, 2 on CHR18, and one on CHR21. Only one medaka chromosome (CHR2) does not share homologous loci with the consensus Fundulus linkage map. A comparative coordinate analysis of loci on the Fundulus linkage map and medaka genome assembly reveal conservation of gene order for approximately 70% of shared loci as well as multiple rearrangements (Figure 3).
 (a)
(a) (b)
(b)
Figure 2. Homologous linkage map-to-chromosomal relationships between F. heteroclitus and medaka (a) or zebrafish (b). In each case, the F. heteroclitus linkage groups (LG) are displayed in rows and the zebrafish/medaka chromosomes are displayed in columns. The numbers in each cell represent number of homologous loci between the F. heteroclitus consensus linkage map and the other model fish species genomes.
 (a)
(a) (b)
(b)
Figure 3. Comparison of gene order between the F. heteroclitus linkage map and the medaka or zebrafish genomes. Map distances (cM) and genome coordinates (Mb) are the accumulated sum of the F. heteroclitus linkage groups (Lg1-Lg24) and additive physical length for medaka chromosomes 1-24 (a) and zebrafish chromosomes 1-25 (b). Each data point represents a gene and its Cartesian coordinates. An abrupt change in coordinates between adjacent genes indicates a rearrangement.
Homologous chromosomes in zebrafish are less apparent. Three killifish linkage groups (12, 8, 22) exhibit 1:1 relationships with zebrafish chromosomes and for 7 other linkage groups (2, 9, 15, 4, 7, 16, 10), >65% of killifish loci fall on a single medaka chromosome with multiple loci located on up to three additional zebrafish chromosomes (Figure 2). Overall, at least 22 of 25 zebrafish chromosomes contain loci from between two and five Fundulus linkage groups. A comparative coordinate analysis of loci of the Fundulus linkage map and zebrafish genome assembly reveals a higher proportion of rearrangements in gene order (Figure 3) than observed when comparing to Medaka.
4. Discussion
The rapid advancement of molecular-genetic technologies has made genome-wide quantitative genetic and population genetic surveys readily assessable. Molecular markers distributed across the genome can be used to assess critical ecological and evolutionary issues such as resilience, local adaptation, population connectivity, and invasion. In this study we report several novel co-dominant microsatellite markers which we have leveraged in combination with many previously published molecular markers to construct the first genetic linkage map for the small estuarine fish model, Fundulus heteroclitus. By integrating independent linkage maps from three F2- intercrosses, we developed a consensus map containing twenty-four linkage groups, consistent with the number of chromosomes (2N = 48) reported for F. heteroclitus [20] [21] . Comparing the individual and consensus linkage maps, there are minor inconsistencies in relative marker position in approximately 4% of loci. The recombinant larvae genotyped from each family to create the linkage maps are descendant from parental generations established by crossing males and females from two different populations. Since the recombination histories of different populations may vary, slight inconsistencies in recombination frequencies when comparing the maps are expected [22] . This is not an uncommon phenomena [23] [24] and does not compromise the utility of a composite map [25] [26] . Differential recombination rates between sexes are also common, with females often having more recombination and longer genetic maps than males [27] . However, the recombinant individuals were not sexually mature when genotyped precluding us from ascertaining male-female linkage variance among loci.
Overall, the 24 Fundulus linkage groups represent 2300 centimorgans (cM) of recombinant genomic space, intermediate in size relative to the current linkage maps for medaka (1401.5 cM) [28] and zebrafish (3192 cM) [29] . A large degree of synteny is observed between the consensus killifish linkage map and the medaka or zebrafish physical genome assemblies with the highest degree of syntenty between medaka and Fundulus (Figures 2-4).
