Open Journal of Physical Chemistry
Vol.05 No.04(2015), Article ID:61046,10 pages
10.4236/ojpc.2015.54013
A Theoretical Study of β-Amino Acid Conformational Energies and Solvent Effect
Victor F. Waingeh1*, Felix N. Ngassa2*, Jie Song3
1School of Natural Sciences, Indiana University Southeast, New Albany, IN, USA
2Department of Chemistry, Grand Valley State University, Allendale, MI, USA
3Department of Chemistry and Biochemistry, University of Michigan-Flint, Flint, MI, USA

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 29 August 2015; accepted 9 November 2015; published 12 November 2015

ABSTRACT
The conformations of four β-amino acids in a model peptide environment were investigated using Hartree-Fock (HF) and density functional theory (DFT) methods in gas phase and with solvation. Initial structures were obtained by varying dihedral angles in increments of 45˚ in the range 0˚ - 360˚. Stable geometries were optimized at both levels of theory with the correlation consistent double-zeta basis set with polarization functions (cc-pVDZ). The results suggest that solvation generally stabilizes the conformations relative to the gas phase and that intramolecular hydrogen bonding may play an important role in the stability of the conformations. The β3 structures, in which the R-group of the amino acid is located on the carbon atom next to the N-terminus, are somewhat more stable relative to each other than the β2 structures which have the R-group on the carbon next to the carbonyl.
Keywords:
Density Functional Theory, β-Amino Acids, Conformational Search

1. Introduction
The functions of numerous biological systems depend on RNA and proteins. The conformation adopted by these biopolymers is linked to their functions in biological systems. As a consequence, there has been an increasing need to identify synthetic polymer backbones that adopt discrete and predictable conformations (“foldamers”) to mimic natural biological systems. Such backbones can serve as tools to probe the functions of large-molecule interactions, such as protein-protein and protein-RNA interactions. In foldamer design, β-amino acids are highly attractive building blocks because the additional carbon confers conformational flexibility to β-amino acids compared to their α-amino acid counterparts.
Early attempts at realizing ordered peptide structures with β-amino acids dated from the end of the 1960s and were continued in the 1970s [1] -[3] . In 1994, Dado and Gellman studied the intramolecular hydrogen bonding properties of β- and γ-amino acid derivatives [4] . They hypothesized that the formation of secondary structures by oligomers of α-amino acids was due in part to the formation of short range hydrogen bonds, such as intramolecular 5- or 7-membered hydrogen bonded rings which were energetically unfavorable among α-amino acid oligomers (Figure 1). Based on their hypothesis, Dado and Gellman reasoned that other oligomers in which the formation of short range hydrogen bonds was unfavorable might also be predisposed to form secondary structures. To test this hypothesis further, Dado synthesized various derivatives of β-alanine as well as γ-isobutyric acid using IR spectroscopy, and examined the intramolecular hydrogen bonding of these molecules. The results demonstrated that while short range hydrogen bonds (7- and 9-membered rings) were common among the diamides of γ-amino-butyric acid, the same was not true for diamides of β-amino acids (Figure 1). Therefore, based on these studies, Dado and Gellman concluded that oligomers of β-amino acids (β-peptides) could potentially form stable secondary structures. The studies carried out by Dado and Gellman did not take into account the substitution on α-, β-, or γ-carbons which could potentially had some constraining effects.
Although, it was predicted that β-peptides were capable of forming stable and compact secondary structures, investigations of the NMR solution structures of poly-β-alanine indicated that the polymer had no folded conformation [2] . In contrast, IR data in the solid state indicated the formation of sheets [5] . Various research groups have studied the polymers of α-isobutyl-L-aspartate by X-ray diffraction, CD, and IR spectroscopy. In 1978, Yuki et al. reported that the polymer of α-isobutyl-L-aspartate formed extended sheets with hydrogen bonding between the strands [6] . In the mid 1980s, Fernández-Santín and co-workers reported that in the solid state, the polymer of α-isobutyl-L-aspartate formed two helical conformations characterized by intramolecular 16- and 20-membered hydrogen bonded rings [7] [8] . In 1995, López-Carrasquero et al. contrasted initial reports by Fernández-Santín and co-workers and instead proposed that the helical conformations formed by the polymer of α-isobutyl-L-aspartate was characterized by 14- and 18-membered hydrogen bonded rings [9] .
