Advances in Biological Chemistry
Vol.3 No.4(2013), Article ID:35680,10 pages DOI:10.4236/abc.2013.34044
In silico analysis of influence of the missense mutation P629S on the molecular interaction and 3D properties of PIK3R5*
![]()
1Department of Genetics, King Faisal Specialist Hospital and Research Centre, Riyadh, KSA
2Department of Neurosciences, King Faisal Specialist Hospital and Research Centre, Riyadh, KSA
Email: †naltassan@kfshrc.edu.sa
Copyright © 2013 Jameela Shinwari et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 12 January 2013; revised 23 February 2013; accepted 2 March 2013
Keywords: PIK3R5; Missense Mutation; Protein Modelling; Protein-Protein Docking; PIK3CG; Gβγ
ABSTRACT
PIK3R5 is the regulatory subunit of Phosphoinositide 3-kinase γ (PI3Kγ) that is responsible for phosphorylating membrane lipids to activate the AKT pathway. PIK3R5 binds Gβγ and facilitates the interaction with p110γ catalytic subunit (PIK3CG) during PI3Kγ activation. The identification of PIK3R5 P629S mutation in AOA2 patients indicated a potential defect in the AKT pathway resulting from impaired PIK3R5 interaction with Gβγ and PIK3CG, defective AKT pathway can result in cerebellar cell death causing neurological symptoms. Our in silico macromolecular docking of the wild type and mutant PIK3R5 protein models with ligand revealed an energy requirement to maintain the mutant complexes compared to no energy required to maintain the wild type complexes, in addition, the mutant structures were loose compared to rigid wild type structures, such structural changes may impair the molecular function of the PIK3R5 and hence affect the AKT pathway.
1. INTRODUCTION
Phosphoinositide 3-kinase γ (PI3Kγ) is a lipid kinase that belongs to class IB subdivision of Phosphoinositide 3- kinases (PI3Ks) [1]. Structurally, PI3Kγ forms a dimeric protein with two subunits; the regulatory p101 (PIK3R5) and catalytic p110γ (PIK3CG) subunits [2]. PI3Kγ is activated via G-protein-coupled receptors (GPCR) through direct interaction with Gβγ [3]. The regulatory p101 subunit binds to Gβγ and recruits the bound p110γ catalytic subunit to the plasma membrane, subsequently, PI3Ks phosphorylate the membrane lipid phosphatidylinositol (PtdIns)-4, 5-P2 to PtdIns-3, 4, 5-P3 and the latter acts as a second messenger required for the translocation of AKT to the plasma membrane so that phosphoinositidedependent kinase (PDK 1 and PDK2) can phosphorylate and activate the AKT [4]. Signals starting from AKT control the initiation of protein synthesis and regulate transcription [5], in general, the collection of PI3K signals are essential for cellular functions such as cell proliferation, survival and apoptosis by phosphorylating a series of substrates [6].
The structure of p110γ includes an amino-terminal adaptor-binding domain (ABD), a Ras-binding domain (RBD), a C2 (protein-kinase-C homology-2) domain, a helical domain which is required as a scaffold for other domains of p110, and a catalytic carboxyl-terminal kinase domain [7]. The work of Voigt and colleagues showed different regions within the p101 primary structure to be responsible for interaction with p110γ and Gβγ, the N terminus of p101 was shown to be important for interaction of p101 with p110γ while C terminus of p101 had a crucial role in the interaction with Gβγ, the binding site on p101 for p110γ spanned at least amino acids 25 - 175 while the binding site on p101 for Gβγ included amino acids 650 - 850. In addition, amino terminal region of p110γ (residues 1 - 144) was found important for interaction with the p101 adaptor [8].
Although p110γ knock-out mice show defective immune responses, yet non-alpha isoforms of p110 (that includes p110γ) were shown to be involved in the development and progression of malignancies, some studies observed an elevated expression of p110γ in chronic myeloid leukemia [9] while other studies suggested an involvement of nonalpha isoforms in cancers of the bladder, brain and colon [10-12]. Although there are no known specific cancer mutations in their genes, wild type nonalpha overexpression genes have been shown to be oncogenic [5].
Recently, four individuals of a native Saudi family from the Southwest region of the Arabian Peninsula with Ataxia Oculomotor Apraxia type 2 (AOA2) traits were identified [13]. Patients were screened for mutations in SETX earlier linked to AOA2 [14], APTX earlier linked to AOA1 [15], and MRE11A linked to AT-like phenotype and oculomotor apraxia [16] but no pathogenic or regulatory mutations in any of these genes were identified. Furthermore, linkage analysis and sequence analysis identified segregating homozygous missense change (c.1885C > T, p.P629S) in exon12 of phosphoinositide-3-kinase, regulatory subunit 5 (PIK3R5). Absence of this change in 477 normal controls and haplotype analysis further suggested its pathogenicity [13].
Information elucidated from the 3 dimensional structure of a protein plays a role in understanding its biological actions; on the other hand, mutations that do not fall in the active site of a particular protein can still change the kinetics of the biochemical reaction triggered by that protein such as the reaction rate or the isoelectric point, a shift in the isoelectric point can affect electrostatic interactions with other proteins and, in addition, a single amino acid substitution at an important folding location can inhibit the formation of the folding nucleus, causing the rest of the structure to quickly condense; such mutations can also affect protein stability, interaction and subcellular localization [17]. This study aims to generate a 3D protein model for the normal and P629S mutant p101 (PIK3R5) to compare their folding patterns and to conduct a macromolecular docking of both normal and mutant p101 to p110γ and Gβγ, then to assess any interaction difference that could consequently influence the AKT signaling pathway and has some biological impacts.
2. METHODS
2.1. Generation of 3D Structures of PIK3R5; PIK3CG and Gβγ
Wild type PIK3R5 structure was predicted using the structure prediction software I-TASSER version 1.1 on Linux 2.6.18 - 92 by multiple threading techniques and identifying the templates from PDB, full length models were assembled through Iterative Monte Carlo simulation [13,18,19] whereas mutant PIK3R5 structure was predicted via LOMETS [13,20]. Best PIK3R5 structure selection was based on the lowest energy level , structures were further validated using NIH-SAVES (http://nihserver.mbi.ucla.edu/SAVES/).
Structure of the region on PIK3CG responsible for binding PIK3R5 (namely the C-terminal amino acids 1 - 144) was predicted using I-TASSER version 1.1 [18,19], meanwhile, the structure of Gβγ was predicted using Modeller 9v10 [21] using multiple templates. Binding sites of Gβγ were predicted based on the van der Waal’s atoms using HOTPOINT [22]. PIK3R5 structure was evaluated using PROVE [23], Verify_3D [24] and PROCHECK [25] server at NIH SAVES. In order to further validate the wild type PIK3R5 and PIK3CG structures, Ramachandran plot was designed using Rampage Server [26].
2.2. Ramachandran Plot and Intramolecular Bonding
Both hydrogen bonding and Ramachandran plot values were computed using Swiss PDB viewer [27] to obtain a detailed analysis of the Mutated and Wild-type PIK3R5 in order to detect any structural difference. S-S bonds were viewed using Rasmol [28].
2.3. Macromolecular Docking
Both wild type and mutant PIK3R5 were docked with PIK3CG using software Hex 6.3 [29], the ligand (PIK3CG) was placed near the binding site of receptor (PIK3R5); the resulting PIK3R5-PIK3CG complex was further interacted with Gβγ using the same software.
3. RESULTS
3.1. Evaluation of PIK3R5, PIK3CG and Gβγ
Ten optimum structures were produced for wild type PIK3R5, the best quality structure among generated models was selected according to the confidence score (Cscore) which was −1.93 with a Template Modeling Score (TM) of 0.48 ± 0.15 and a Root-Mean-Square Deviation (RMSD) of 13.4 ± 4.1. Disulphide Bridges were calculated for PIK3R5 using an online tool Disulfide By Design [30], a total of 100 S-S bonds were created and the structure was viewed in Rasmol [28,31] (Figure 1).
Mutant PIK3R5 also generated 10 structures, the DOPE Score for wild type PIK3R5 structure was −71815.453125 while DOPE score for mutant selected structure was −74452. The DOPE graph indicated a slight variance in energy level compared to the wild-type PIK3R5 (Supplementary Figure 1), Figure 2 illustrates both wild type and mutant predicted protein models and the Image was created by PyMOL. Superimposition was carried out using Swiss PDB Viewer (Supplementary Figure 2), superimposed structure of wild type and mutant PIK3R5 provided an RMSD value of 2.01 Å. Ten structures were predicted for PIK3CG and the best one was selected on the basis of the C-score which was −2.61 (Figure 3) while DOPESCORE was −13656.806641
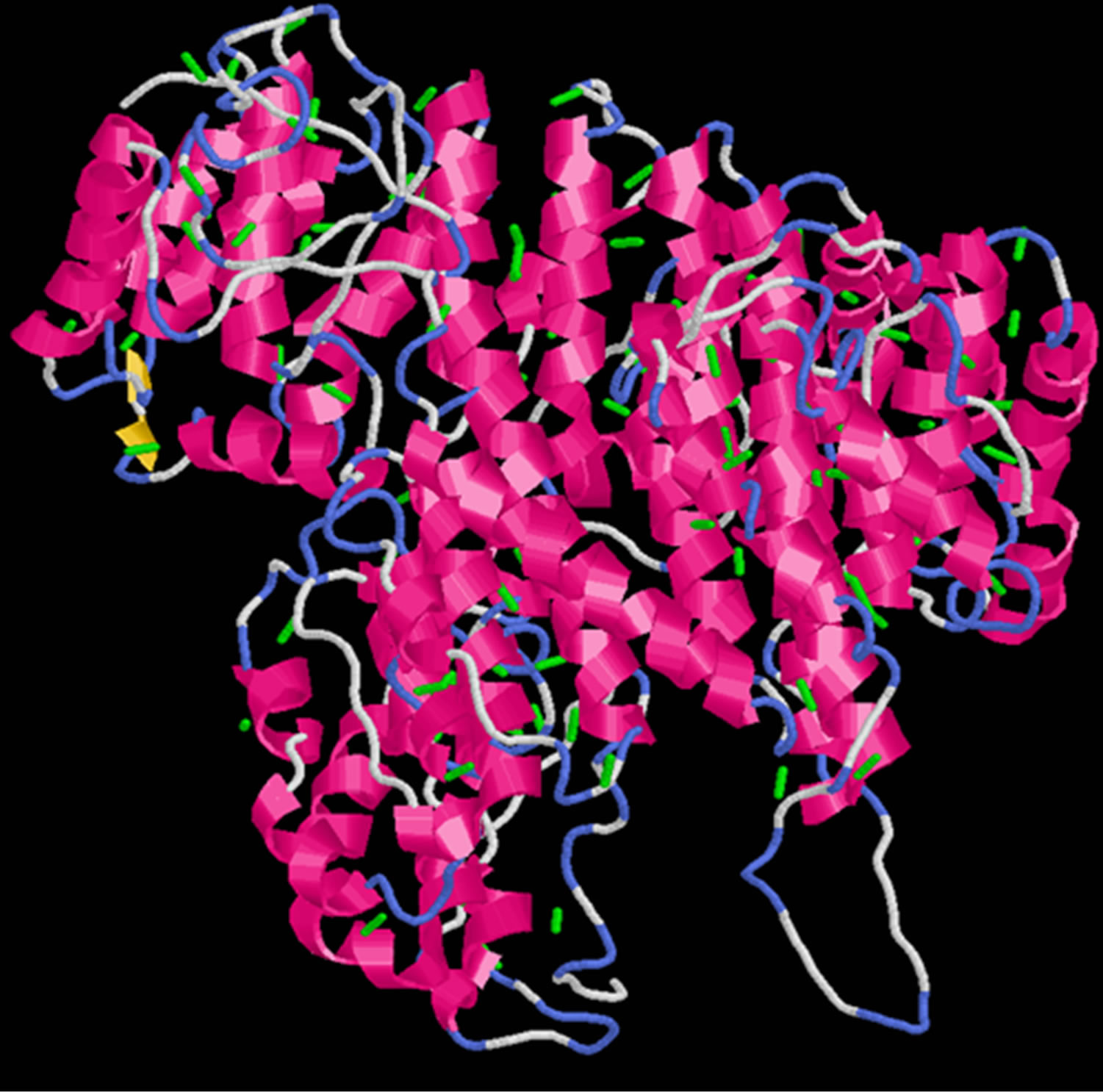
Figure 1. Disulphide bridges of PIK3R5 (The green bars indicate Disulphide bridges).