The higher degree of synteny between medaka and Fundulus genome organization is consistent with their taxonomic classifications. All three teleost species are euteleostein with Fundulus and medaka containing 24 chromosomes (2N = 48) and representing the subdivision neoteleostei versus zebrafish with 25 chromosomes (2N = 50) in the subdivision ostariophysi. Several comparative phylogenetic [30] and phylogenomic [31] [32] assessments of teleost fish support this taxonomic structure and relative evolutionary distances. Earlier theories explaining genome evolution suggest synteny is an artifact of ancestral linkages that have not yet been disrupted by random micro- and macro-rearrangements such as indels, inversions and translocations [33] . It has since become apparent that gene distribution in eukaryotic genomes is not always random [34] . For example, co-regu- lated genes and their regulatory elements often exhibit strict conservation of both gene order and linkage [35] - [37] .
Teleost fish species provide an outstanding model to study evolution at the genetic and genomic levels. Their genomes exhibit considerable plasticity in size, chromosome number [38] [39] , and in the expression of phenotypic traits [4] [15] [40] [41] . An extensive molecular tool-box exists for zebrafish and the Japanese medaka including annotated genome assemblies, genetic maps [28] [29] , and commercially available microarray technologies. In both species, gene function can be studied routinely in vivo via trans-genesis, “morpholino” gene- knockdowns, and large-scale mutagenesis [42] [43] . Like zebrafish and medaka, genetic and genomic resources for F. heteroclitus are emerging at rapid pace. The novel polymorphic microsatellite loci and genetic map reported herein will contribute to the growing F. heteroclitus molecular toolbox. For example, the microsatellites

Figure 4. An example of syntenic relationships between F. heteroclitus linkage groups and their respective homologous chromosomes in the zebrafish and medaka genomes.
have utility for population genetic studies and the genetic map can serve as a tool for quantitative trait analysis (QTL) of the adaptive variation observed in F. heteroclitus [1] by localizing chromosomal regions containing candidate genes contributing to phenotypic traits.
5. Conclusion
In conclusion, our genetic linkage map, in conjunction with other emerging genetic and genomic tools, will elevate the status of F. heteroclitus within the ranks of small fish genetic models.
Acknowledgements
We thank Marjorie Oleksiak for contributing single nucleotide polymorphism reagents for inclusion on the linkage map. We also thank Jeffery Markert for thoughtful comments which lead to the improvement of this manuscript. Data interpretation was aided by reference to a preliminary draft of the F. heteroclitus genome sequence which was supported by funding from the National Science Foundation (DEB-1120512). The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
Cite this paper
Eric R. Waits,John Martinson,Brian Rinner,Stephen Morris,Dina Proestou,Denise Champlin,Diane Nacci, (2016) Genetic Linkage Map and Comparative Genome Analysis for the Atlantic Killifish (Fundulus heteroclitus). Open Journal of Genetics,06,28-38. doi: 10.4236/ojgen.2016.61004
References
- 1. Burnett, K.G., Bain, L.J., Baldwin, W.S., Callard, G.V., Cohen, S., Di Giulio, R., et al. (2007) Fundulus as the Premier Teleost Model in Environmental Biology: Opportunities for New Insights Using Genomics. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 2, 257-286.
http://dx.doi.org/10.1016/j.cbd.2007.09.001 - 2. Whitehead, A., Lindmeier, J.B., Duvernell, D.D. and Faust, K.E. (2008) Relative Influences of Historical and Contemporary Forces Shaping the Distribution of Genetic Variation in the Atlantic Killifish, Fundulus heteroclitus. Molecular Ecology, 17, 1344-1360.
http://dx.doi.org/10.1111/j.1365-294X.2007.03648.x - 3. Nacci, D., Coiro, L., Champlin, D., Jayaraman, S., McKinney, R., Gleason, T., et al. (1999) Adaptation of Wild Fish Populations to Dioxin-Like Environmental Contamination. Marine Biology, 134, 9-17.
http://dx.doi.org/10.1007/s002270050520 - 4. Nacci, D., Champlin, D. and Jayaraman, S. (2010) Adaptation of the Estuarine Fish Fundulus heteroclitus to Toxic Pollutants. Estuaries and Coasts, 33, 853-864.
http://dx.doi.org/10.1007/s12237-009-9257-6 - 5. Van Veld, P.A. and Nacci, D. (2008) Chemical Tolerance: Acclimation and Adaptations to Chemical Stress. In: Di Giulio, R.T. and Hinton, D.E., Eds., The Toxicology of Fishes, Taylor and Francis, USA.