The biggest breakthrough in the synthesis β-peptides of defined sequence that allowed crystallographic and high-resolution NMR data to be obtained was achieved by the research groups of Gellman [10] -[15] and Seebach [16] -[19] . Gellman and co-workers have synthesized and characterized the oligomers of trans-2-amino- cyclo-pentanecarboxylic acid (ACPC) [12] , trans-2-aminocyclohexanecarboxylic acid (ACHC) [11] and trans- 3-aminopyrrolidine-4-carboxylic acid (ACP) [15] . Gellman et al. showed that while ACHC adopted a 14-helical conformation in organic solvent, ACPC adopted a 12-helical conformation. The Oligomers containing both ACPC and ACP residues were also shown to form 12-helical conformations in aqueous solution [15] . Seebach and co-workers showed that β-peptides composed of acyclic residues with side chains derived from α-amino acids also formed 14-helical conformations in organic solvent [16] .
In polypeptides of α-amino acids, the backbone conformations are defined by three sets of torsion angles and (Figure 2) [20] . Generally in peptides and proteins, the torsion angle about the peptide bond is restricted to a trans geometry (β = 180). Backbone conformation of β-amino acid residues in peptides is determined by four main chain torsional variables q and using the convention of Balaram (Figure 2) [20] . By this convention, the
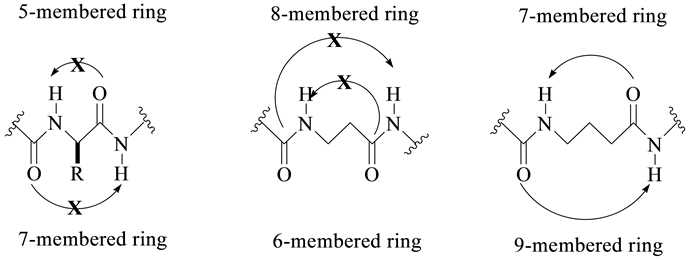
Figure 1. Favorable and non-favorable intramolecular hydrogen bonding in α-, β-, and γ-amino acid peptides.
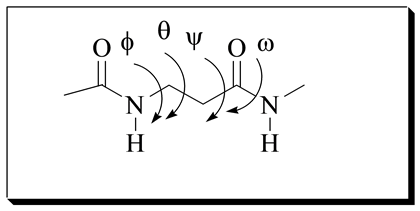
Figure 2. Backbone conformation of β-amino acid.
backbone conformation is read sequentially from the N- to the C-terminus with the torsional variables defined as (N-Cβ), q (Cβ-Cα), (Cα-CO), and (CO-N) (Figure 2). In β-amino acids, the torsion angle about the C-C bond (q) can lie close to the gauche (q = ±60˚) and trans (q = 180˚) conformations [20] . A predominance of gauche conformations can easily lead to the formation of folded structures.
The interactions between oligomers and solvent can affect the conformations adopted by oligomers. The ability for an oligomer to fold can be favored or disfavored by such solvent properties as dielectric constant, solubility, and hydrogen bonding capability. Solvent effects on the conformations of phenylacetylene oligomers have been reported by Nelson and co-workers [21] . Based on NMR and UV spectroscopic studies, they concluded that a well-ordered conformation was observed in deuterated acetonitrile, but not in deuterated chloroform. The difference in behavior of the oligomer in the different solvents could be a result of difference in solubility. The oligomer was found to be completely soluble in chloroform irrespective of the number of residues and had many conformations. In acetonitrile, however, the oligomer became less soluble as the chain length increased resulting in a single, well-defined conformation.
Like natural peptides and proteins, β-peptides can adopt folded conformations including common secondary structures such as helices [11] -[13] [22] -[28] , turns [18] [29] and sheets [14] [30] . Compared to α-peptides, β-peptides have the advantage of increased conformational stability in an aqueous environment. [15] The β- peptide backbone compared to natural peptides is resistant to protease degradation and has the potential for a great variety of substitution patterns [31] . In addition β-peptides form more stable helices in solution compared to α-peptides; β-peptides can form secondary structures with as few as four to six residues in solution [13] [16] [19] , compared to over 30 residues needed for stability of the natural analogs. The stability of β-peptides, important for biological activity, makes them good candidates for useful drugs. [18] It has been shown that foldamers comprising a mixture of α-, β-, and g-amino acid residues are not degraded by proteases which bodes well for biological application [26] [27] .
