Open Journal of Genetics
Vol.2 No.1(2012), Article ID:17766,8 pages DOI:10.4236/ojgen.2012.21004
The mus309 mutation, defective in DNA double-strand break repair, increases the frequency of X-ray-induced somatic crossing over in Drosophila melanogaster, but the effect is not dose-rate dependent
![]()
Laboratory of Genetics, Department of Biology, University of Turku, Turku, Finland
Email: petter.portin@utu.fi
Received 9 November 2011; revised 16 December 2011; accepted 17 January 2012
Keywords: Ionizing Radiation; Mitosis; Mutagenic Sensitivity; RecQ Protein; X Chromosome
ABSTRACT
Effect of a 1000 R dose of hard X-rays, with two different dose-rates viz. 300 and 1000 R/min on somatic crossing over in the X chromosome of Drosophila melanogaster was studied in two different genotypes. Irradiation was given during the first-instar larval stage of the development. In the control crosses the flies carried wild-type autosomes, but in the experimental crosses the 3rd chromosomes carried a DNA double-strand break repair deficient mus309 mutant gene constitution. As expected, the frequency of X-rayinduced somatic crossing over increased in the mutant flies with both dose-rates of irradiation. As also expected, in the control flies irradiation given with the 300 R/min dose-rate caused more somatic crossovers than irradiation given with the 1000 R/min rate. However, rather unexpectedly, in the experimental flies there was no significant difference in the frequency of somatic crossing over between the two dose-rates of irradiation. The results can be explained by assuming that X-ray-induced somatic crossing over is a two-step event, and that the mechanism which repairs the lesion caused by the irradiation is controlled by the mus309 gene. In the control flies the repairing mechanism is capable to recover if the irradiation is given with a short term high dose-rate, but is not capable to recover if the irradiation is given with a long lasting low dose-rate. However, in the experimental mutant flies the repairing mechanism is only poorly recovered irrespective of the doserate.
1. INTRODUCTION
1.1. General Introduction
DNA double-strand breaks (DSBs) are considered the most lethal form of DNA damage. They can result from either endogenous or exogenous sources. Naturally occurring DSBs are generated spontaneously during DNA synthesis when the replication fork encounters a damaged template, and during certain cellular processes. Exogenous factors which cause DSBs are for example ionizing radiation, UV light and radiomimetic drugs [1,2].
Failure to repair DSBs, or their misrepair, may result in cell death or chromosomal rearrangements, including deletions and translocations or genome instability in general. Two major pathways have evolved to repair DSBs and thereby suppress genomic instability. These are the non-homologous end-joining (NHEJ) pathway and homologous recombination (HR), also called homology-directed repair (HDR) pathway [1,2].
The HDR pathway is widely regarded as an accurate error-free form of repair, which requires the presence of a homologous template, such as a sister chromatid, and functions only after DNA replication [3-5]. The NHEJ pathway joins the two ends of a DSB through a process largely independent of homology. In its simplest form it entails straightforward ligation of DNA ends [1,2]. In contrast to HDR, NHEJ is active throughout the cell cycle [6], and it is also considered the major pathway for the repair of irradiation-induced DSBs at least in human cells [7]. Of these two major pathways, however, only the HDR pathway can lead to crossing over.
Two alternative pathways for the repair of the DSBs by homologous recombination are known. They are the synthesis-dependent strand annealing (SDSA) pathway and the double-strand-break repair (DSBR) pathway [8]. The former pathway leads exclusively to non-crossover products and the latter to both crossover and non-crossover products [9,10].
1.2. The mus309 Locus of Drosophila melanogaster and Its Role in DSB Repair
The mus309 locus, also known e.g. as DmBlm and Ku70, on the right arm of chromosome three of Drosophila melanogaster has been identified as a mutagen sensitive locus [11]. It encodes, in a manner similar to its orthologues in other organisms, the mammalian BLM locus included, a RecQ helicase [12-15] and, accordingly, is involved in DSB repair [9,10,16]. Among other mutagens, mus309 mutants are, of course, sensitive to ionizing radiation as well [17]. In humans, gene defects in BLM cause Bloom’s syndrome (BS), a rare, autosomal recessive disorder characterized for example by an increased incidence of many types of cancer [12].
It is known that in the female meiosis of Drosophila melanogaster, the product of the mus309 locus is involved in the SDSA pathway of the repair of the DSBs [18, 19]. More specifically, it is also known that in mus309 mutants the SDSA pathway is blocked, while the DSBR pathway remains functional [20]. Thus, the mus309 gene seems to control the choice made by the oocyte between the two alternative pathways of DSB repair. The same is also true for the mus309 orthologue, the Sgs1 locus, in meiocytes of yeast [21].
Two different models of mitotic DSB repair by homologous recombination which are not mutually exclusive have been proposed for Drosophila melanogaster. They are the “dissolution model” [22] and the “disruptase model” [23-25]. These models are based on studies of the effect of the mus309 gene on gap repair and on mitotic exchange in the germ line of the males, or on the interaction of mus309 with other genes being involved in DSB repair. Both models are modified versions of the DSBR model of meiotic crossing over presented by Szostak et al. [26], and the dissolution model was originally presented by Ira et al. [27] and Wu and Hickson [28].