- 6. Whitehead, A., Pilcher, W., Champlin, D. and Nacci, D. (2012) Common Mechanism Underlies Repeated Evolution of Extreme Pollution Tolerance. Proceedings of the Royal Society B: Biological Sciences, 279, 427-433.
http://dx.doi.org/10.1098/rspb.2011.0847 - 7. Adams, A.M., Oleksiak, M.F. and Duvernell, D. (2005) Microsatellite Primers for the Atlantic Coastal Killifish, Fundulus heteroclitus, with Applicability to Related Fundulus Species. Molecular Ecology Notes, 5, 275-277.
http://dx.doi.org/10.1111/j.1471-8286.2005.00898.x - 8. Li, C., Bessert, M.L., Macrander, J. and Orti, G. (2007) Microsatellite Loci for the Plains Topminnow. (Fundulus sciadicus, Fundulidae). Molecular Ecology Notes, 7, 691-693.
http://dx.doi.org/10.1111/j.1471-8286.2007.01679.x - 9. Jackson, S.A., Bagley, M., Whitehead, A., Roberts, D.A., Duvernell, D., Nacci, D. and Wang, R.-L. (2010) Isolation and Characterization of Novel Microsatellite Loci for Atlantic Coastal Killifish (Fundulus heteroclitus) from an Expressed Sequence Tag (EST) Library and Cross-Species Amplification. Molecular Ecology Resources, 10, 1098-1105.
- 10. Williams, L.M. and Oleksiak, M.F. (2011) Selectively Important SNPs Identified in Natural Populations. Molecular Biology and Evolution, 28, 1817-1826.
http://dx.doi.org/10.1093/molbev/msr004 - 11. Proestou, D., Flight, P., Champlin, D. and Nacci, D. (2014) Targeted Approach to Identify Genetic Loci Associated with Evolved Dioxin Tolerance in Atlantic Killifish (Fundulus heteroclitus). BMC Evolutionary Biology, 14, 7.
http://dx.doi.org/10.1186/1471-2148-14-7 - 12. Hamilton, M.B., Pincus, E.L., Di-Fiore, A. and Fleischer, R.C. (1999) Universal Linker and Ligation Procedures for Construction of Genomic DNA Libraries Enriched for Microsatellites. Biotechniques, 27, 500-507.
- 13. Glenn, T.C. and Schable, N.A. (2005) Isolating Microsatellite DNA Loci. In: Zimmer, E.A. and Roalson, E., Eds., Molecular Evolution: Producing the Biochemical Data, Part B, Academic, USA, 202-222.
http://dx.doi.org/10.1016/s0076-6879(05)95013-1 - 14. Rozen, S. and Skaletsky, H.J. (2000) Primer3 on the WWW for General Users and for Biologist Programmers. In: Krawetz, S. and Misener, S., Eds., Bioinformatics Methods and Protocols: Methods in Molecular Biology, Humana Press, USA.
- 15. Waits, E.R. and Nebert, D.W. (2011) Genetic Architecture of Susceptibility to PCB126-Induced Developmental Cardiotoxicity in Zebrafish. Toxicological Sciences, 122, 466-475.
http://dx.doi.org/10.1093/toxsci/kfr136 - 16. De Givry, S., Bouchez, M., Chabrier, P., Milan, D. and Schiex, T. (2005) CARTHAGENE: Multipopulation Integrated Genetic and Radiation Hybrid Mapping. Bioinformatics, 21, 1703-1704.
http://dx.doi.org/10.1093/bioinformatics/bti222 - 17. Kasahara, M., Naruse, K., Sasaki, S., Nakatani, Y., Qu, W., Ahsan, B., et al. (2007) The Medaka Draft Genome and Insights into Vertebrate Genome Evolution. Nature, 447, 714-719.