Investigating the 3-D structure of β-peptides is critical to understanding their biological functions. Although crystal structures and NMR spectroscopy can provide adequate structural information for β-peptides, these methods still have some limitations. Crystallography only provides solid-state structural information and in most cases obtaining a good crystal structure for X-ray crystallography is often difficult. NMR spectroscopy can provide structural information corresponding to solution structures. However, limitations exist in the size of the peptide; NMR works best for relatively small peptides.
Computational modeling can provide structural information, at the atomic level, for β-peptides from the sequence of β-amino acids [32] . A good computational method depends on the ability to reproduce the structures and energies of β-amino acid conformations in a target molecule. Herein, the conformations of some selected β-peptides resulting from rotation along their backbones are studied theoretically. The conformations are first calculated in the gas phase at the HF and DFT level to determine their relative stabilities. Then, solvent effect is included by applying the continuum solvation model and comparisons of the two models are made.
2. Computational Details
Several conformations of selected β-amino acids were computed. To mimic the environment in longer peptide chains, the amino-end of the each amino acid was capped with an acetyl group and the carboxylic end was capped with methylamine. The resulting structure template and torsion angle labels are as shown in Figure 3.
Selected β-amino acids for this study include β-alanine (R = −CH3), β-cysteine (R = −CH2SH), β-leucine
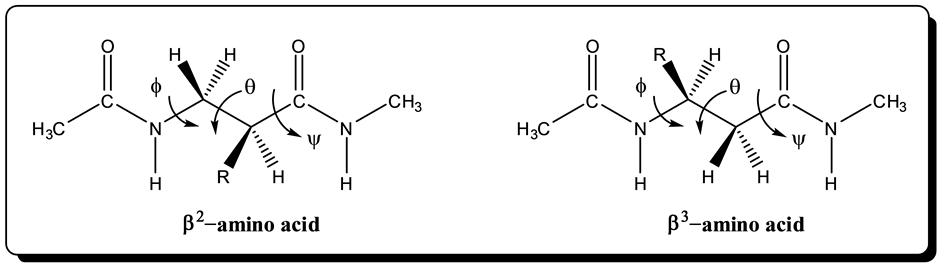
Figure 3. Structure of β-amino acid in peptide environment.
(R= −CH2CH(CH3)2), and β-serine (R = −CH2SH). In the β2 structure, the R-group of the amino acid is on the carbon attached to the carbonyl function at the C-end while in β3 the R-group is closer to the N-end. The β3 conformation is included in this study because studies have shown that β3-peptides can populate a secondary structure known as a 14-helix, which is characterized by 14-membered ring hydrogen bonds between the amide at potion i and the carbonyl at position i + 2, a left-handed helical twist with three discrete faces [33] -[35] .
Initial conformations were obtained by rotating θ angles by 45˚ increments in the range 0˚ to 360˚. The structures were then optimized at HF/cc-pVDZ and DFT/cc-pVDZ levels of theory in the gas phase, without any restraints. Calculations were then repeated to include solvation effects by employing the Polarizable Continuum Model (PCM) for water at the DFT level. For comparisons with gas-phase conformational energies, calculations with solvation were all done with a constant dielectric of 1.0. In all instances, the estimation of relative conformational energies was concluded by performing single point energy calculations at both the HF and DFT levels for each conformation. All calculations were performed using GAMESS suite program [36] and Avogadro [37] was used for visualization.
3. Results and Discussions
HF and DFT were used to study the conformations of selected β-amino acids in both the gas phase and with solvation (DFT only). Stable conformations were identified and the relative energies calculated as the difference in energy between each identified conformation and the lowest energy conformation.
3.1. β-Alanine
Gas phase calculations for β-alanine predicted 5 stable conformations for both HF and DFT. The optimized dihedral angles and relative energies of conformers are given in Table 1. The minimum energy conformation occurred at a dihedral angle of 57˚ in the HF calculations and at −64˚ in the DFT calculations (Figure 4).