The aim of the present study was to investigate the explanatory power of these two models of mitotic DSB repair by studying the effect of certain mus309 mutants, defective in DSB repair, on X-ray induced somatic crossing over.
The results support the hypothesis that X-ray-induced somatic crossing over is a two-step event both of the mechanisms of HDR pathway proposed being involved. The results, combined with the data of others, also suggest that the mechanisms for the repair of induced DSBs work differently in the somatic and germ line cells. Further, it is proposed that in the wild-type flies the repairing mechanism is capable to recover if the irradiation is given with a short term high dose-rate, but is not capable to recover if the irradiation is given with a long lasting low dose-rate. In the mus309 mutant flies, however, the repairing mechanism is only poorly recovered irrespective of the dose-rate.
2. MATERIALS AND METHODS
2.1. Principle of the Investigation of Somatic Crossing Over in Drosophila
Curt Stern discovered in 1936 [29] that crossing over was not restricted to meiosis but also occurred during mitosis. Mitotic crossing over may occur in somatic cells, where its consequences may be visible as somatic mosaicism, because heterozygous recessive marker genes can become homozygous and give rise to mutant clones as shown in Figure 1. Irradiation will induce mitotic crossing over in the soma, leading to an elevation of the frequency of mosaicism for recessive markers [30]. This is now a widespread method for the generation of somatic mosaicism. For practical purposes, an X-ray dose of the order of 1000 R is used, usually for first-instar larvae [31].
Mitotic crossing over in somatic cells in the X chromosome most often occurs proximally to the singed locus (sn, 1 - 21.0) [32]. Provided that the homologous X chromosome is marked with the yellow marker (y, 1 - 0.0) and given a certain type of the segregation of chromatids in mitosis illustrated in Figure 1 and called x-segregation by Stern [29], two cells homozygous for different marker genes are born. Regarding cuticular cells, this result of somatic crossing over could in principle be detected as a mutant twin spot on the otherwise wild-type cuticulum. This is so, because the cuticulum is a one-cell layer, and thus both results of somatic crossing over are visible. Always, as in the present study as well, however, more single spots than twin spots are found, and among the singles usually more sn than y [32]. The causes of these phenomena are obviously manifold [32-35], but they are irrelevant for the conclusions of this study.
2.2. Description of the mus309 Mutants Used
Two alleles of the mus309 locus were used. They were mus309D2 and mus309D3 both of which were coupled with a second site lethal gene and balanced with the TM6 balancer marked with the Tubby (Tb, 3 - 90.6) marker in the stocks used.
Both the mus309 alleles used carry mutational changes that could potentially impair or abolish at least the helicase function of the MUS309 protein. In mus309D2, there
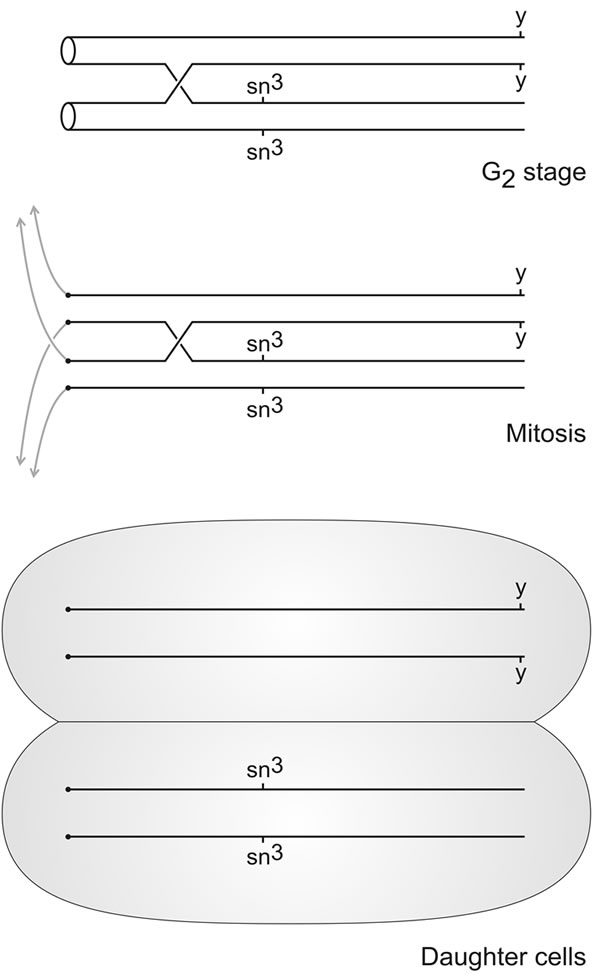
Figure 1. Principle of the study of somatic crossing over in the X chromosome of Drosophila melanogaster. The X chromosomes are marked each with a different recessive marker gene, yellow (y) and singed-3 (sn3) in this case. If somatic crossing over occurs between the proximal sn3 marker and the centromere during the G2-stage of the cell cycle in a cell of the thoracic imaginal disc of a female larva, and if the centromeres orientate in the following mitosis in the way indicated, homozygous yellow and singed-3 daughter cells will be born. These will develop into yellow and singed-3 clones respectively, which can be detected as mutant spots on the otherwise wild-type cuticulum of the adult female individuals in which somatic crossing over occurred during the larval stage.
is a stop codon between the sequence motifs encoding the third and fourth helicase motif of the protein. mus309D3, for its part, has a glutamic acid to lysine substitution in the conserved helicase II motif, in addition to another amino acid substitution close to the C terminus [35]. It has been demonstrated that the genotype mus309D2/ mus309D3 is semi-sterile [11,35,36].