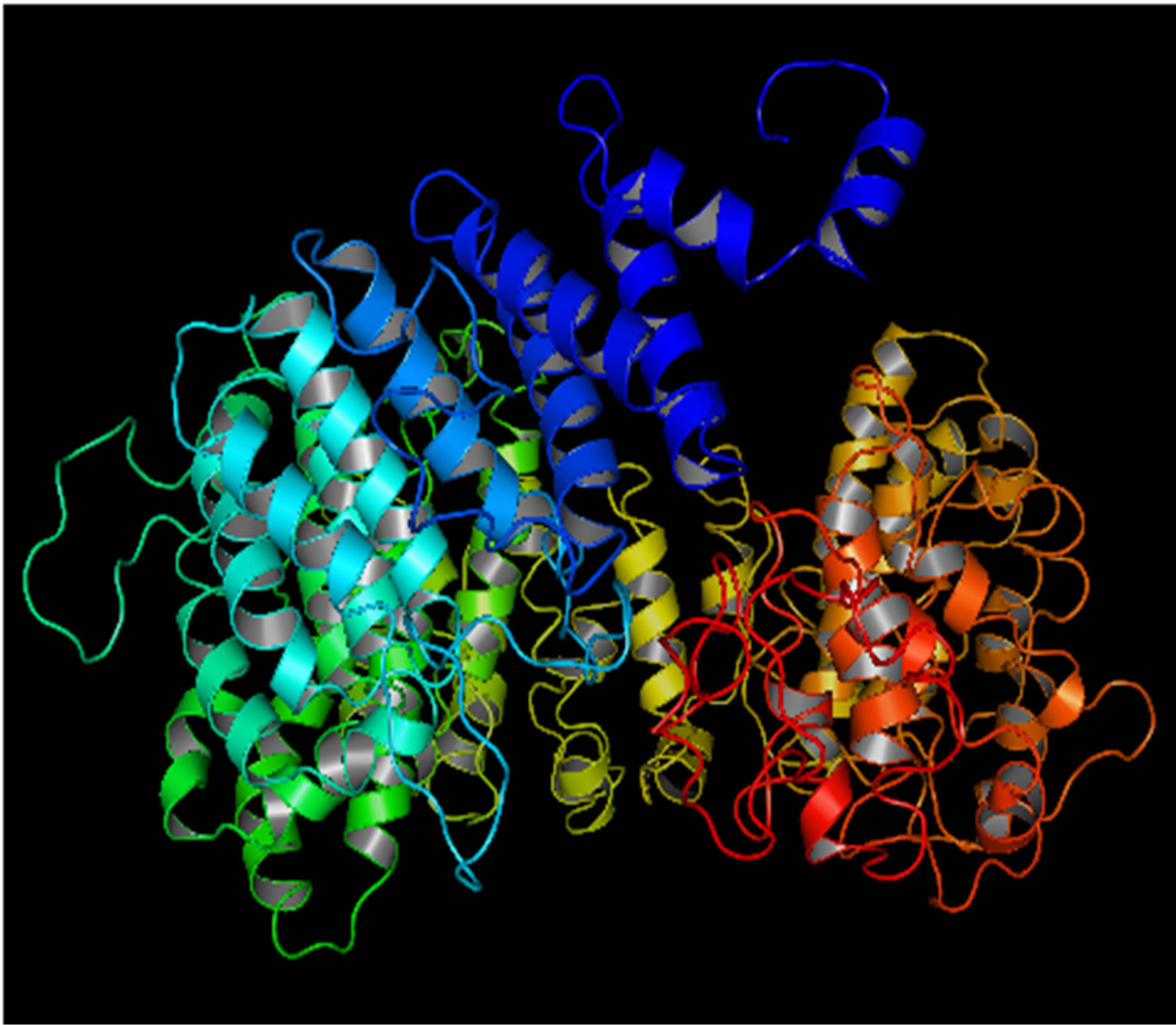 (a)
(a)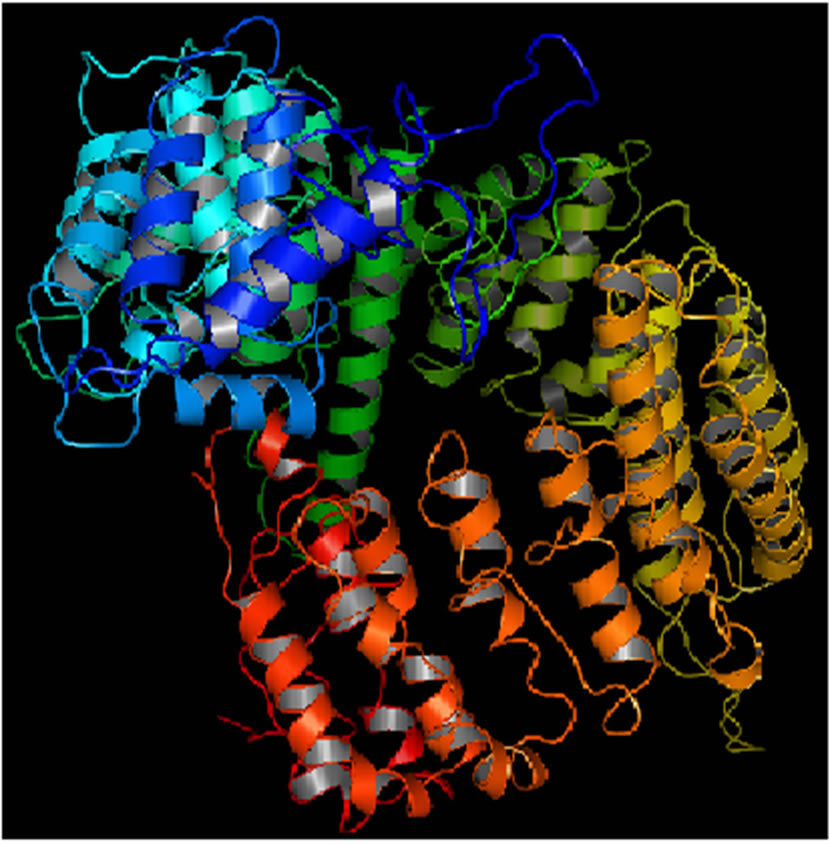 (b)
(b)
Figure 2. PIK3R5 structure (a) Mutant PIK3R5 (b) Wild-Type PIK3R5.
and the TM score was 0.41 ± 0.14. The major domain of Gβγ was already predicted in RCSB protein data bank. The structure was modified using Homology Modelling (Figure 4); the binding sites of Gβγ are shown in (Supplementary Figure 3).
3.2. Structures Evaluation
Moreover, Ramachandran plot represented for PIK3R5 and PIK3CG showed a fairly good quality plot of 91.2% approximately and 86.6% respectively in favorable regions [26,32,33] (Supplementary Figures 4(a) and (b)). PROVE output for the protein was also good and lead to
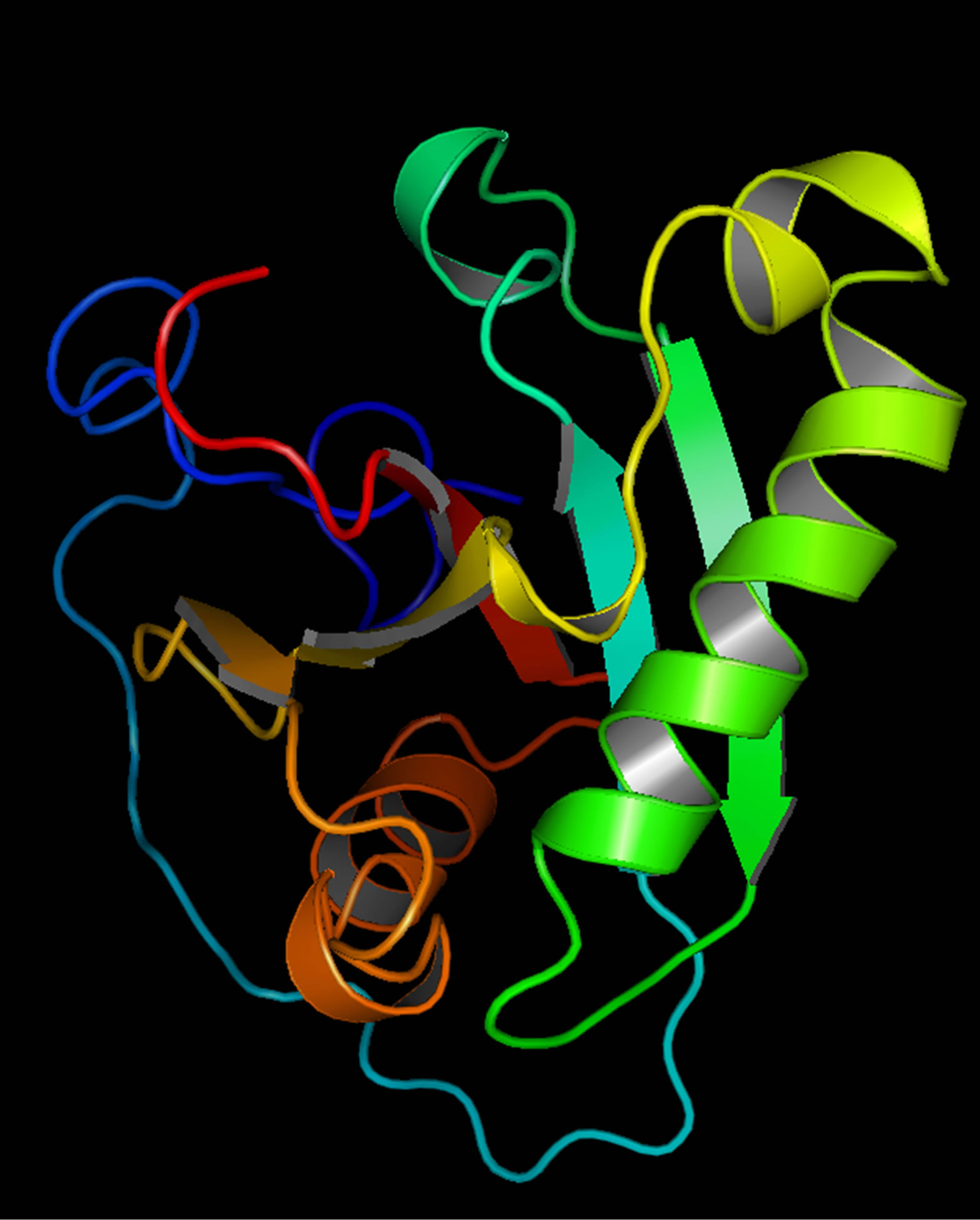
Figure 3. PIK3CG structure (The protein domain that interacts with PIK3R5).