The highest energy conformation in both cases is one with an optimized dihedral angle closer to 180˚ and is about 17 kJ/mol higher in energy than the minimum. When solvation is included, calculations yield seven stable conformations, with a 17 kJ/mol energy difference between the most and the least stable conformations. The β3 structure of alanine shows a lot more variation in relative stabilities of its conformations with the lowest energy conformation at least 5 kJ/mol lower that the second lowest conformation as opposed to the smaller differences of 1 - 2 kJ/mol seen with the β2 structures. Solvation seems to stabilize the β3 structure to some extent as the relative energies of the identified conformations above the minimum are all within 2 kJ/mol of each other.
3.2. β-Cysteine
The presence of the ?CH2SH side chain in cysteine would be expected to introduce additional degrees of freedom and conformational flexibility when compared to alanine. The calculated relative energies (Table 2) show a wide variation ranging from 0 - 29 kJ/mol. As was the case with β-alanine, solvation seems to introduce to form of stabilization for both the β2 and β3 structures. Figure 5 shows the minimum energy conformations in gas phase.
Unlike in the corresponding β-amino acid, in β-cysteine, the positioning of ?CH2SH group also allows for the

Table 1. β-Alanine conformations and relative energies.
Table 2. β-Cysteine conformations and relative energies.
possibility of weak intramolecular hydrogen bonding between the side chain ?SH and the backbone ?C=O groups.
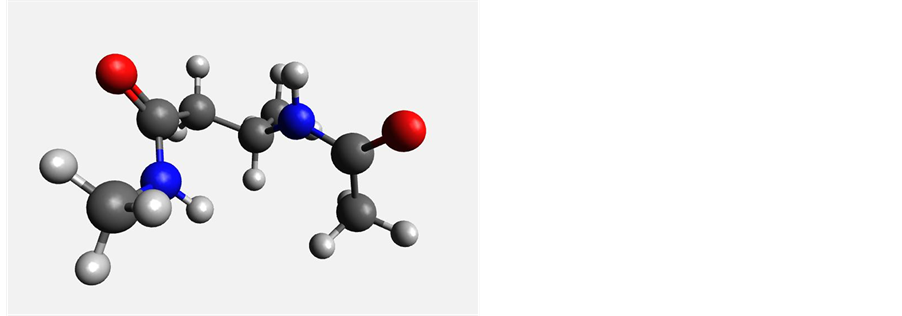
Figure 4. Most stable gas-phase conformers of β2-alanine (left) and β3-alanine (right).
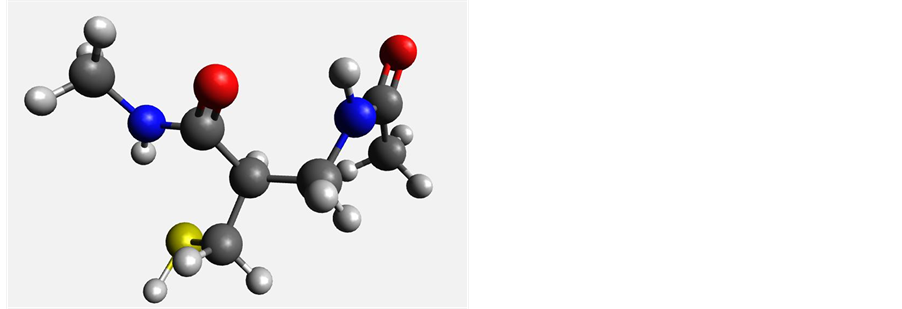

Figure 5. Most stable conformers of β2-cysteine (left) and β3-cysteine (right).
3.3. β-Serine
The ?CH2OH side chain in serine is similar to that of cysteine, and would introduce the same conformational flexibility. However, the replacement of the sulfur in cysteine with the oxygen in serine results in less polarizability and more hydrogen bonding capability. The conformational search and single point energy calculations resulted in three stable conformations in the gas phase for both the β2 and β3 structures in both HF and DFT. The β3 structures are more stable relative to each other than are the β2 structures as evident from the calculated relative energies as seen in Table 3.