2.3. Experimental Procedures
In the P-generation y; mus309D2/TM6, Tb and sn3; mus309D2/TM6, Tb females were crossed with y/Y; mus309D3/TM6, Tb and sn3/Y; mus309D3/TM6, Tb males respectively. (y, yellow 1 - 0.0; sn3, singed 1 - 21.0; mus309, mutagen-sensitive309 3 - 86E17). In the resulting F1-generation y; mus309D2/mus309D3 females were crossed with sn3/Y; mus309D2/mus309D3 males. These females and males were identified on the basis of their non-Tubby phenotype. The resulting F2-generation was exposed to X-ray irradiation during the first-instar larval stage for the induction of somatic crossing over, or they were left without irradiation. After eclosion the F2-females with the y +/+ sn3; mus309D2/mus309D3 genotype were investigated for the existence of yellow and singed macrochaeta on the otherwise wild-type cuticula of their mesonota. These are called experimental females, and the vast majority of all the F2-females are of this genotype. (The rest constituted of the very few mus309D2 and mus309D3 homozygotes which were recombinants for the second site lethal genes). For a control, the respective irradiation and investigation procedure was made with the females of the y +/+ sn3; +/+ genotype.
2.4. Details of the Irradiation Procedure
In the experimental F1-generation crosses and in the respective control crosses the flies were allowed to mate in culture bottles for 2 - 3 days. After that the flies were transferred to Petri dishes containing standard Drosophila medium consisting of semolina, syrup, agar-agar and both dried and fresh yeast using very light ether narcosis, ten females and males to each dish. After a 24 h period of egg laying, the flies were discarded. After another 24 h interval the Petri dishes containing the developing larvae were irradiated with 1000 R of hard X-rays using a linear accelerator (Clinac 2100 C/D, Varian Medical Systems, CA) with a 6 MeV photon irradiation at the dose-rate of 3 Gy/min. Thus during the irradiation the larvae were in the mid first-instar stage of development. Two different sets were made: the distance of the focus to the Petri dishes was either 1 m or 77.5 cm the first giving the irradiation to the larvae with a slow doserate of 300 R/min and the second with a fast dose-rate of 1000 R/min. After irradiation, the medium cake containing the larvae was transferred from each Petri dish to a culture bottle containing fresh Drosophila medium, and the cultures were incubated in 25˚C.
After eclosion the females of the irradiated and nonirradiated progeny generations were carefully inspected under a dissecting microscope for mutant clones involving the mesonotal macrochaeta: yellow or singed single spots or yellow-singed twin spots, and notes were taken for the existence of the spots.
2.5. Statistical Methods
In the statistical analyses of the results Chi-square test, Fisher’s exact test and logistic regression analysis [37] were used. All analyses were conducted with SAS statistical software version 9.22 (SAS Institute Inc., Cary, North Carolina, USA
3. RESULTS
No mutant spots were observed either in control or experimental females if they were not exposed to irradiation (Tables 1 and 2). Thus it seems that the mus309 mutations investigated do not increase the frequency of spontaneous somatic crossing over. This is in contrast with the observation of Johnson-Schlitz and Engels [22] and McVey et al. [24] who found that in mus309 mutants the frequency of spontaneous mitotic crossing over in the germ line of the males was increased by several orders of magnitude.
On the other hand, the mus309 mutations investigated significantly increased the frequency of X-ray induced somatic crossing over with both 300 R/min and 1000 R/ min dose-rate (Tables 1 and 2). The increase in the frequency of mutant spots in experimental mus309 females as compared to wild-type control females irradiated with the 300 R/min dose-rate was 1.80-fold (P = 0.0405; Fisher’s exact test). The corresponding increase in females irradiated with the 1000 R/min dose-rate was 2.52- fold (P = 0.0160). The difference between the two factors of increase was significant (χ2 = 3.95, d.f. = 1, P = 0.0468; multiple logistic regression model).
The distribution of the different types of spots was similar in the control and experimental females irrespective of the dose-rate. The significances of difference were as follows: 300 R/min set: χ2 = 1.18, d.f. = 2, P = 0.5543; 1000 R/min set: χ2 = 2.54, d.f. = 2, P = 0.2808. This observation indicates that the actual mechanism of X-ray induced somatic crossing over is the same in mus309 mutant and wild-type flies.
There was an overwhelming majority of the frequency of single spots over the frequency of twin spots, and of singed spots over yellow spots among the singles in every irradiation set. In addition to this the frequency of yellow single spots was never higher than the frequency of twin spots (Tables 1 and 2). Similar results have earlier been obtained by several authors [29,38-40].