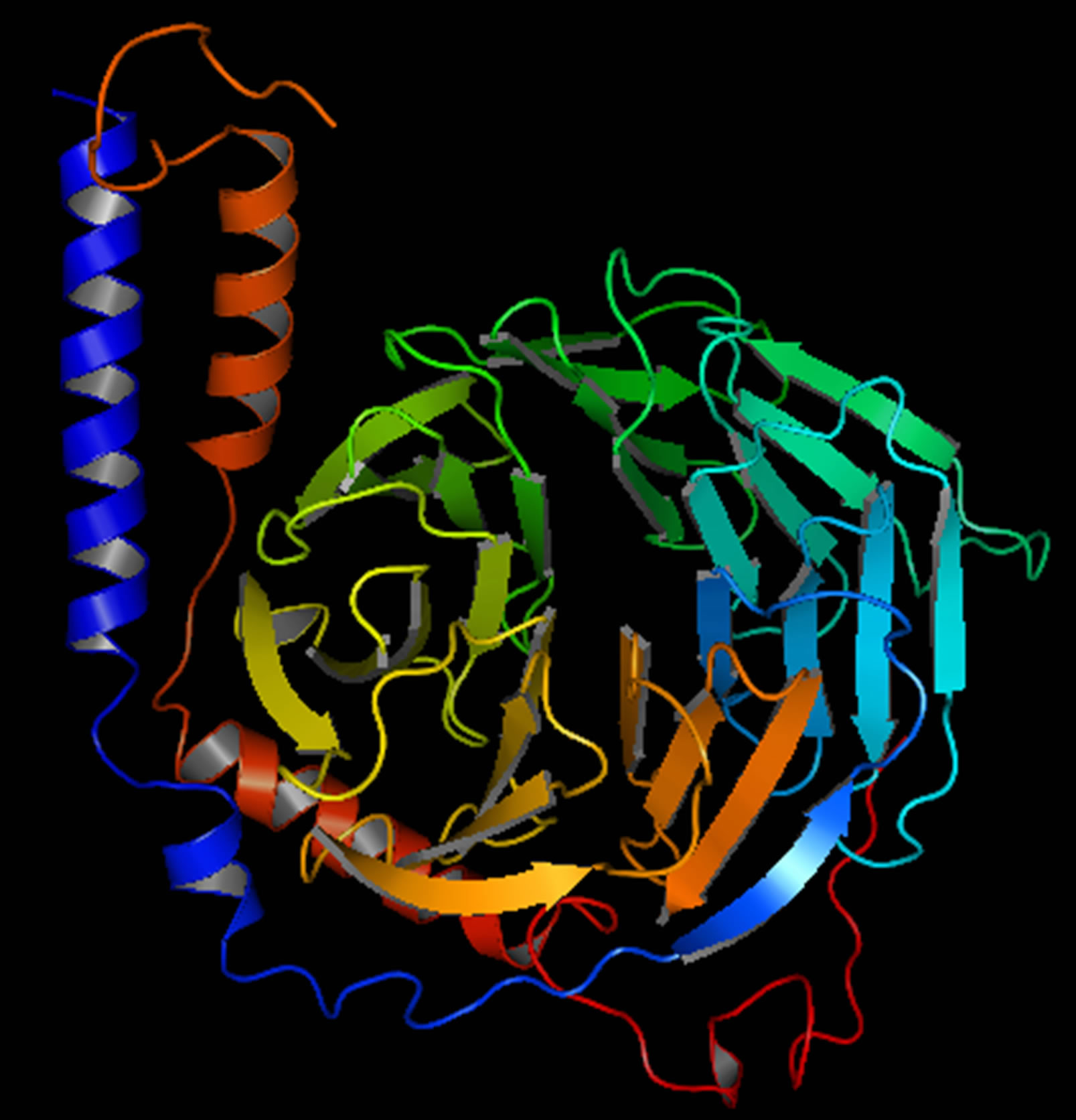
Figure 4. G-Beta gamma 3D structure.
the structure acceptance (Supplementary Figure 5). The average Z-score was laying near to the defined region thus this structure was selected. For G-Beta Gamma Structure, the overall quality factor produced from ER-RAT [34] was 82.663 (Supplementary Figure 6). Ramachandran plot was created using PROCHECK, which showed 86.7% of residues in favorable regions (Supplementary Figure 7).
3.3. Assessment of Wild Type vs. Mutant PIK3R5 Differences via Ramachandran Plot
Ramachandran plot value for Proline 629 in normal PIK3R5 was (−50.959φ, −45.026ψ) while Ramachandran plot value for Serine 629 in mutant PIK3R5 was (−59.095φ, −38.288ψ) (Supplementary Figure 8).
3.4. Assessment of Wild Type vs. Mutant PIK3R5 Differences via Protein Docking with Ligand
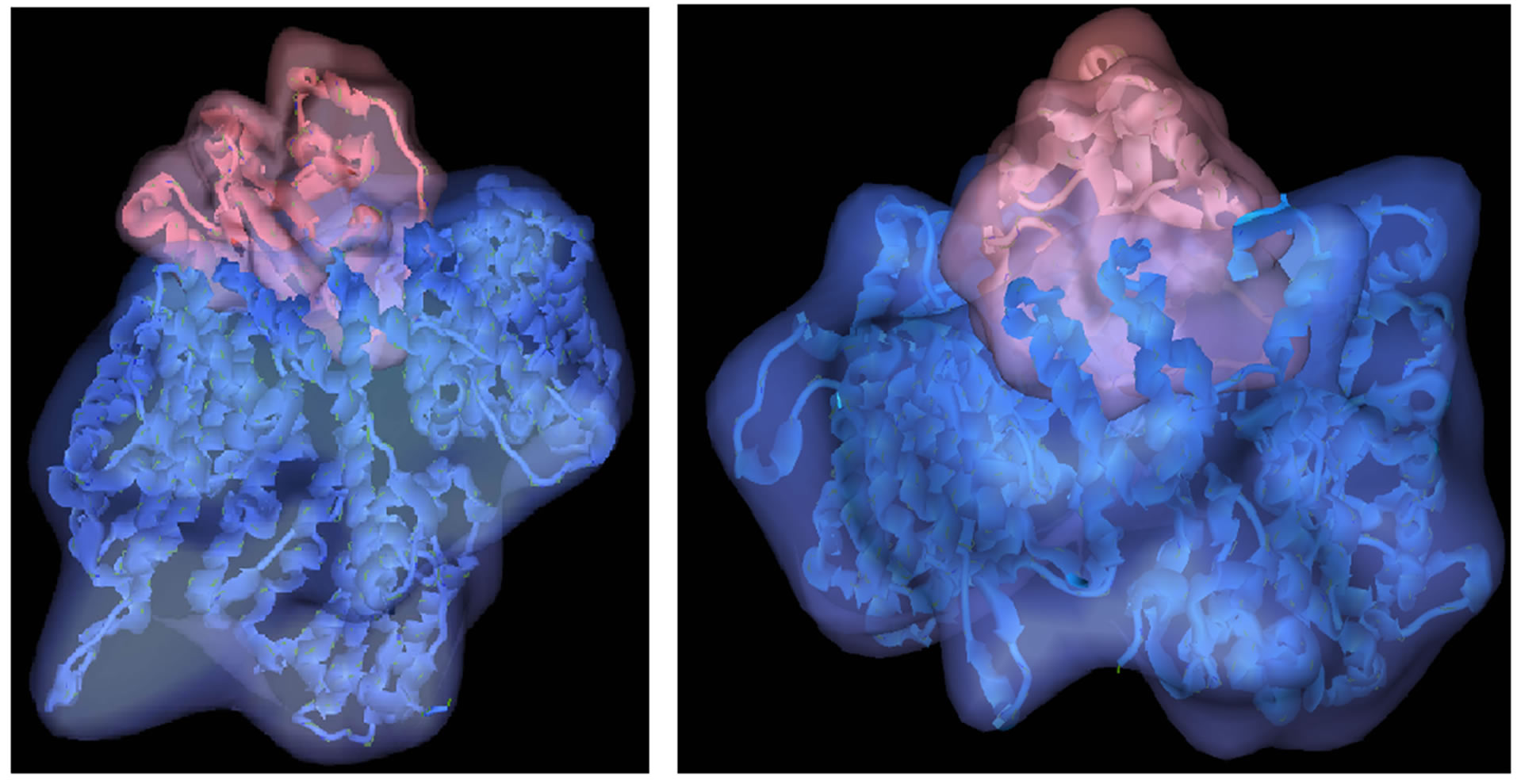
Wild type PIK3R5 docking with PIK3CG produced 2000 solutions with different energies at different positions. The best among them was selected based on more interactions and less energy, the total energy of the model was −587.65 and the Eshape (shape energy) was −587.65, additionally, the difference in bumps demonstrating the rigidity of the protein was −1 and the protein ligand interaction force (Eforce) was 0 [35], further interaction of PIK3R5-PIK3CG complex with Gβγ revealed a total energy of −242.24 and an Eshape of −212.35, bumps difference and Eforce were −1 and 0 respectively. Docking of Mutant PIK3R5 with PIK3CG was performed in a similar way as the wild type docking. The total energy of the model was −1432.22 and the Eshape (shape energy) was −1395, bumps difference was 0 and the protein ligand interaction force (Eforce) was −36.97. Figure 5 shows the wild type against Mutant PIK3R5-PIK3CG complex structures. The total energy for Mutant PIK3R5- PIK3CG complex interaction with Gβγ was −290.41 and the Eshape was −336.69, bumps difference was 0 and
 (a) (b)
(a) (b)
Figure 5. PIK3R5-PIK3CG docking. (a) Wild-type PIK3R5 (Blue) with PIK3CG (Pink) (b) Mutant PIK3R5 (Blue) with PIK3CG (Pink).