In both HF and DFT, the next lowest energy conformation is at least 14 kJ/mol higher than the minimum, however for the β3 structures, the higher energy conformations are all within ~1 kJ/mol of each other. This would suggest that the possibility of favorable and strong intramolecular hydrogen bonding in the β3 structure helps to restrict the molecule. Figure 6 shows the optimized structures of the most stable conformers of β2 and β3-serine. When solvation is included in the calculations, there is evidence of some substantial stabilization in solution with a mean deviation of about 22.9 ± 2.5 kJ/mol for the β2 structure and 11.2 ± 1.9 kJ/mol for β3 structures.
3.4. β-Leucine
In the gas phase, the lowest energy conformation for β2-leucine occurs at a optimized dihedral angle of 58˚ in both HF and DFT with relative energies of higher energy conformations ranging from 13 - 26 kJ/mol in HF and 17 - 29 kJ/mol in DFT. For β3-leucine, the most favorable conformation occurs at −63˚ (Figure 7) with relative energies in the range 6 - 23 kJ/mol in HF and 8 - 23 kJ/mol in DFT. As was the case with other β-amino acids, solvation stabilizes the conformations relative to each other, with relative energies for the β2-leucine conformation in the range 3 - 18 kJ/mol and those for β3-leucine in the 2 - 11 kJ/mol range (Table 4). In general, the β3
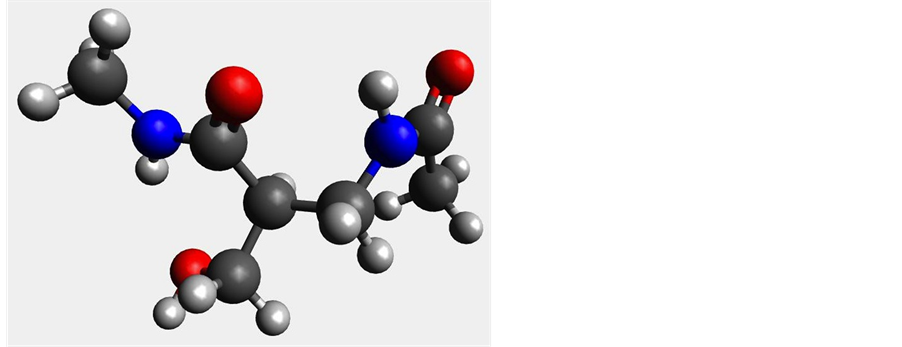

Figure 6. Most stable conformers of β2-serine (left) and β3-serine (right).
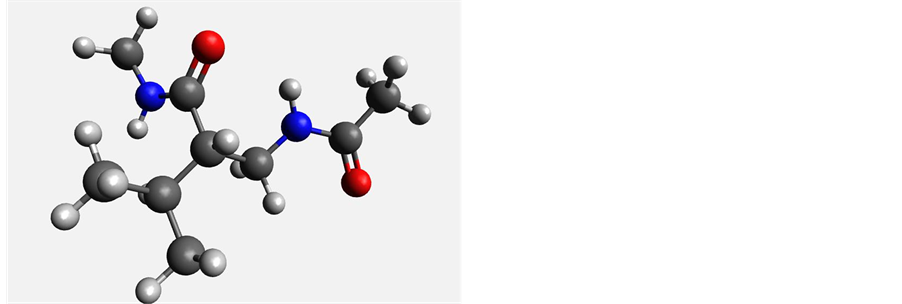

Figure 7. Most stable conformers of β2-leucine (left) and β3-leucine (right).
Table 3. β-Serine conformations and relative energies.
Table 4. Optimized dihedral angles and relative energies of β-Leucine conformations.
conformations seem to show slightly more solvent stabilization than the β2 conformations.