However, the distribution of the different types of spots was similar with both dose-rates regardless of the genotype of the females. The significances of difference were as follows: control females: χ2 = 2.58, d.f. = 2, P = 0.2753; experimental females: χ2 = 0.1125, d.f. = 2, P = 0.9453. This latter observation indicates that the causes of the apparent non-reciprocality of somatic crossing over are not dependent of the dose-rate.
As appears from Table 3, in the control females irradiated with the 300 R/min dose-rate the frequency of mosaic mesonota was significantly higher than in females irradiated with the 1000 R/min dose-rate (P = 0.0477; Fisher’s exact test). In the experimental females, however, the difference was not significant (P = 0.0702). Thus, the frequency of somatic crossing over in the wildtype flies was, as expected, dependent on the dose-rate of the irradiation. On the other hand, in the mus309 mutant flies the dose-rate effect was absent, or it was at most very weak, which has not been observed before.
4. DISCUSSION
4.1. General Discussion
It has been known since the pioneering work of Jutta Haendle [41-44] that the frequency of the X-ray induced somatic crossing over in Drosophila melanogaster increases with the increasing dose of the irradiation, and that irradiation given with hard X-rays and a slow doserate induces more exchanges than the same dose given
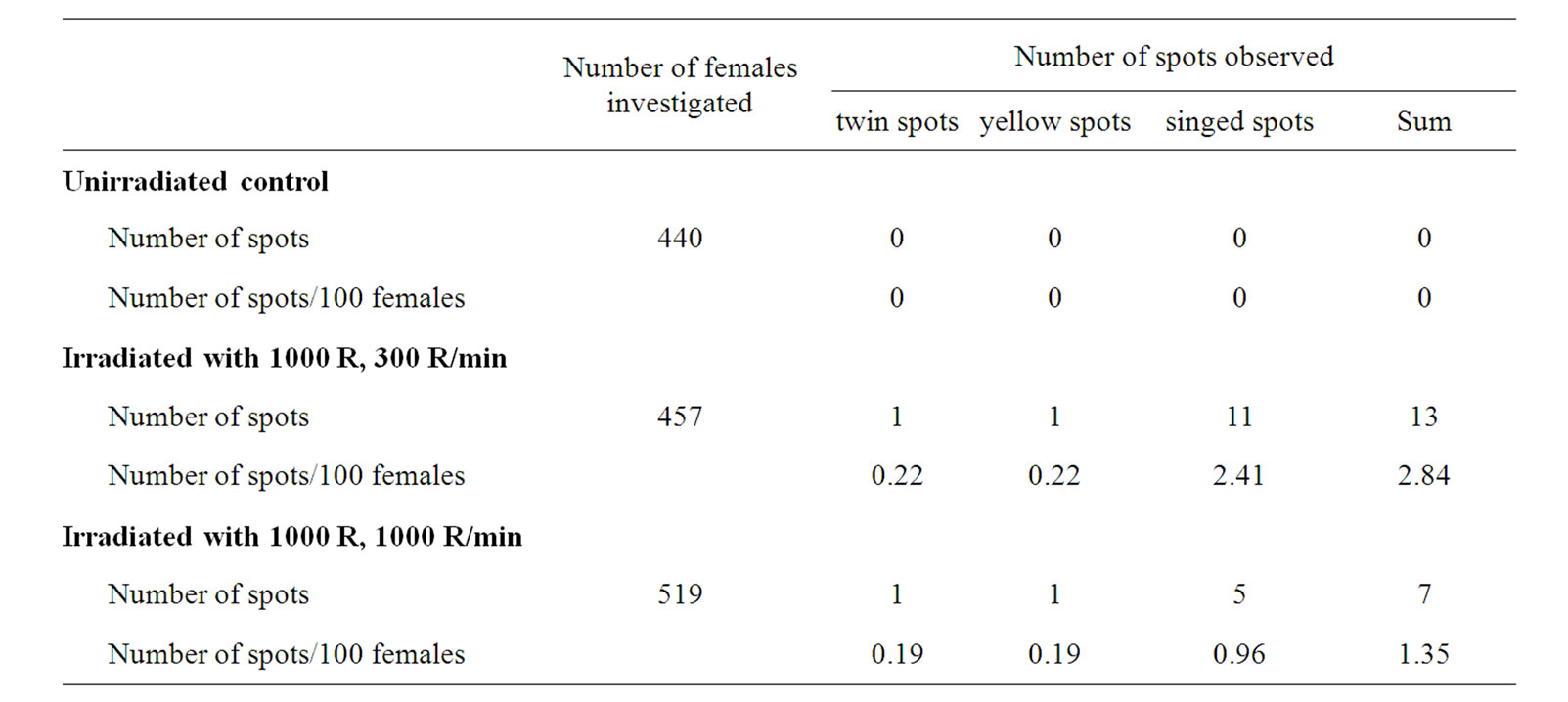
Table 1. Results from the control irradiation sets: distribution of different mosaic spots on the mesonota of the females of the genotype y +/+ sn3; +/+ exposed to 1000 R of X-ray irradiation during the first-instar larval stage of development given with two different dos rates, 300 R and 1000 R/minute.