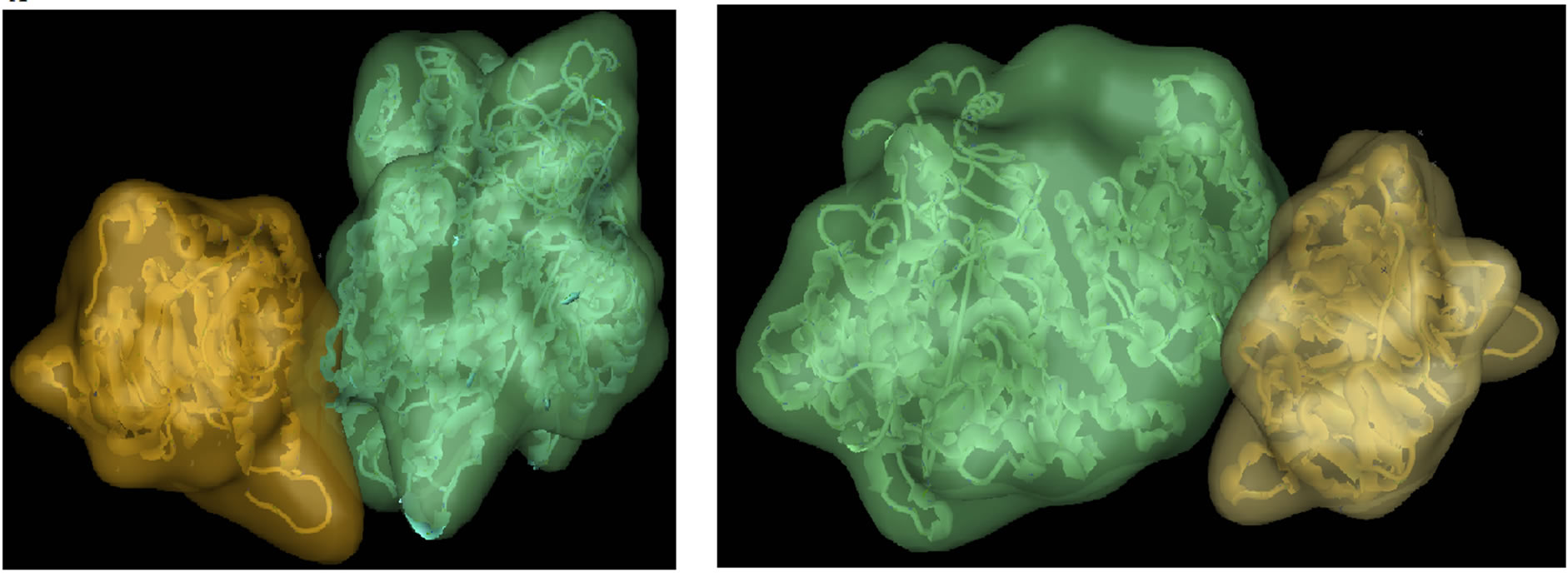
Eforce was −46.28, Figure 6 shows the wild type against Mutant PIK3R5-PIK3CG-Gβγ complex structures, and protein docking statistics are represented in Supplementary Table 1.
4. DISCUSSION
Complex protein stability, flexibility, interaction, subcellular localization and expression are important biological aspects that can be influenced by amino acids substitutions. A single mutation in a protein can result in replacing a small side chain amino acid for an amino acid with a large side chain hence troubling the conformational limit and stability of the protein [36]. On the other hand, a mutation can affect protein stability by causing substitutions between amino acids with hydrophilic and hydrophobic properties, masking of charged residues, destabilizing and loss of hydrogen bonds, or S–S bonds [17]. Commonly, biochemical reactions involve conformational changes in specific protein’s functional domains and some mutations can make a protein more or less rigid [37] thereby affecting its flexibility and causing protein aggregation and formation of fibrils [38]. Moreover, access of the binding group to a tight binding pocket can be completely or partially prevented by missense mutations that cause the substitution of a small side chain for a larger side chain in the binding site and therefore affect the molecular interaction [17].
P629S is a missense mutation identified in the phosphoinositide-3-kinase, regulatory subunit 5 gene (PIK 3R5) and it involves a change from a non-polar Proline to a polar Serine; the change is located adjacent to the C-terminus Gβγ binding domain which spans amino acids 650 - 850. P629S was predicted to decrease the protein stability as indicated by in silico analysis using IMutant software [13]; it is well known that 80% of missense mutations that cause disease are substitutions that shift the proteins stability by several kcal/mol [17].
Proline is a very unique amino acid as it plays an important role in protein folding, fiber formation and protein-protein interaction [39]. The unique structure of the amino acid Proline makes it unable to act as a hydrogenbond donor because the cyclic side chain fixes the back
 (a) (b)
(a) (b)
Figure 6. PIK3R5-PIK3CG-Gβγ docking. (a) Wild-type PIK3R5-PIK3CG (Green) with Gβγ (Yellow) (b) Mutant PIK3R5-PIK3CG (Green) with Gβγ (Yellow).
bone dihedral φ angle and the amide proton is replaced by CH2 group. Proline also has the ability to undergo cis-trans isomerization that plays an important role in protein folding; also the size of the N-CH2 group limits the conformation of the residue ahead of Proline thereby prohibiting the α-helix conformation [40].
3D structures that were predicted for PIK3R5 wild type protein and PIK3CG by multiple-threading alignments had C-scores of −1.93 and −2.61, respectively, such scores signify a model with a high confidence since C-score has an acceptable range of (−5.2). To validate the structures designed, the difference between native protein and designed protein structures (TM score) were obtained for the two models by scores in (0 - 1) range, where 1 implies a perfect match between two structures, scores below 0.20 suggest randomly chosen unrelated proteins and scores higher than 0.5 assume almost the same fold, the obtained TM score of 0.48 ± 0.15 for wild type PIK3R5 and 0.41 ± 0.14 for PIK3CG were accepted values in correspondence with the C-scores, in addition, the variability of differences in the distance between backbone atoms of theoretical protein and actual measurement of template when they are superimposed (RMSD values) conducted for both wild type PIK3R5 and PIK3CG presented acceptable values in correspondence with the C-scores, further analyses for the compatibility of PIK3R5 and PIK3CG atomic 3D model with their own amino acid sequence via Verify3D resulted in positive values indicating no potential problems in the structures. Moreover, the position of an amino acid after rotation of dihedral angles [ψ and φ] of that amino acid was conducted by means of Ramachandran plot and showed most of the amino acids to be in favorable regions for both PIK3R5 and PIK3CG. Taken together, validation data suggested satisfactory structures for wild type PIK3R5 and PIK3CG that were suitable to process further.
The 3D structure for Mutant PIK3R5 by LOMETS web tool was selected according to the confidence score, z-score and coverage length. For the mutant PIK3R5 validation DOPE values and graph were obtained, DOPE graph indicated a slight variance between wild type PIK3R5 and mutant PIK3R5. Superimposed structure of wild type PIK3R5 and mutant PIK3R5 provided an RMSD value of 2.01 Å. Thus, mutant structure was suitable to carry on with the docking process.
As indicated by the DOPE graph, substituting Proline for Serine at position 629 of PIK3R5 involves an energy change and therefore further comparison through Ramachandran plot shows the conformations of the dihedral angles [ψ and φ] that are possible for an amino-acid residue in a protein, a change in dihedral angle can be seen in Ramachandran plot of Proline 629 in normal PIK3R5 and Serine 629 in mutated PIK3R5. Although the residues are in the acceptable region but a slight change in their angle indicates a change in structure and bonding, moreover, the change in hydrogen bond length within the structure gave it a narrower structure at one side and disrupted helix on the other side that is converted into coil.
Proline is the most common residue found adjacent to protein-protein interaction sites [41], most protein interaction domains favor ligand sequences that are Proline rich or at least contain a single Proline residue [42]. Proline also facilitates formation of elements for proteins secondary structure such as turns and polyproline II helix, therefore it is possible that if this mutation is in a scaffold domain for the C-terminus or N-terminus interaction domains it may influence p101 interaction to other important proteins including Gβγ or p110γ (PIK3CG ), it is clear from the docking results that when wild type PIK3R5 was docked with PIK3CG and further with Gβγ protein the total protein energies and Eshape energy were negative and low and the PIK3CG was fully fitted in the cavity of PIK3R5. Furthermore, the bumps value of −1 indicated rigid complexes and E force of 0 indicated that no force was needed for interacting. On the contrary when mutant PIK3R5 was docked with PIK3CG and further with Gβγ protein the total protein energies and Eshape energy were lower than wild type and the mutation caused the cavity (which will interact with the PIK3CG) to become slightly expanded so unable to fit properly. Thus the structure of PIK3CG was titled to fit in to a large cavity while finding a suitable place for interaction and unable to interact properly to form a dimer or perform its normal function. In addition, the bumps value of 0 indicated the loss of rigidity in these complexes and E force of −36.97 and −46.28 for mutant PIK3R5-PIK3CG and mutant PIK3R5-PIK3CG-Gβγ indicated an extra energy to be used by the mutated PIK3R5 in order to get interacted with the PIK3CG and Gβγ afterwards.
5. CONCLUSION
In summary, our in-silico analysis demonstrated that although this missense variant which is reported in the SNP database is not in a defined functional domain or a well characterized binding domain of the protein, it has a deleterious effect on the binding of PIK3R5-PIK3GCGβγ complex, thus disturbing the activation and the function of PI3Kγ. The involvement of this new gene in this heterogeneous disorder emphasizes the role of the AKT pathway in the development of this disease. Screening for mutations in PIK3R5 in similar disorders is of great importance. Our analysis highlights the importance of investigating the role of rare variants in the development of diseases.
6. AUTHOR’S CONTRIBUTIONS
NA conceived and designed the study. AIT performed the modeling and Docking. JS and AIT analyzed the data. SB contributed to analysis tools. All authors contributed in writing the paper. NA was the principal investigator and oversaw experimental design and analysis. JS and AIT contribute equally to this work.