4. Conclusion
In this study, the conformations of β-amino acids were investigated in the gas phase by HF and DFT calculations. The PCM water model was employed with DFT to investigate the solvation effects. Relative energies were computed for both gas phase and solution structures. The results suggest that solvation generally stabilizes the conformations relative to the gas phase, with smaller energy differences between the conformations in solution than in gas phase. It is also likely that intramolecular hydrogen bonding may play an important role in the stability of the conformations. The β3 structures, in which the R-group of the amino acid is located on the carbon atom next to the N-terminus, are somewhat more stable relative to each other than the β2 structures which have the R-group on the carbon next to the carbonyl. These results provide insight to the conformational structures of β-amino acids and may be useful in establishing the potential use of β-amino acids in the backbone of polypeptide chain that will be less susceptible to degradation. However, further work will need to be done, with a larger collection of β-amino acids. Also the use of a continuum solvent model to describe solvation of these systems is reasonable and provides a basis for qualitative comparison. However, to obtain a more quantitatively accurate description of the system, future work may employ explicit solvation models.
Cite this paper
Victor F.Waingeh,Felix N.Ngassa,JieSong, (2015) A Theoretical Study of β-Amino Acid Conformational Energies and Solvent Effect. Open Journal of Physical Chemistry,05,122-131. doi: 10.4236/ojpc.2015.54013
References
- 1. Bestian, H. (1968) Poly-β-Amides. Angewandte Chemie International Edition in English, 7, 278-285.
http://dx.doi.org/10.1002/anie.196802781 - 2. Glickson, J.D. and Applequist, J. (1971) The Conformation of Poly-β-Alanine in Aqueous Solution from Proton Magnetic Resonance and Deuterium Exchange Studies. Journal of the American Chemical Society, 93, 3276-3281.
http://dx.doi.org/10.1021/ja00742a030 - 3. Kovacs, J., Ballina, R., Rodin, R.L., Balasubramanian, D. and Applequist, J. (1965) Poly-β-L-Aspartic Acid. Synthesis through Pentachlorophenyl Active Ester and Conformational Studies. Journal of the American Chemical Society, 87, 119-120.
http://dx.doi.org/10.1021/ja01079a022 - 4. Dado, G.P. and Gellman, S.H. (1994) Redox Control of Secondary Structure in a Designed Peptide. Journal of American Chemical Society, 115, 12609-12610.
- 5. Narita, M., Doi, M., Kudo, K. and Terauchi, Y. (1986) Conformations in the Solid State and Solubility Properties of Protected Homooligopeptides of Glycine and Beta-Alanine. Bulletin of Chemical Society of Japan, 59, 3553-3557.
- 6. Yuki, H., Okamoto, Y., Taketani, Y., Tsubota, T. and Marubayshi, Y. (1978) Poly(β-Amino Acid)s. IV. Synthesis and Conformational Properties of Poly(α-Isobutyl-L-Aspartate). Journal of Polymer Science Part A: Polymer Chemistry, 16, 2237-2251.
http://dx.doi.org/10.1002/pol.1978.170160913 - 7. Fernandez-Santin, J.M., Aymami, J., Rodrigues-Galan, A., Munoz-Guerra, S. and Subirana, J.A. (1984) Pseudo α-Helix from Poly(α-Isobutyl-L-aspartate), a Nylon-3 Derivative. Nature, 311, 53-54.
http://dx.doi.org/10.1038/311053a0 - 8. Fernandez-Santin, J.M., Munoz-Guerra, S., Rodrigues-Galan, A., Aymami, J., Lloveras, J., Subrina, J.A., Giralt, E. and Ptak, M. (1987) Helical Conformations in Polyamide of the Nylon-3 Family. Macromolecules, 20, 62-68.
http://dx.doi.org/10.1021/ma00167a013 - 9. Lopez-Carrasquero, F., Aleman, C. and Munoz-Guerra, S. (1995) Conformational Analysis of Helical Poly(β-L-Aspartate)s by IR Dichroism. Biopolymers, 36, 263-271.
http://dx.doi.org/10.1002/bip.360360302 - 10. Appella, D.H., Barchi Jr., J.J., Durell, S.R. and Gellman, S.H. (1999) Formation of Short, Stable Helices in Aqueous Solution by β-Amino Acid Hexamers. Journal of American Chemical Society, 121, 2309-2310.
http://dx.doi.org/10.1021/ja983918n - 11. Appella, D.H., Christianson, L.A., Karle, I.L., Powell, D.R. and Gellman, S.H. (1999) Synthesis and Characterization of Trans-2-Aminocyclohexanecarboxylic Acid Oligomers: An Unnatural Helical Secondary Structure and Implications for β-Peptide Tertiary Structure. Journal of the American Chemical Society, 121, 6206-6212.