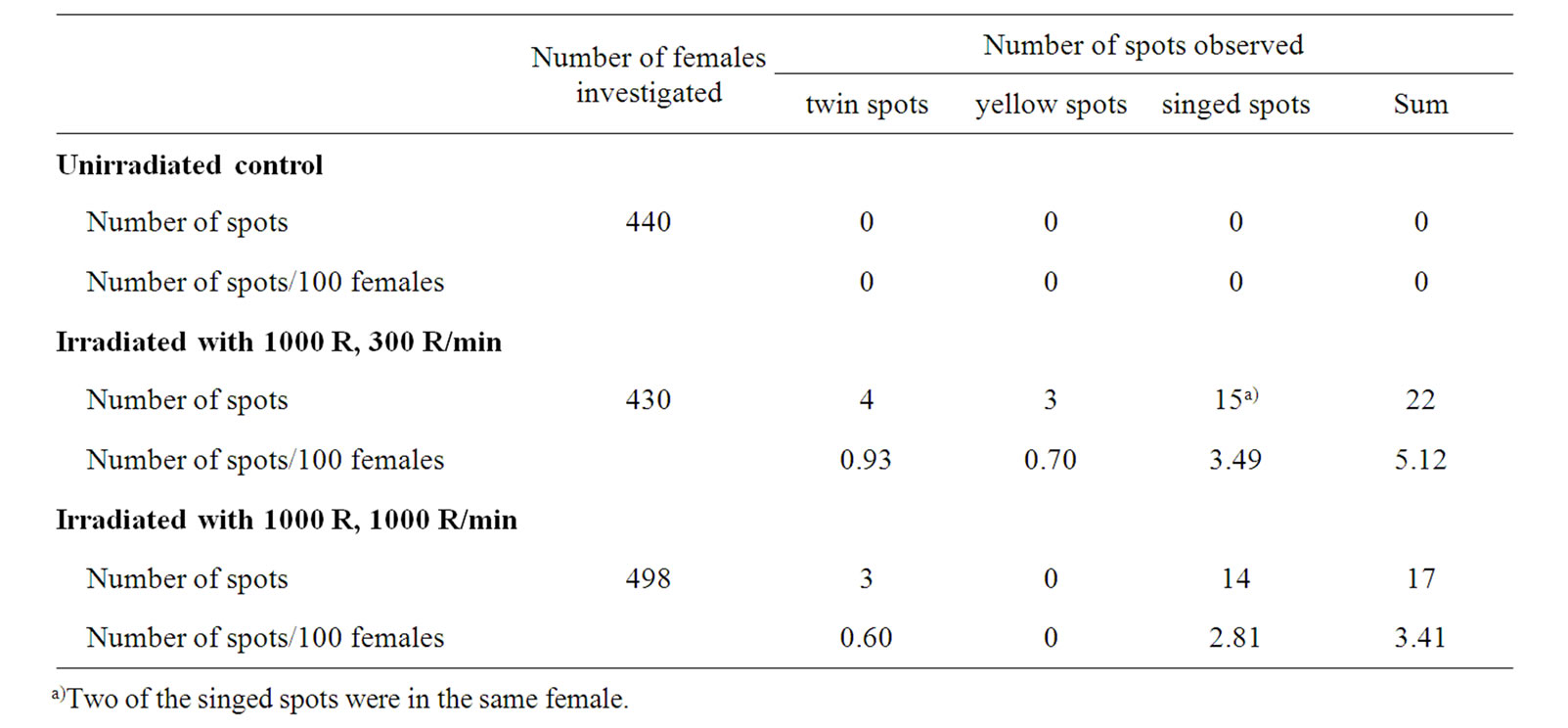
Table 2. Results from the experimental irradiation sets: distribution of different mosaic spots on the mesonota of the females of the genotype y +/+ sn3; mus309D2/mus309D3 exposed to 1000 R of X-ray irradiation during the first-instar larval stage of development given with two different dose rates, 300 R and 1000 R/minute.
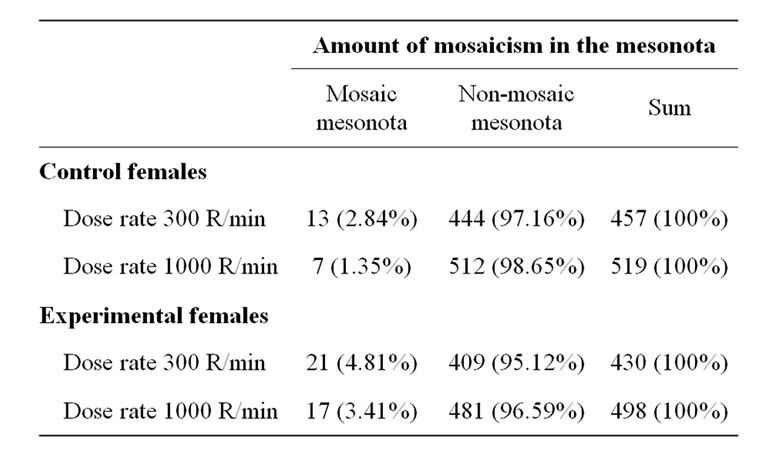
Table 3. Comparison of the different irradiation sets: amount f mosaicism in the control (y +/+ sn3; +/+) and experimental (y +/+ sn3; mus309D2/mus309D3) females exposed to 1000 R of X-ray irradiation during the first-instar larval stage of development given with two different dose rates, 300 R and 1000 R/minute.