REFERENCES
- Chen, K., Iribarren, P., Gong, W. and Wang, J.M. (2005) The essential role of phosphoinositide 3-kinases (PI3Ks) in regulating pro-inflammatory responses and the progression of cancer. Cellular & Molecular Immunology, 2, 241-252.
- Shymanets, A., Ahmadian, M.R., Kossmeier, K.T., Wetzker, R., Harteneck, C. and Nurnberg, B. (2012) The p101 subunit of PI3Kgamma restores activation by Gbeta mutants deficient in stimulating p110gamma. The Journal of Biological Chemistry, 441, 851-858. doi:10.1042/BJ20111664
- Brock, C., Schaefer, M., Reusch, H.P., Czupalla, C., Michalke, M., Spicher, K., Schultz, G. and Nurnberg, B. (2003) Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3- kinase gamma. The Journal of Cell Biology, 160, 89-99. doi:10.1083/jcb.200210115
- Osaki, M., Oshimura, M. and Ito, H. (2004) PI3K-Akt path- way: Its functions and alterations in human cancer. Apoptosis, 9, 667-676. doi:10.1023/B:APPT.0000045801.15585.dd
- Zhao, L. and Vogt, P.K. (2008) Class I PI3K in oncogenic cellular transformation. Oncogene, 27, 5486-5496. doi:10.1038/onc.2008.244
- Vanhaesebroeck, B. and Alessi, D.R. (2000) The PI3KPDK1 connection: More than just a road to PKB. The Journal of Biological Chemistry, 346, 561-576. doi:10.1042/0264-6021:3460561
- Walker, E.H., Perisic, O., Ried, C., Stephens, L. and Williams, R.L. (1999) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature, 402, 313- 320. doi:10.1038/46319
- Voigt, P., Brock, C., Nurnberg, B. and Schaefer, M. (2005) Assigning functional domains within the p101 regulatory subunit of phosphoinositide 3-kinase gamma. The Journal of Biological Chemistry, 280, 5121-5127. doi:10.1074/jbc.M413104200
- Skorski, T., Bellacosa, A., Nieborowska-Skorska, M., Majewski, M., Martinez, R., Choi, J.K., Trotta, R., Wlodarski, P., Perrotti, D., Chan, T.O., et al (1997) Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO Journal, 16, 6151-6161. doi:10.1093/emboj/16.20.6151
- Benistant, C., Chapuis, H. and Roche, S. (2000) A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene, 19, 5083-5090. doi:10.1038/sj.onc.1203871
- Knobbe, C.B., Trampe-Kieslich, A. and Reifenberger, G. (2005) Genetic alteration and expression of the phosphorinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathology and Applied Neurobiology, 31, 486-490. doi:10.1111/j.1365-2990.2005.00660.x
- Mizoguchi, M., Nutt, C.L., Mohapatra, G. and Louis, D.N. (2004) Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathology, 14, 372-377. doi:10.1111/j.1750-3639.2004.tb00080.x
- Al Tassan, N., Khalil, D., Shinwari, J., Al Sharif, L., Bavi, P., Abduljaleel, Z., Abu Dhaim, N., Magrashi, A., Bobis, S., Ahmed, H., et al. (2012) A missense mutation in PIK3R5 gene in a family with ataxia and oculomotor apraxia. Human Mutation, 33, 351-354. doi:10.1002/humu.21650
- Moreira, M.C., Klur, S., Watanabe, M., Nemeth, A.H., Le Ber, I., Moniz, J.C., Tranchant, C., Aubourg, P., Tazir, M., Schols, L., et al. (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nature Genetics, 36, 225-227. doi:10.1038/ng1303
- Amouri, R., Moreira, M.C., Zouari, M., El Euch, G., Barhoumi, C., Kefi, M., Belal, S., Koenig, M. and Hentati, F. (2004) Aprataxin gene mutations in Tunisian families. Neurology, 63, 928-929. doi:10.1212/01.WNL.0000137044.06573.46
- Fernet, M., Gribaa, M., Salih, M.A., Seidahmed, M.Z., Hall, J. and Koenig, M. (2005) Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Human Molecular Genetics, 14, 307-318. doi:10.1093/hmg/ddi027
- Zhang, Z., Miteva, M.A., Wang, L. and Alexov, E. (2012) Analyzing effects of naturally occurring missense mutations. Computational and Mathematical Methods in Medicine, 2012, 805827. doi:10.1155/2012/805827
- Roy, A., Kucukural, A. and Zhang, Y. (2010) I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocols, 5, 725-738. doi:10.1038/nprot.2010.5
- Zhang, Y. (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins: Structure, Function, and Bioinformatics, 69, 108-117. doi:10.1002/prot.21702
- Wu, S. and Zhang, Y. (2007) LOMETS: A local metathreading-server for protein structure prediction. Nucleic Acids Research, 35, 3375-3382. doi:10.1093/nar/gkm251
- Sali, A. and Blundell, T.L. (1994) Comparative protein modelling by satisfaction of spatial restraints. Protein Structure by Distance Analysis, 64, C86. doi:10.1006/jmbi.1993.1626
- Tuncbag, N., Gursoy, A. and Keskin, O. (2009) Identification of computational hot spots in protein interfaces: Combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics, 25, 1513- 1520. doi:10.1093/bioinformatics/btp240
- Pontius, J., Richelle, J. and Wodak, S.J. (1996) Deviations from standard atomic volumes as a quality measure for protein crystal structures. Journal of Molecular Biology, 264, 121-136. doi:10.1006/jmbi.1996.0628
- Luthy, R., Bowie, J.U. and Eisenberg, D. (1992) Assessment of protein models with three-dimensional profiles. Nature, 356, 83-85. doi:10.1038/356083a0
- Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26, 283-291. doi:10.1107/S0021889892009944
- Lovell, S.C., Davis, I.W., Arendall, W.B., de Bakker, P.I., Word, J.M., Prisant, M.G., Richardson, J.S. and Richardson, D.C. (2003) Structure validation by Calpha geometry: Phi,psi and Cbeta deviation. Proteins, 50, 437-450. doi:10.1002/prot.10286
- Guex, N. and Peitsch, M.C. (1997) SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis, 18, 2714-2723. doi:10.1002/elps.1150181505
- Roger, S. and Milner-White, E.J. (1995) RasMol: Biomolecular graphics for all. Trends in Biochemical Sciences (TIBS), 20, 374. doi:10.1016/S0968-0004(00)89080-5
- Ritchie, D.W. and Venkatraman, V. (2010) Ultra-fast FFT protein docking on graphics processors. Bioinformatics, 26, 2398-2405. doi:10.1093/bioinformatics/btq444
- Dombkowski, A.A. (2003) Disulfide by Design™: A computational method for the rational design of disulfide bonds in proteins. Bioinformatics, 19, 1852-1853. doi:10.1093/bioinformatics/btg231
- Herbert, J.B. (2000) Recent changes to RasMol, recombining the variants. Trends in Biochemical Sciences, 25, pp. 453-455. doi:10.1016/S0968-0004(00)01606-6
- Chen, V.B., Arendall 3rd, W.B., Headd, J.J., Keedy, D.A., Immormino, R.M., Kapral, G.J., Murray, L.W., Richardson, J.S. and Richardson, D.C. (2010) MolProbity: Allatom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography, 66, 12-21. doi:10.1107/S0907444909042073
- Chen, V.B., Davis, I.W. and Richardson, D.C. (2009) KING (Kinemage, Next Generation): A versatile interactive molecular and scientific visualization program. Protein Science, 18, 2403-2409. doi:10.1002/pro.250
- Colovos, C. and Yeates, T.O. (1993) Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Science, 2, 1511-1519. doi:10.1002/pro.5560020916
- Olson, A.J. and Goodsell, D.S. (2007) A semiempirical free energy force field with charge-based desolvation. Journal of Computational Chemistry, 28, 1145-1152. doi:10.1002/jcc.20634
- Koukouritaki, S.B., Poch, M.T., Henderson, M.C., Siddens, L.K., Krueger, S.K., VanDyke, J.E., Williams, D.E., Pajewski, N.M., Wang, T. and Hines, R.N. (2007) Identification and functional analysis of common human flavincontaining monooxygenase 3 genetic variants. Journal of Pharmacology and Experimental Therapeutics, 320, 266- 273. doi:10.1124/jpet.106.112268
- Tang, K.E. and Dill, K.A. (1998) Native protein fluctuations: The conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. Journal of Biomolecular Structure & Dynamics, 16, 397-411. doi:10.1080/07391102.1998.10508256
- Board, P.G., Pierce, K. and Coggan, M. (1990) Expression of functional coagulation factor XIII in Escherichia coli. Journal of Thrombosis and Haemostasis, 63, 235- 240.
- Deber, C.M., Brodsky, B. and Rath, A. (2010) Proline residues in proteins. John Wiley & Sons Ltd., Chichester.
- Williamson, M.P. (1994) The structure and function of proline-rich regions in proteins. Biochemical Journal, 297, 249-260.
- Kini, R.M. and Evans, H.J. (1995) A hypothetical structural role for proline residues in the flanking segments of protein-protein interaction sites. Biochemical and Biophysical Research Communications, 212, 1115-1124. doi:10.1006/bbrc.1995.2084
- Kay, B.K., Williamson, M.P. and Sudol, M. (2000) The importance of being proline: The interaction of prolinerich motifs in signaling proteins with their cognate domains. The FASEB Journal, 14, 231-241.