http://dx.doi.org/10.1021/ja990748l - 12. Appella, D.H., Christianson, L.A., Klein, D.A., Richards, M.R., Powell, D.R. and Gellman, S.H. (1999) Synthesis and Structural Characterization of Helix-Forming β-Peptides: Trans-2-Aminocyclopentanecarboxylic Acid Oligomers. Journal of the American Chemical Society, 121, 7574-7581.
http://dx.doi.org/10.1021/ja991185g - 13. Barchi Jr., J.J., Huang, X., Appella, D.H., Christianson, L.A., Durell, S.R. and Gellman, S.H. (2000) Solution Conformations of Helix-Forming β-Amino Acid Homooligomers. Journal of the American Chemical Society, 122, 2711-2718.
http://dx.doi.org/10.1021/ja9930014 - 14. Krauthauser, S., Christianson, L.A., Powell, D.R. and Gellman, S.H. (1997) Antiparallel Sheet Formation in β-Peptide Foldamers: Effects of β-Amino Acid Substitution on Conformational Preference. Journal of the American Chemical Society, 119, 11719-11720.
http://dx.doi.org/10.1021/ja9730627 - 15. Wang, X., Espinosa, J.F. and Gellman, S.H. (2000) 12-Helix Formation in Aqueous Solution with Short β-Peptides Containing Pyrrolidine-Based Residues. Journal of the American Chemical Society, 122, 4821-4822.
http://dx.doi.org/10.1021/ja000093k - 16. Seebach, D., Abele, S., Gademann, K., Guichard, G., Hintermann, T., Juan, B., Mathews, J.L. and Schreiber, J.V. (1998) Beta2- and Beta3-Peptides with Proteinaceous Side Chains: Synthesis and Solution Structures of the Constitutional Isomers, a Novel Helical Secondary Structure and the Influence of Solvation and Hydrophobic Interactions on Folding. Helvetica Chimica Acta, 81, 932-982.
http://dx.doi.org/10.1002/hlca.19980810513 - 17. Seebach, D., Ciceri, P., Overhand, M., Juan, B., Rigo, D., Oberer, L., Hommel, U., Amstutz, R. and Widmer, H. (1996) Probing the Helical Secondary Structure of Short-Chain-Beta-Peptides. Helvetica Chimica Acta, 79, 2043-2066.
http://dx.doi.org/10.1002/hlca.19960790802 - 18. Seebach, D. and Mathews, J.L. (1997) Beta-Peptides: A Surprise at Every Turn. Chemical Communications, No. 21, 2015-2022.
http://dx.doi.org/10.1039/a704933a - 19. Seebach, D., Schreiber, J.V., Abele, S., Daura, X. and van Gunsteren, W.F. (2000) Structure and Conformation of β-Oligopeptide Derivatives with Simple Proteinogenic Side Chains: Circular Dichroism and Molecular Dynamics Investigations. Helvetica Chimica Acta, 83, 34-57.
http://dx.doi.org/10.1002/(SICI)1522-2675(20000119)83:1<34::AID-HLCA34>3.0.CO;2-B - 20. Banerjee, A. and Balaram, P. (1997) Stereochemistry of Peptides and Polypeptides Containing Omega Amino Acids. Current Science, 73, 1067-1077.
- 21. Nelson, J.C., Saven, J.G., Moore, J.S. and Wolynes, P.G. (1997) Solvophobically Driven Folding of Nonbiological Oligomers. Science, 277, 1793-1796.
http://dx.doi.org/10.1126/science.277.5333.1793 - 22. Gellman, S.H. (1998) Foldamers: A Manifesto. Accounts of Chemical Research, 31, 173-180.
http://dx.doi.org/10.1021/ar960298r - 23. Hayen, A., Schmitt, M.A., Ngassa, F.N., Thomasson, K.A. and Gellman, S.H. (2004) Two Helical Conformations from a Single Foldamer Backbone: “Split Personality” in Short Alpha/Beta-Peptides. Angewandte Chemie, 43, 505-510.