http://dx.doi.org/10.1038/nature05846 - 18. Howe, K., Clark, M.D., Torroja, C.F., Torrance, J., Berthelot, C., Muffato, M., et al. (2013) The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature, 496, 498-503.
http://dx.doi.org/10.1038/nature12111 - 19. Dakin, E.E. and Avise, J.C. (2004) Microsatellite Null Alleles in Parentage Analysis. Heredity, 93, 504-509.
http://dx.doi.org/10.1038/sj.hdy.6800545 - 20. Chen, T.R. and Ruddle, F.H. (1969) A Chromosome Study of Four Species and a Hybrid of the Killifish Genus Fundulus (Cyprinodontidae). Chromosoma, 29, 255-267.
http://dx.doi.org/10.1007/BF00325942 - 21. Chen, T.R. (1971) A Comparative Chromosome Study of Twenty Killifish Species of the Genus Fundulus (Teleostei: Cyprinodontidae). Chromosoma, 32, 436-453.
http://dx.doi.org/10.1007/BF00285254 - 22. Beavis, W.D. and Grant, D. (1991) A Linkage Map Based on Information from Four F2 Populations of Maize (Zea mays L.). Theoretical and Applied Genetics, 82, 636-644.
http://dx.doi.org/10.1007/BF00226803 - 23. Lombard, V. and Delourme, R. (2001) A Consensus Linkage Map for Rapeseed (Brassica napus L.): Construction and Integration of Three Individual Maps from DH Populations. Theoretical and Applied Genetics, 103, 491-507.
http://dx.doi.org/10.1007/s001220100560 - 24. Loridon, K., McPhee, K., Morin, J., Dubreuil, P., Pilet-Nayel, M., Aubert, G., et al. (2005) Microsatellite Marker Polymorphism and Mapping in Pea (Pisum sativum L.). Theoretical and Applied Genetics, 111, 1022-1031.
http://dx.doi.org/10.1007/s00122-005-0014-3 - 25. Lespinasse, D., Rodier-Goud, M., Grivet, L., Leconte, A., Legnate, H. and Seguin, M. (2000) A Saturated Genetic Linkage Map of Rubber Tree (Hevea spp.) Based on RFLP, AFLP, Microsatellite, and Isozyme Markers. Theoretical and Applied Genetics, 100, 127-138.
http://dx.doi.org/10.1007/s001220050018 - 26. N’Diaye, A., Van de Weg, W.E., Kodde, L.P., Koller, B., Dunemann, F., Thiermann, M., et al. (2008) Construction of an Integrated Consensus Map of the Apple Genome Based on Four Mapping Populations. Tree Genetics & Genomes, 4, 727-743.
http://dx.doi.org/10.1007/s11295-008-0146-0 - 27. Hedrick, P.W. (2007) Sex: Differences in Mutation, Recombination, Selection, Gene Flow, and Genetic Drift. Evolution, 61, 2750-2771.
http://dx.doi.org/10.1111/j.1558-5646.2007.00250.x - 28. Naruse, K., Tanaka, M., Mita, K., Shima, A., Postlethwait, J. and Mitani, H. (2004) A Medaka Gene Map: The Trace of Ancestral Vertebrate Proto-Chromosomes Revealed by Comparative Gene Mapping. Genome Research, 14, 820-828.
http://dx.doi.org/10.1101/gr.2004004 - 29. Woods, I.G., Wilson, C., Friedlander, B., Chang, P., Reyes, D.K., Nix, R., et al. (2005) The Zebrafish Gene Map Defines Ancestral Vertebrate Chromosomes. Genome Research, 15, 1307-1314.
http://dx.doi.org/10.1101/gr.4134305 - 30. Yan, J. and Cai, Z. (2010) Molecular Evolution and Functional Divergence of the Cytochrome P450 3 (CYP3) Family in Actinopterygii (Ray-Finned Fish). PLoS ONE, 5, e14276.