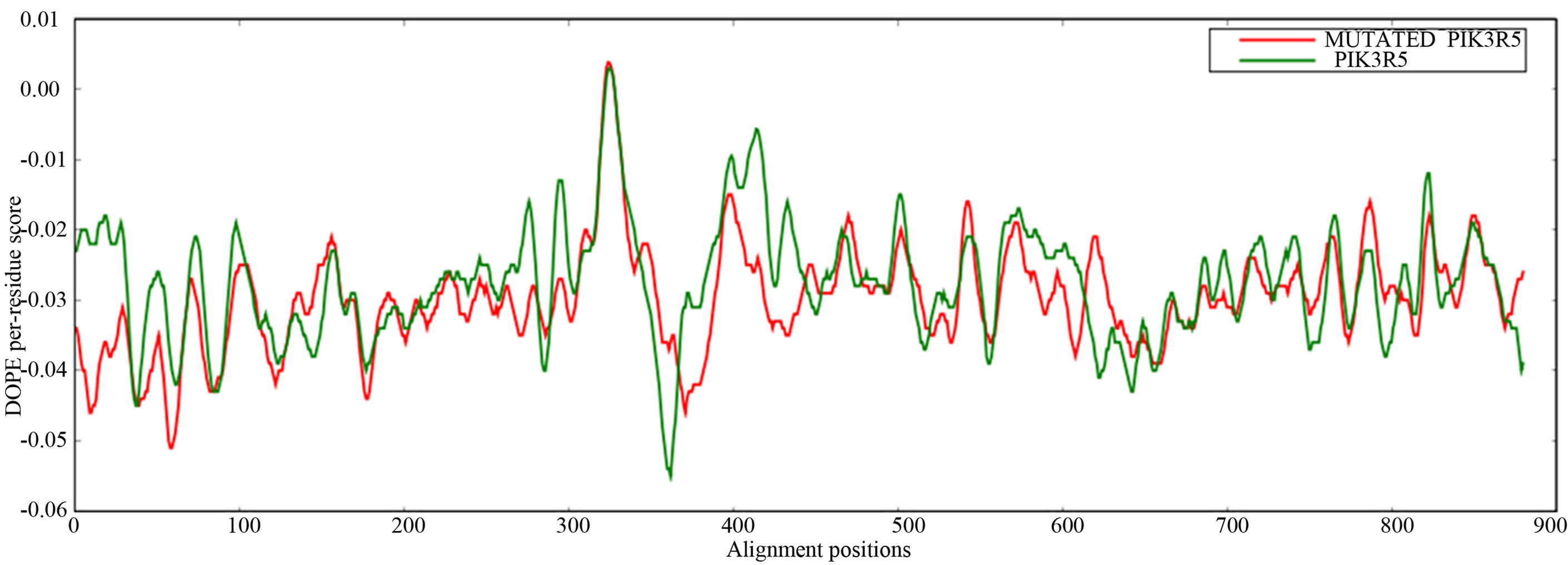
- Supplementary Figure 1. DOPE graph. In general wild type PIK3R5 residues in most of the alignment positions are conserved in the mutated model (X-axis: residues of PIK3R5 (880a.a), Y-axis: energy per residue).
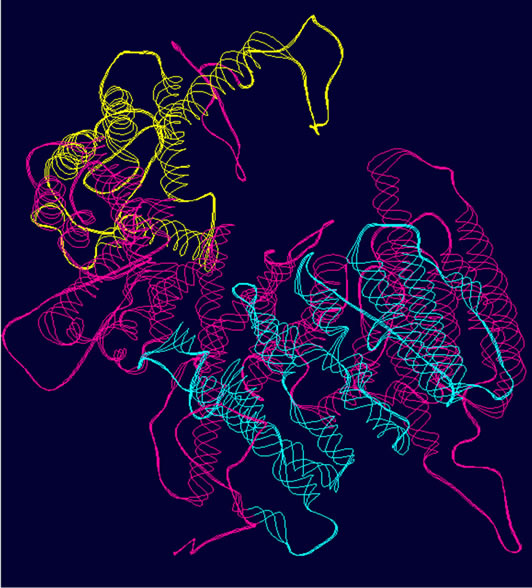
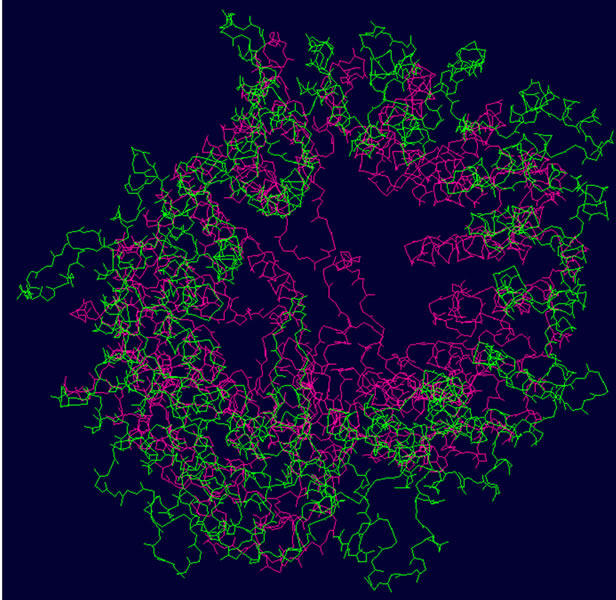
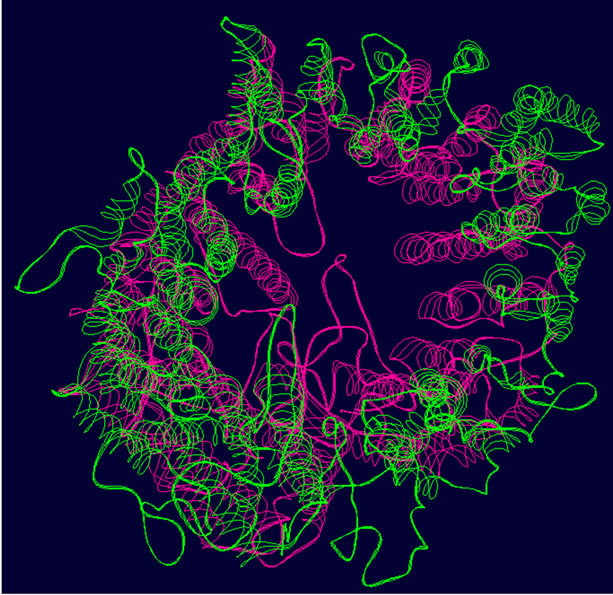
- (a) (b) (c)
- Supplementary Figure 2. WT and Mutant PIK3R5 superimposed structures. (a) Binding sites of PIK3CG on PIK3R5 (yellow), binding site of Gβγ on PIK3R5 (blue); (b) Backbone superimpose structure of Wild-type PIK3R5 (pink) and mutant PIK3R5 (green); (c) Superimposed structure of Wild-type PIK3R5 and mutated PIK3R5, Pink structure shows the wild type PIK3R5 and green structure shows mutated PIK3R5.
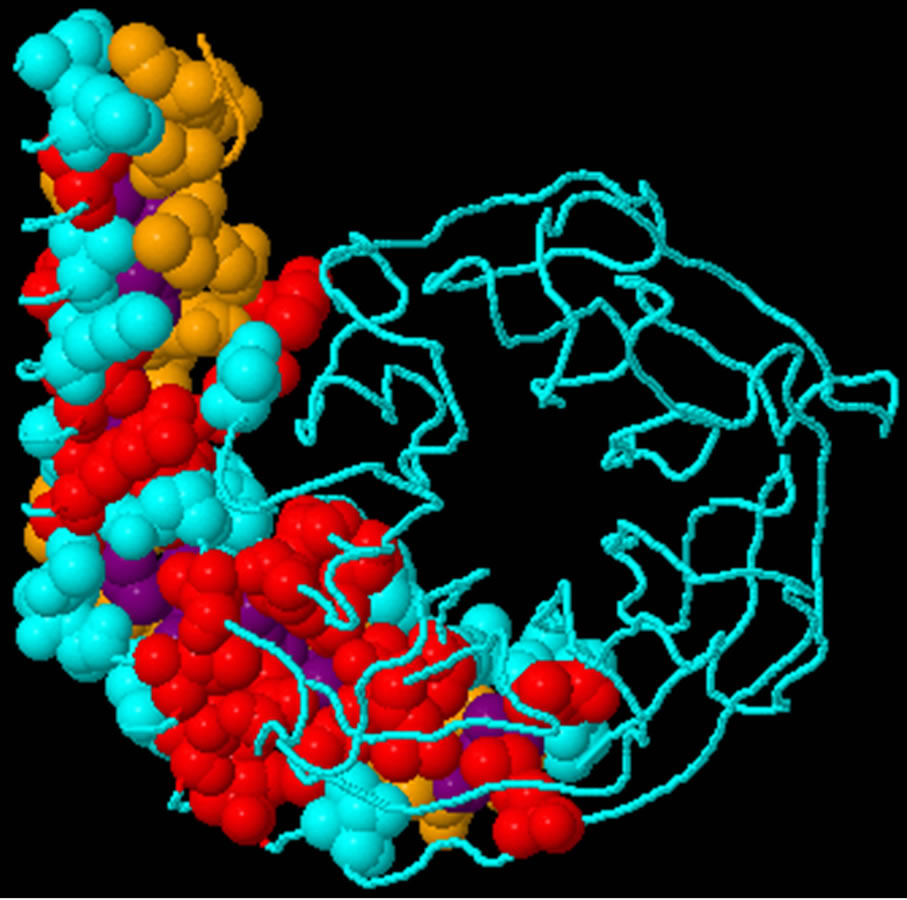
- Supplementary Figure 3. Predicted binding sites of Gβγ. Blue; Interface residues on chain (a) Red; Predicted Hot spots in interface (Chain A). Yellow; Interface residues on chain (b) Purple; Predicted Hot spots in interface (Chain B).
- (a) (b)
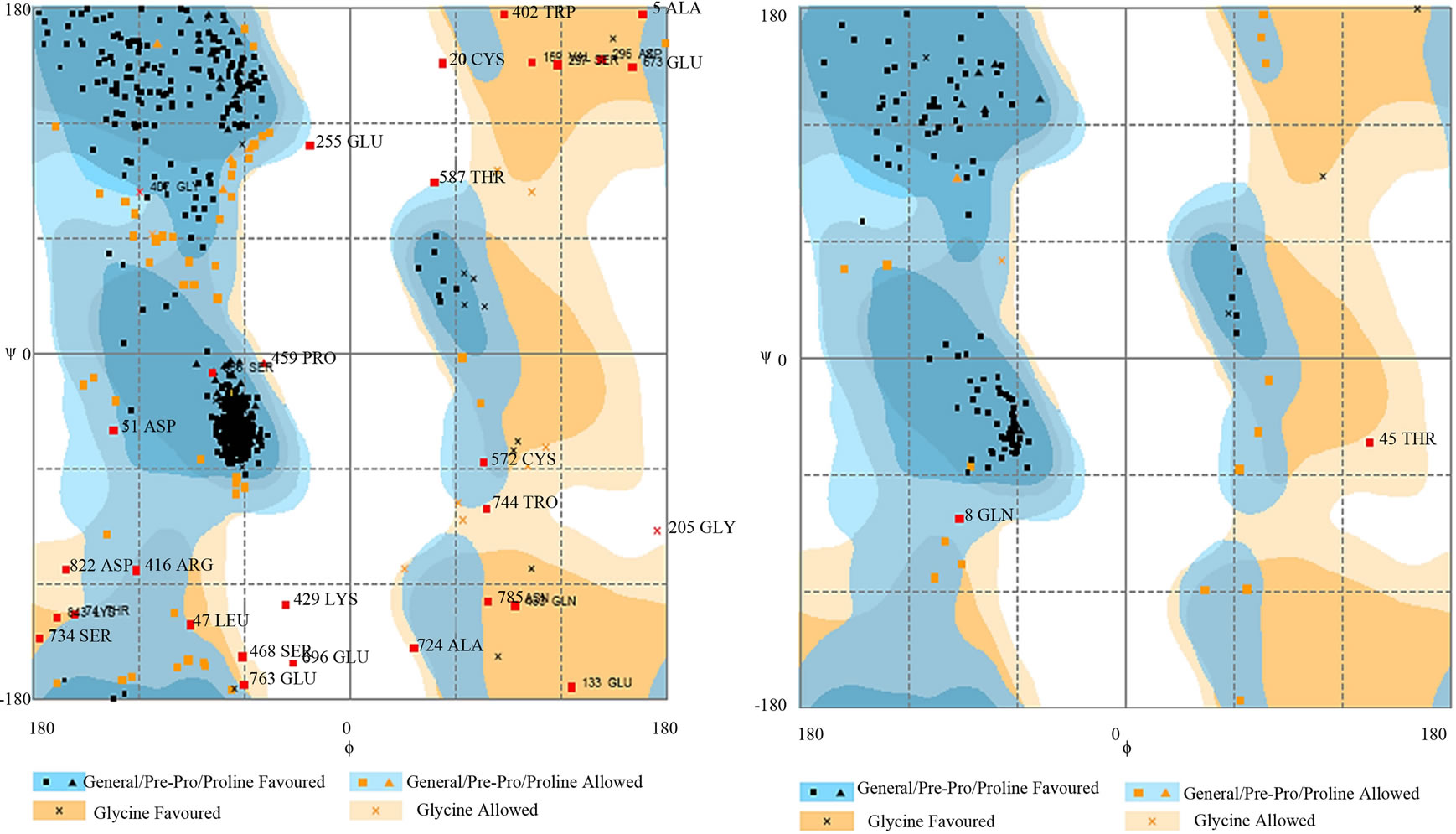
- Supplementary Figure 4. Ramachandran plot of PIK3R5 and PIK3CG. (a) PIK3R5 showing most of the amino acids in favorable regions. Number of Residues in favored region: 788 (91.2%), Number of residues in allowed region: 60 (6.4%), Number of residues in outlier region: 30 (2.4%); (b) PIK3CG. Number of Residues in favored region: 123 (86.6%), Number of residues in allowed region: 17 (12.0%) Number of residues in outlier region: 2 (1.4%). X-axis: Phi-angle. Y-Axis: Psi-angle.