with a fast dose-rate. With hard X-rays delivered at high dose-rates, the frequency of somatic crossing over increases nonlinearly with dose. (There is a marked shoulder at about 1000 R). With lower dose-rates or with softer X-rays the nonlinearity is less apparent. There is a very marked loss of efficiency in the induction of somatic crossing over with hard X-rays at dose-rates below 3000 R/min. These results were interpreted to mean that somatic crossing over induced with hard X-rays is a multihit event. One of these events is due to the soft component of irradiation and leads to induction of somatic crossing over by soft X-rays. This event is characterized by the fact that its recovery shows no time dependence. The other event is unique to hard X-rays and is distinguished by the fact that it is rapidly—within 30 - 150 seconds—repaired thus explaining the dose-rate effect of hard X-rays [31,32].
4.2. The Difference between Somatic and Germ Line Cells
The result that no spontaneous somatic crossing over was observed either in the control or experimental females, as compared with the results of Johnson-Schlitz and Engels [22] and McVey et al. [24] that the frequency of mitotic crossing over in the germ line cells was greatly increased in the mus309 mutant males, suggests that the repairing mechanism controlled by the mus309 gene works differently in the male germ line and the female soma. This difference may be due to the rapid cell division rate of the spermatogonial cells as compared to the somatic cells in the imaginal discs. Based on the time table of the duration of different stages of spermatogenesis in Drosophila melanogaster [45] it can be calculated that the four synchronous spermatogonial mitotic cycles last ca. 4 h each. On the other hand, the doubling of the cell number in the wing imaginal disc is about 7.5 h [46]. The spermatogonial cells may be more sensitive to the lack of repair of the DSBs either because there are more DSBs in them or because the repairing mechanism has less time to be recovered.
4.3. Somatic Crossing Over Is More Frequent in mus309 Mutant Flies than in Wild-Type Flies
As could be expected, somatic crossing over is more frequent in the mus309 mutant flies, deficient in DSB repair, than in the wild-type flies. Contrarily with the expectations, however, X-ray irradiation given with the dose-rate of 300 R/min induced more somatic crossing over than irradiation given with a dose-rate of 1000 R/ min both in the wild-type and mus309 mutant flies. This result is a reflection of the dose-rate independence of somatic crossing over in the mutant flies, which will be discussed in the following section.
4.4. X-Ray Induced Somatic Crossing Over in the mus309 Mutants is Independent on the Dose-Rate
The most important result of this study was the unexpected observation that the dose-rate effect, found in the control flies, was absent or very weak in the mus309 mutant flies. In this respect the mus309 mutants are similar to the c(3)G homozygote flies, in which meiotic crossing over is completely suppressed (Haendle, quoted by Becker [32]). This suggests that both the mus309 and the c(3)G mutations affect the rapidly repaired breakage mechanism found by Haendle [41,42] (cf. [31,32]).
The results can be explained by assuming that X-rayinduced somatic crossing over is a two-step event, and that the mechanism which repairs the lesion caused by the irradiation is controlled by the mus309 gene. In the control flies the repairing mechanism is capable to recover if the irradiation is given with a short term high dose-rate, but is not capable to recover if the irradiation is given with a long lasting low dose-rate. However, in the experimental mutant flies the repairing mechanism is only poorly recovered irrespective of the dose-rate.
It is hypothesized that the two steps involved are the dissolvase and disruptase activities of the MUS309 protein, which are sub sequential [24]. One or the other of these activities is poorly recovered in the mus309 mutants, but the other one is normally functional. In the wild-type control flies both components act normally. These suggestions are supported by the fact that, based on the results concerning the distribution of mosaic spots, both genotypes studied have in principle the same repairing mechanism irrespective of the dose-rate; it is only the recovering time which differs between genotypes.
4.5. Suggestions for Further Studies
It remains to be studied in details what is the relation of the two steps of somatic crossing over observed in this study and characterized in terms of molecular genetics to the respective steps observed earlier by Jutta Haendle and characterized in terms of cellular and chromosomal genetics. It would be particularly interesting to know whether the rapidly repaired mechanism found by Haendle in actual fact is identical with the dissolvase or disruptase activity of the MUS309 protein. Specifically the dose-rate independence observed should, if possible, be studied in amorphic null mutants of the mus309 locus. If the hypothesis of two steps presented is correct, no dose-rate dependence at all should be observed in such mutants. Unfortunately, however, the present author, being already retired, has no resources available any more to conduct these experiments.
5. ACKNOWLEDGEMENTS
Thanks are given to Professor Janos Szabad (Szeged, Hungary) for introducing me to the mus309 gene, and the generous donation of the mutant stocks which, however, are readily available in stock centers. Nuclear physicist, Docent Jarmo Kulmala, Ph.D. from the Cancer clinic of University Central Hospital in Turku took care of the X-ray machine and made the irradiations, lecturer Heikki Ruskeepää, Ph.D. from The Department of mathematics and Docent Samuli Helle, Ph.D. from The Section of ecology of The Department of biology helped me by conducting the statistical analyses, and museum technician Veikko Rinne, M.Sc. from The Zoological museum of The Department of biology by drawing the figure. To all of them I am very grateful. Special thanks are given to Marja Vieno, M.Sc. for checking the language.