http://dx.doi.org/10.1371/journal.pone.0014276 - 31. Thomas-Chollier, M. and Ledent, V. (2008) Comparative Phylogenomic Analyses of Teleost Fish Hox Gene Clusters: Lessons from the Cichlid Fish Astatotilapia burtoni. BMC Genomics, 9, 35.
http://dx.doi.org/10.1186/1471-2164-9-35 - 32. Cheung, N.K., Cheung, A.C., Ye, R.R., Ge, W., Giesy, J.P. and Au, D.W. (2013) Expression Profile of Oestrogen Receptors and Oestrogen-Related Receptors Is Organ Specific and Sex Dependent: The Japanese Medaka Oryzias latipes Model. Journal of Fish Biology, 83, 295-310.
http://dx.doi.org/10.1111/jfb.12164 - 33. Nadeau, J.H. and Taylor, B.A. (1984) Lengths of Chromosomal Segments Conserved since Divergence of Man and Mouse. Proceedings of the National Academy of Sciences of the United States of America, 81, 814-818.
http://dx.doi.org/10.1073/pnas.81.3.814 - 34. Hurst, L.D., Pal, C. and Lercher, M.J. (2004) The Evolutionary Dynamics of Eukaryotic Gene Order. Nature Reviews Genetics, 5, 299-310.
http://dx.doi.org/10.1038/nrg1319 - 35. Santini, S., Boore, J.L. and Meyer, A. (2003) Evolutionary Conservation of Regulatory Elements in Vertebrate Hox Gene Clusters. Genome Research, 13, 1111-1122.
http://dx.doi.org/10.1101/gr.700503 - 36. Engstrom, P.G., Sui, S.J.H., Drivenes, O., Becker, T.S. and Lenhard, B. (2007) Genomic Regulatory Blocks Underlie Extensive Microsynteny Conservation in Insects. Genome Research, 17, 1898-1908.
http://dx.doi.org/10.1101/gr.6669607 - 37. Kikuta, H., Laplante, M., Navratilova, P., Komisarczuk, A.Z., Engstrom, P.G., Fredman, D., et al. (2007) Genomic Regulatory Blocks Encompass Multiple Neighboring Genes and Maintain Conserved Synteny in Vertebrates. Genome Research, 17, 545-555.
http://dx.doi.org/10.1101/gr.6086307 - 38. Venkatesh, B. (2003) Evolution and Diversity of Fish Genomes. Current Opinion in Genetics & Development, 13, 588-592.
http://dx.doi.org/10.1016/j.gde.2003.09.001 - 39. Volff, J.N. (2004) Genome Evolution and Biodiversity in Teleost Fish. Heredity, 94, 280-294.
http://dx.doi.org/10.1038/sj.hdy.6800635 - 40. Mitton, J.B. (1994) Molecular Approaches to Population Biology. Annual Review of Ecology and Systematics, 25, 45-69.
http://dx.doi.org/10.1146/annurev.es.25.110194.000401 - 41. Wirgin, I. and Waldman, J.R. (2004) Resistance to Contaminants in North American Fish Populations. Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis, 552, 73-100.
http://dx.doi.org/10.1016/j.mrfmmm.2004.06.005 - 42. Driever, W., Solnica-Krezel, L., Schier, A.F., Neuhauss, S.C., Malicki, J., Stemple, D.L., et al. (1996) A Genetic Screen for Mutations Affecting Embryogenesis in Zebrafish. Development, 123, 37-46.
- 43. Furutani-Seiki, M., Sasado, T., Morinaga, C., Suwa, H., Niwa, K., Yoda, H., et al. (2004) A Systematic Genome-Wide Screen for Mutations Affecting Organogenesis in Medaka, Oryzias latipes. Mechanisms of Development, 121, 647-658.
http://dx.doi.org/10.1016/j.mod.2004.04.016
Supplementary Data
Supplementary Data Table 1(a) and Table 1(b) can be viewed online at: https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/JNZKCJ
NOTES

*Corresponding author.