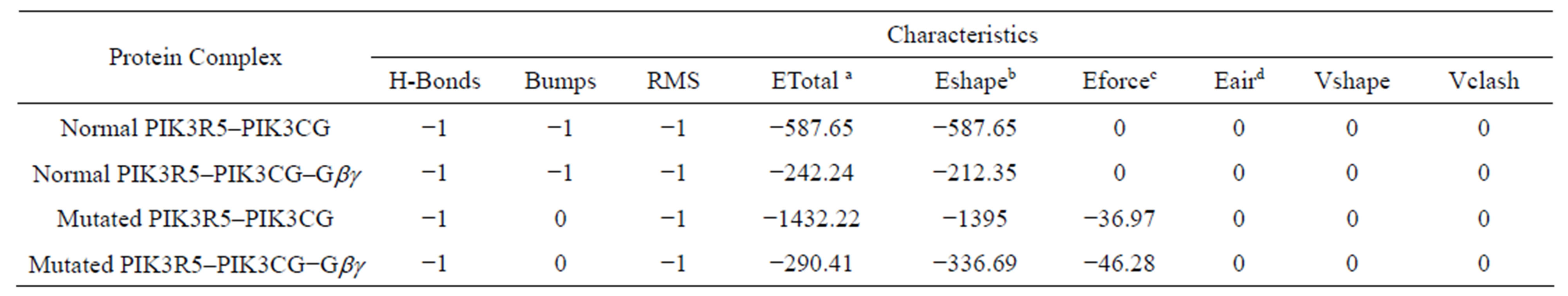
- Supplementary Table 1. Docking energies and statistics.
- aETotal-Total energy of the system. bEshape = Hex shape-based pseudo energy. cEforce = force field energy. dEair = (Ambiguous Interaction Restraints).
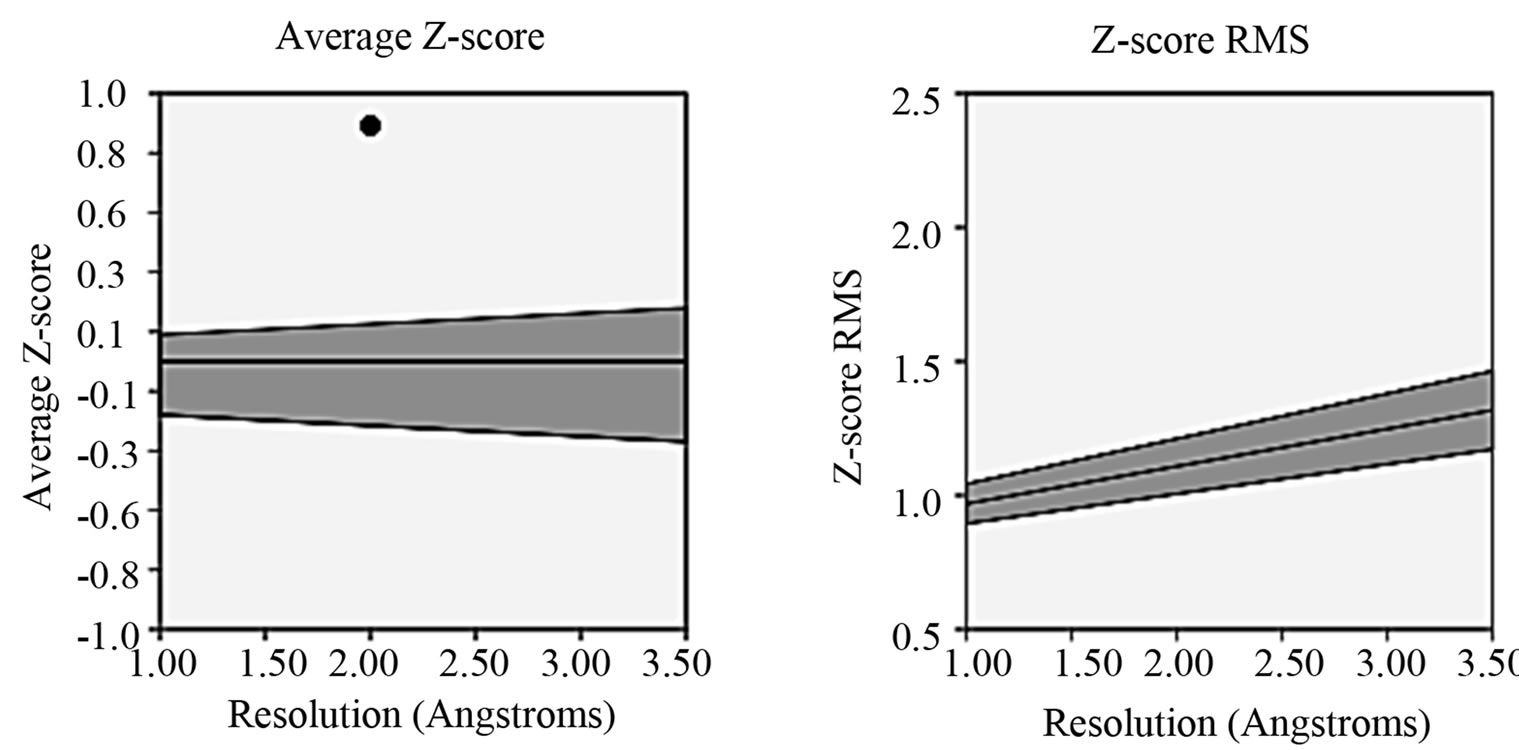
- Supplementary Figure 5. PROVE PLOT for PIK3R5 (Z-score and root mean square score).
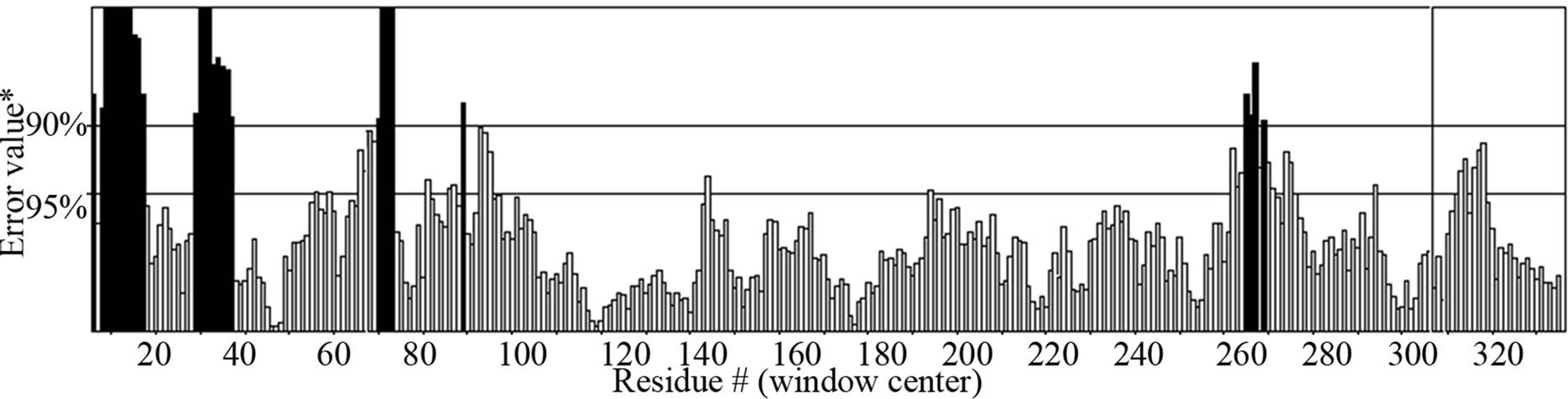
- Supplementary Figure 6. ERRAT graph for Gβγ.