REFERENCES
- Mahaney, B.L., Meek, K. and Lees-Miller, S.P. (2009) Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochemical Journal, 417, 639-650.
- Hartlerode, A.J. and Scully, R. (2009) Mechanisms of double-strand break repair in somatic mammalian cells. Biochemical Journal, 423, 157-168. doi:10.1042/BJ20090942
- Paques, F. and Haber, J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews, 63, 349-404.
- Sung, P. and Klein, H. (2006) Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nature Reviews Molecular Cell Biology, 7, 739-750. doi:10.1038/nrm2008
- Helleday, T., Lo, J., van Gent, D.C. and Engelward, B.P. (2007) DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair, 6, 923-935. doi:10.1016/j.dnarep.2007.02.006
- Rothkamm, K., Kruger, I., Thompson, L.H. and Lobrich, M. (2007) Pathways of DNA double-strand break repair during the mammalian cell cycle. Molecular and Cellular Biology, 23, 5706-5715. doi:10.1128/MCB.23.16.5706-5715.2003
- Branzei, D. and Foiani, M. (2008) Regulation of DNA repair throughout the cell cycle. Nature Reviews Molecular Cell Biology, 9, 297-308. doi:10.1038/nrm2351
- Symington, L.S. (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiology and Molecular Biology Reviews, 66, 630-670. doi:10.1128/MMBR.66.4.630-670.2002
- Heyer, W.-D., Ehmsen, K.T. and Solinger, J.A. (2003) Holliday junctions in eukaryotic nucleus: Resolution in sight? Trends in Biochemical Sciences, 28, 548-557. doi:10.1016/j.tibs.2003.08.011
- Heyer, W.-D. (2004) Recombination: Holliday junction resolution and crossover formation. Current Biology, 14, R56-R58. doi:10.1016/j.cub.2003.12.043
- Boyd, J.B., Golino, M.D., Shaw, K.E.S., Osgood, C.J. and Green, M.M. (1981) Third-chromosome mutagen-sensitive mutants of Drosophila melanogaster. Genetics, 97, 607- 623
- Ellis, N.A., Groden, J., Ye, T-Z., Staughen, J., Lennon, D.J., Ciocci, S., Proytcheva, M. and German, J. (1995) The Bloom’s syndrome gene-product is homologous to RecQ helicases. Cell, 83, 655-666. doi:10.1016/0092-8674(95)90105-1
- Karow, J.K., Chakraverty, R.K. and Hickson, J.D. (1997) The Bloom’s syndrome gene product is a 3’- 5’ DNA helicase. Journal of Biological Chemistry, 272, 30611- 30614. doi:10.1074/jbc.272.49.30611
- Mohaghegh, P., Karow, J.K., Brosh, R.M. Jr., Bohr, V.A. and Hickson, I.D. (2001) The Bloom’s and Werner’s syndrome proteins are DNA structure-specific homologues. Nucleic Acids Research, 29, 2843-2849. doi:10.1093/nar/29.13.2843
- Wu, L., Davies, S.L., Levitt, N.C. and Hickson, I.D. (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD5. Journal of Biological Chemistry, 276, 19375-19381. doi:10.1074/jbc.M009471200
- Van Brabant, A.J., Stan, R. and Ellis, N.A. (2000) DNA helicases, genome instability, and human genetic disease. Annual Reviews of Genomics and Human Genetics, 1, 409-459. doi:10.1146/annurev.genom.1.1.409
- Kooistra, R., Pastink, A., Zonneveld, J.B.M., Lohman, P.H.M. and Eeken, J.C.J. (1999)The Drosophila melanogaster DmRAD54 gene plays a crucial role in doublestrand break repair after P-element excision and acts synergistically with Ku70 in the repair of X-ray damage. Molecular and Cellular Biology, 19, 6269-6275.