http://dx.doi.org/10.1002/anie.200352125 - 24. Hill, D.J., Mio, M.J., Prince, R.B., Hughes, T.S. and Moore, J.S. (2001) A Field Guide to Foldamers. Chemical Reviews, 101, 3893-4012.
http://dx.doi.org/10.1021/cr990120t - 25. Porter, E.A., Wang, X., Lee, H.S., Weisblum, B. and Gellman, S.H. (2000) Non-Haemolytic Beta-Amino-Acid Oligomers. Nature, 404, 13.
http://dx.doi.org/10.1038/35003742 - 26. Porter, E.A., Weisblum, B. and Gellman, S.H. (2002) Mimicry of Host-Defense Peptides by Unnatural Oligomers: Antimicrobial Beta-Peptides. Journal of the American Chemical Society, 124, 7324-7330.
http://dx.doi.org/10.1021/ja0260871 - 27. Raguse, T.L., Porter, E.A., Weisblum, B. and Gellman, S.H. (2002) Structure-Activity Studies of 14-Helical Antimicrobial Beta-Peptides: Probing the Relationship between Conformational Stability and Antimicrobial Potency. Journal of the American Chemical Society, 124, 12774-12785.
http://dx.doi.org/10.1021/ja0270423 - 28. Werder, M., Hauser, H., Abele, S. and Seebach, D. (1999) Beta-Peptides as Inhibitors of Small-Intestinal Cholesterol and Fat Absorption. Helvetica Chimica Acta, 82, 1774-1783.
http://dx.doi.org/10.1002/(SICI)1522-2675(19991006)82:10<1774::AID-HLCA1774>3.0.CO;2-O - 29. DeGrado, W.F., Schneider, J.P. and Hamuro, Y. (1999) The Twists and Turns of Beta-Peptides. The Journal of Peptide Research: Official Journal of the American Peptide Society, 54, 206-217.
http://dx.doi.org/10.1034/j.1399-3011.1999.00131.x - 30. Gung, B.W., Zou, D. and Miyahara, Y. (2000) Synthesis of a Hybrid Peptide with Both α- and β-Amino Acid Residues: Toward a New β-Sheet Nucleator. Tetrahedron, 56, 9739-9746.
http://dx.doi.org/10.1016/S0040-4020(00)00881-4 - 31. Hintermann, T. and Seebach, D. (1997) The Biological Stability of Beta-Peptides: No Interactions between Alpha- and Beta-Peptide Structures. Chimia, 51, 244-247.
- 32. Kaminsky, J. and Jensen, F. (2007) Force Field Modeling of Amino Acid Conformational Energies. Journal of Chemical Theory and Computation, 3, 1774-1788.
http://dx.doi.org/10.1021/ct700082f - 33. Cheng, R.P., Gellman, S.H. and DeGrado, W.F. (2001) Beta-Peptides: From Structure to Function. Chemical Reviews, 101, 3219-3232.
http://dx.doi.org/10.1021/cr000045i - 34. Seebach, D., Beck, A.K. and Bierbaum, D.J. (2004) The World of Beta- and Gamma-Peptides Comprised of Homologated Proteinogenic Amino Acids and Other Components. Chemistry & Biodiversity, 1, 1111-1239.
http://dx.doi.org/10.1002/cbdv.200490087 - 35. Qiu, J.X., Petersson, E.J., Matthews, E.E. and Schepartz, A. (2006) Toward Beta-Amino Acid Proteins: A Cooperatively Folded Beta-Peptide Quaternary Structure. Journal of the American Chemical Society, 128, 11338-11339.
http://dx.doi.org/10.1021/ja063164+ - 36. Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S., Windus, T.L., Dupuis, M. and Montgomery, J.A. (1993) General Atomic and Molecular Electronics Structure System. Journal of Computational Chemistry, 14, 1347-1363.
http://dx.doi.org/10.1002/jcc.540141112 - 37. Hanwell, M.D., Curtis, D.E., Lonie, D.C., Vandermeersch, T., Zurek, E. and Hutchison, G.R. (2012) Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. Journal of Cheminformatics, 4, 17.
http://dx.doi.org/10.1186/1758-2946-4-17
NOTES

*Corresponding author.