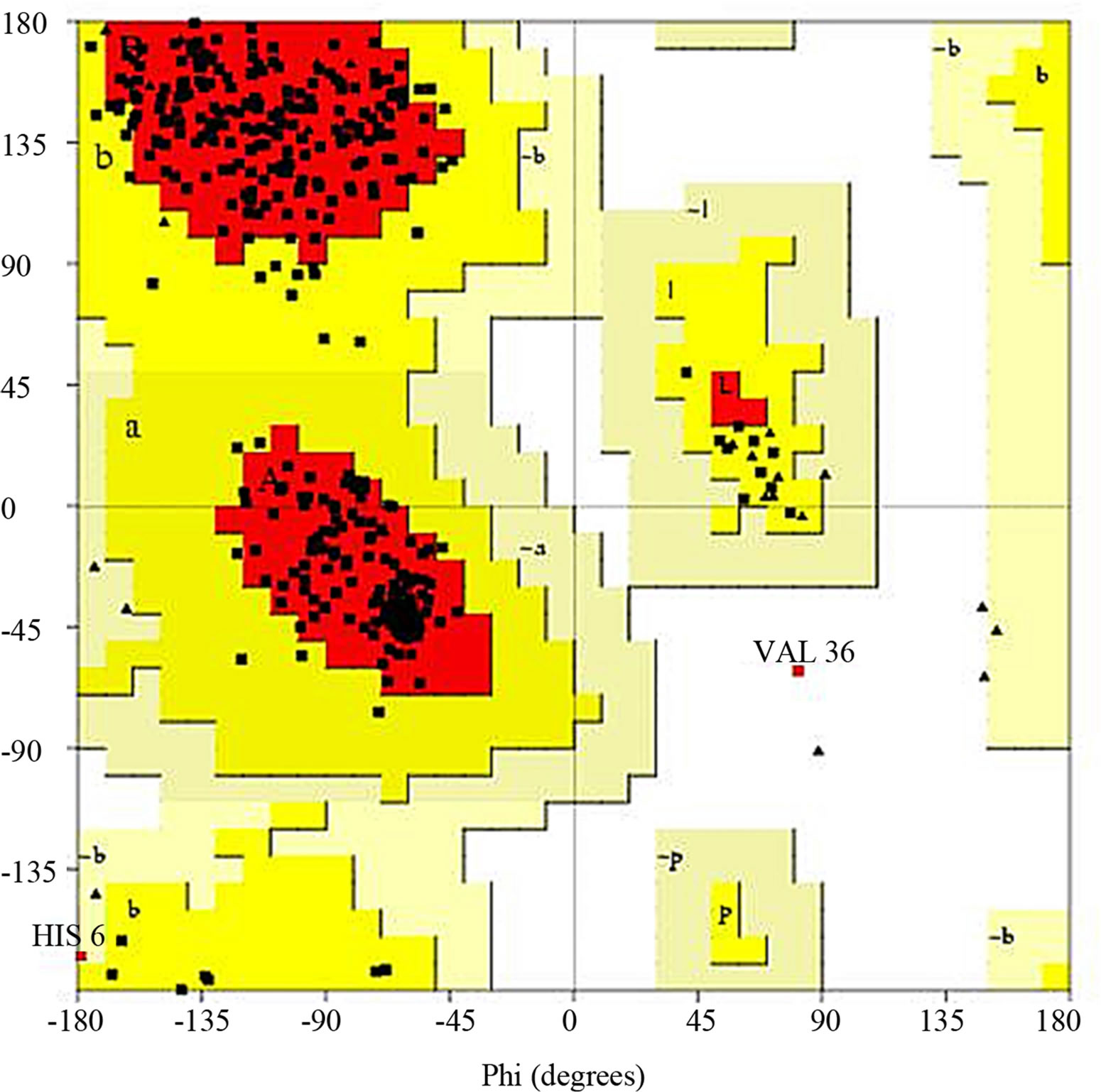
- Supplementary Figure 7. Ramachandran plot for Gβγ.
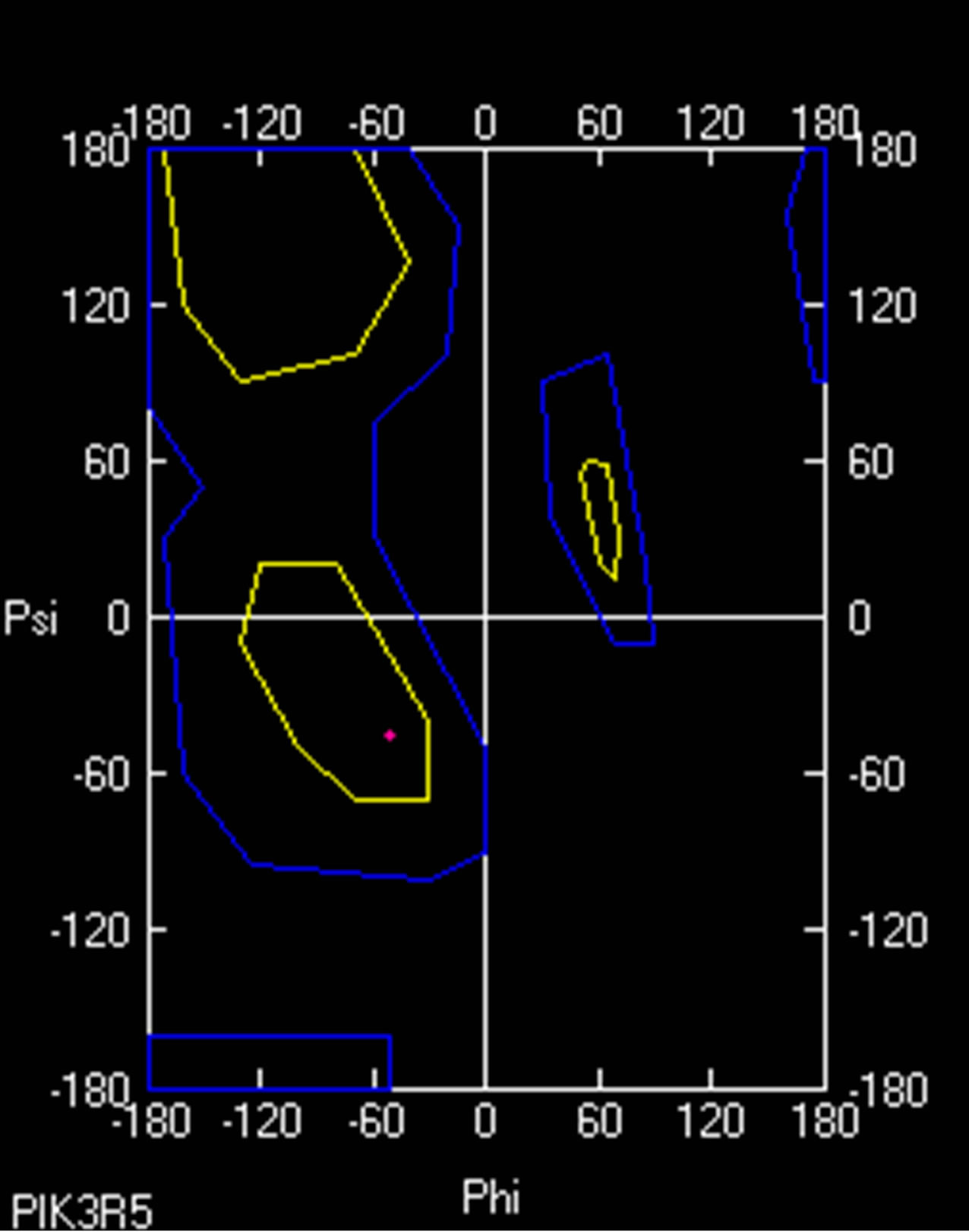
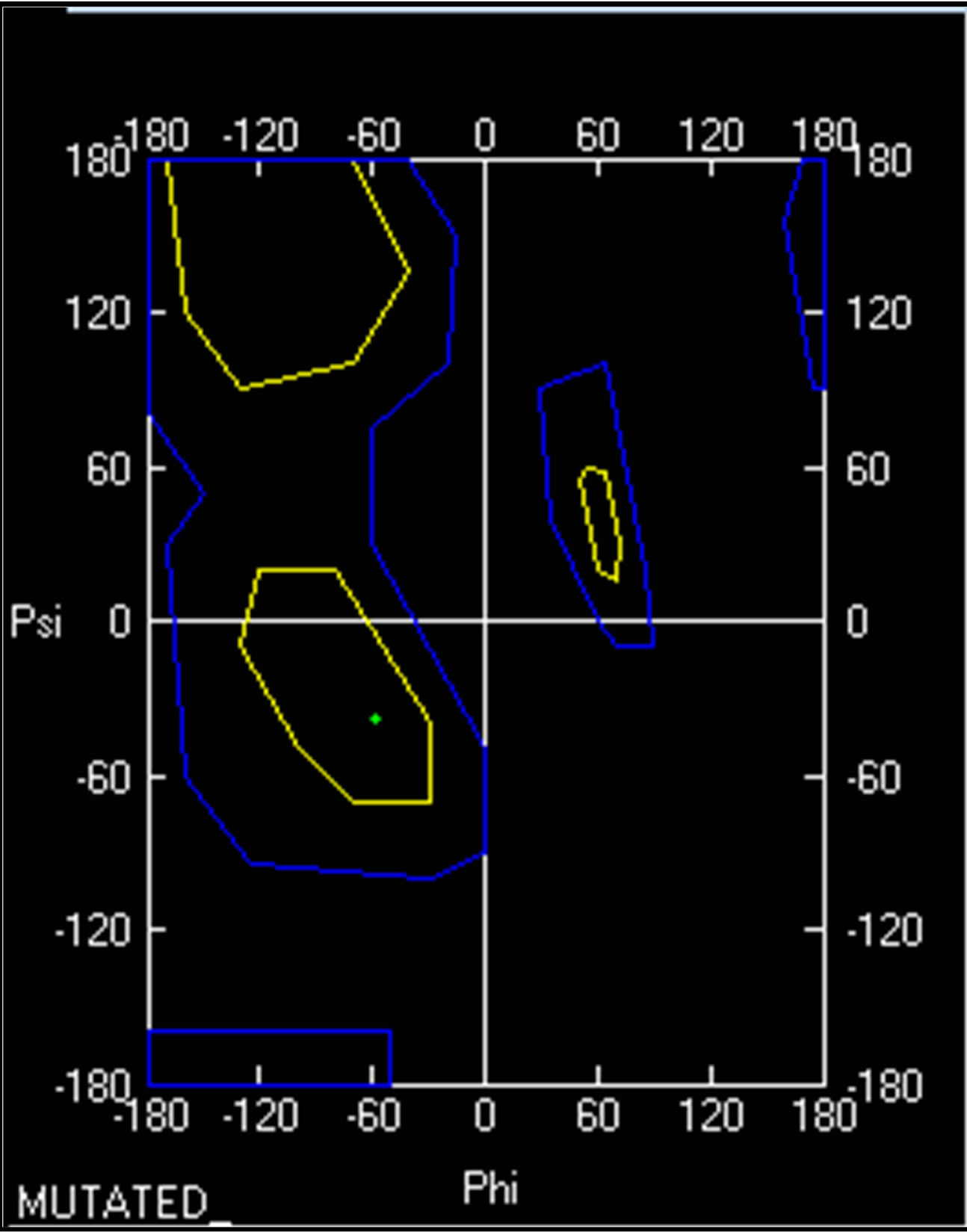
- (b) (b)
- Supplementary Figure 8. Ramachandran plot for PIK3R5. A small change in dihedral angle between wild type (Red) and mutant PIK3R5 (Green) is visible.
NOTES
*Competing Interests: None of the authors have any financial interest related to the work presented in this manuscript and they declare no competing interests.
#Authors contributed equally to manuscript.
†Corresponding author.