- Adams, M.D., McVey, M. and Sekelsky, J.J. (2003) Drosophila BLM in double-strand break repair by synthesisdependent strand annealing. Science, 299, 265-267. doi:10.1126/science.1077198
- Laurencon, A., Orme, C.M., Peters, H.K., Boulton, C.L., Vladar, E.K., Langley, S.A., Bakis, E.P., Harris, D.T., Harris, N.J., Wayson, S.M., Hawley, R.S. and Burtis, K.C. (2004) A large-scale screen for mutagen sensitive loci in Drosophila. Genetics, 167, 217-231. doi:10.1534/genetics.167.1.217
- Portin, P. (2005) mus309 mutation, defective in DNA double-strand break repair, affects intergenic but not intragenic meiotic recombination in Drosophila melanogaster. Genetical Research, 86, 185-191. doi:10.1017/S0016672305007883
- Rockmill, B., Fung, J.C., Branda, S.S. and Roeder, G.S. (2003) The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Current Biology, 13, 1954- 1962. doi:10.1016/j.cub.2003.10.059
- Johnson-Schlitz, D. and Engels, W.R. (2003) Template disruption and failure of double Holliday junction dissolution during double-strand break repair in Drosophila BLM mutants. Proceedings of the National Academy of Sciences USA, 103, 16840-16845. doi:10.1073/pnas.0607904103
- McVey, M., Larocque, J.R., Adams, M.D. and Sekelsky, J.J. (2004) Formation of deletions during double-strand break repair in Drosophila DmBlm mutants occurs after strand invasion. Proceedings of the National Academy of Sciences USA, 101, 15694-15699. doi:10.1073/pnas.0406157101
- McVey, M. andersen, S.L., Broze, J. and Sekelsky, J. (2007) Multiple functions of Drosophila BLM helicase in maintenance of genome stability. Genetics, 176, 1979- 1992. doi:10.1534/genetics.106.070052
- Trowbridge, K., McKim, K., Brill, S.J. and Sekelsky, J. (2007) Synthetic lethality of Drosophila in the absence of the MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics, 176, 1993- 2001. doi:10.1534/genetics.106.070060
- Szostak, J.W., Orr-Weaver, T.L., Rothstein, R.J. and Stahl, F.W. (1983) The double-strand-break repair model for recombination. Cell, 33, 25-35. doi:10.1016/0092-8674(83)90331-8
- Ira, G., Malkova, A., Liberi, G., Foiani, M. and Haber, J.E. (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell, 115, 401-411. doi:10.1016/S0092-8674(03)00886-9
- Wu, L. and Hickson, I.D. (2003) The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature, 426, 870-874. doi:10.1038/nature02253
- Stern, C. (1936) Somatic crossing over and segregation in Drosophila melanogaster. Genetics, 21, 625-730.
- Patterson, J.T. (1929) The production of mutations in somatic cells of Drosophila melanogaster by means of X-rays. Journal of Experimental Zoology, 53, 327-372. doi:10.1002/jez.1400530302
- Ashburner, M. (1989) Drosophila. A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
- Becker, H.J. (1976) Mitotic recombination. In: Ashburner, M. and Novitki, E., Eds., The Genetics and Biology of Drosophila Vol. 1 c, Academic Press, London, 1019- 1087.
- Garcia-Bellido, A. (1972) Some parameters of mitotic recombination in Drosophila melanogaster. Molecular and General Genetics, 115, 54-72. doi:10.1007/BF00272218
- Ayaki, T., Fujikawa, K., Ryo, H., Itoh, T. and Kondo, S. (1990) Induced rates of mitotic crossing over and possible mitotic gene conversion per wing anlage cell in Drosophila melanogaster by X rays and fission neutrons. Genetics, 126, 157-166.
- Kusano, K., Johnson-Schlitz, D.M. and Engels, W.R. (2001) Sterility of Drosophila with mutations in the Bloom syndrome gene—Complementation by Ku70. Science, 291, 2600-2602. doi:10.1126/science.291.5513.2600
- Beal, E.L. and D.C. Rio, D.C. (1996) Drosophila IRBP/ Ku p70 corresponds to the mutagen-sensitive mus309 gene and is involved in P-element excision in vivo. Genes and Development, 10, 921-933. doi:10.1101/gad.10.8.921
- Allison, P.S. (1999) Logistic regression using the SAS® system: Theory and applications. SAS Institute Inc., Cary.
- Kaplan, W.D. (1953) The influence of Minutes upon somatic crossing-over Drosophila melanogaster. Genetics, 38, 630-651.
- Walen, K.H. (1964) Somatic crossing over in relationship to heterochromatin in Drosophila melanogaster. Genetics, 49, 905-923.
- Ronen, M. (1964) Interchromosomal effects on somatic recombination in Drosophila melanogaster. Genetics, 50, 649-658.
- Haendle, J. (1971) Röntgeninduzierte mitotische Rekombination bei Drosophila melanogaster. I. Ihre Abhängigkeit von der Dosis, der Dosisrate und vom Spektrum. Molecular and General Genetics, 113, 114-131.
- Haendle, J. (1971) Röntgeninduzierte mitotische Rekombination bei Drosophila melanogaster. II. Beweis der Existenz und Charakterisierung zweier von der Art des Spektrums abhängiger Reaktionen. Molecular and General Genetics, 113, 132-149.
- Haendle, J. (1974) X-ray induced mitotic recombination in Drosophila melanogaster. III. Dose dependence of the “pairing” component. Molecular and General Genetics, 128, 233-239. doi:10.1007/BF00267112
- Haendle, J. (1979) X-ray induced mitotic recombination in Drosophila melanogaster. IV. Distribution within euand heterochromatin. Mutation Research, 62, 467-475. doi:10.1016/0027-5107(79)90042-3
- Lindsley, D.L. and Tokuyasu, K.T. (1980) Spermatogenesis. In: Ashburner, M. and Wright, T.R.F., Eds., The Genetics and Biology of Drosophila Vol. 2 d, Academic Press, London, 225-294.
- Madhavan, M.M. and Schneiderman, H.A. (1977) Histological analysis of the dynamics of growth of imaginal discs and histoblast nests during the larval development of Drosophila melanogaster. Wilhelm Roux’s Archives of Developmental Biology, 183, 269-305. doi:10.1007/BF00848459