Open Journal of Physical Chemistry
Vol. 2 No. 3 (2012) , Article ID: 22106 , 13 pages DOI:10.4236/ojpc.2012.23021
Aminolysis of 1-(1-Hydroxybenzotriazolyl)-2,4-dinitrobenzene and 2-(1-Hydroxybenzotriazolyl)-5-nitropyridine
1Department of Chemistry, Faculty of Science, Alexandria University, Ibrahimia, Egypt
2Department of Chemistry and Physics, Faculty of Education, Alexandria University, Alexandria, Egypt
3Institute for Research in Biomedicine, Barcelona Science Park, Baldiri Reixac, Spain
4Department of Chemistry, College of Science, King Saud University, Riyadh, KSA
Email: ezzatah2@hotmail.com
Received April 11, 2012; revised May 15, 2012; accepted June 17, 2012
Keywords: Nucleophilic Aromatic Substitution Reactions; Kinetic Measurements, 1-Hydroxybenzotriazole; Solvent Effect; Morpholine; Cyclohexylamine and Aniline; Uncatalyzed and Specific Base Catalyzed Mechanisms
ABSTRACT
The reaction 1-(1-hydroxybenzotriazolyl)-2,4-dinitrobenzene 1 and 2-(1-hydroxybenzotriazolyl)-5-nitro-pyridine 2 with amines undergoes amination followed by elimination of the 1-hydroxyl benzotriazolyl anion. The kinetic data for the reaction of 1 and 2 with Mo, CHA and An in MeOH and AN proceeded by uncatalysed mechanism in which the rate limiting step is the leaving group departure, whereas the reaction with Mo in toluene proceeded by uncatalysed mechanism in which the formation of the zwitterionic intermediate is the rate determining step. While the reactions of 1 with CHA and An and the reaction of 2 with CHA in toluene proceeded by SB mechanism in which the rate determining step is the proton transfer process. The reactions of 1 and 2 with Mo in the three solvents and with CHA and An in MeOH and AN is greatly depended on the stability of the zwitterionic intermediate. The effect of ring activation is due to the ground state stabilization and the more efficient delocalization of the negative charge with a nitro group than with a ring-nitrogen in the transition state. The low activation enthalpies ΔH# and the highly negative activation entropies ΔS# are due to the intramolecular hydrogen bonding with the ammonio hydrogen present in the transition state.
1. Introduction
Nucleophilic aromatic substitution (SNAr) reaction constitutes an active field of organic chemistry, which is very important from viewpoints of both practical [1-3], computational [4-6] and theory of nucleophilic aromatic substitution reactions [7-10]. Different mechanisms were suggested for unimolecular nucleophilic aromatic substitution [11], aryne mechanism [12], nucleophilic aromatic substitution with rearrangement [13-15], nucleophilic aromatic substitution involving hydrogen substitution and nucleophilic aromatic substitution via addition-elimination process (SNAr). Kinetic studies involving primary and secondary amines as nucleophiles, in solvents of different polarities, play a central role not only in firmly establishing the two or multi-step nature of the mechanism, but also in answering questions regarding the relative rates of intermediate complex formation and decomposition [16-24]. Possible pathway mechanism for this type of reactions are specific base (SB) [22-26], specific basegeneral acid (SB-GA) [27], or dimer-mechanism [28,29].
In continuation of our studies in the field of nucleophilic aryl and acyl reactions [30-45], we addressed kinetic studies of the leaving group ability of 1-hydroxybenzotriazole (HOBt) in two example: 1-(1-hydroxybenzotriazolyl)-2,4-dinitro-benzene 1 and 2-(1-hydroxy-benzotriazolyl) 5-nitro-pyridine 2. The kinetic studies of 1 and 2 with different amines such as morpholine, cyclohexylamine and aniline in methanol (MeOH), acetonitrile (AN) and toluene (Tol) will be measured. Other parameters such as ring activation, steric effect at the reaction centre, ground state stabilization of substrates 1 and 2 and finally, a plausible mechanism for each substrate with the titled amine and solvent of different polarity will be discussed.
2. Results and Discussion
2.1. Synthesis of the Starting Materials and Reaction Products
1-(1-hydroxybenzotriazolyl)-2,4-dinitrobenzene 1 and 2-(1-hydroxybenzotriazolyl) 5-nitropyridine 2 were readily prepared by the reaction of 1-hydroxy benzotriazole (HOBt) with 1-chloro-2,4-dinitrobenzene and 2-chloro-5-nitropyridine, in the presence of 2 equivalents of triethylamine (Et3N), Scheme 1. The structure of compounds 1 and 2 was confirmed by Elemental analysis, IR and NMR spectroscopy (Exp. section).
The reaction of 1 and 2 with cyclohexylamine (CHA), morpholine (Mo) and aniline (An) in methanol (MeOH), acetonitrile (AN) and toluene (Tol) undergoes amination followed by elimination of the 1-hydroxyl benzotriazolyl anion as indicated from the isolated products namely Ncyclohexyl-2,4-dinitroaniline 3 [46], 4-(2,4-dinitrophenyl) morpholine 4 [47], 2,4-dinitro-N-phenylaniline 5 [48], Ncyclohexyl-5-nitropyridin-2-amine 6 [49], 4-(5-nitropyridin-2-yl) morpholine 7 [50] and 5-nitro-N-phenylpyridin-2-amine 8 [51] respectively, Scheme 1. The purity of the products was checked by comparing their melting point with the previously reported ones in the literature.
2.2. Kinetics and the Reaction Mechanism
In general, the reaction of 1 and 2 with Mo, CHA, An in MeOH exhibited lower rates than those in AN and has different kinetic behavior in Tol. Also the order of reactivity of amines towards compounds 1 and 2 followed the order Mo > CHA > An while their pKa values are in the order CHA > Mo > An, Tables 1 and 4. This is due to 1) that MeOH molecules reduce the nucleophilicity of the amines by solvation through hydrogen bonding; 2) the formation of less polar transition state which is favored in less polar solvent; 3) the pKa values of the amines, which may be responsible for the rate differences; 4) the stability of the zwitterionic or the Meisenheimer intermediates involved in the reaction pathway(s). This stabilization arises from the intramolecular and intermolecular interactions; 5) the difference in nature of the electronwithdrawing power of the substituent in the substrate.
2.2.1. Reactions of (1) with Mo, CHA and An in MeOH, AN and Tol
1) In MeOH and AN The rate of the reactions of 1 with Mo, CHA and An are followed spectrophotomertrically to 90% completion of the products. The condition of measurements ensuring first-order kinetics.
The pseudo-first order rate constants observed (kobs) obeyed Equation (1) with negligible ko (~0) in MeOH and AN indicate that general base catalysis is absent in the present aminolysis. The apparent second-order rate constants kA (l·mol−1·s−1) are summarized in Table 1.
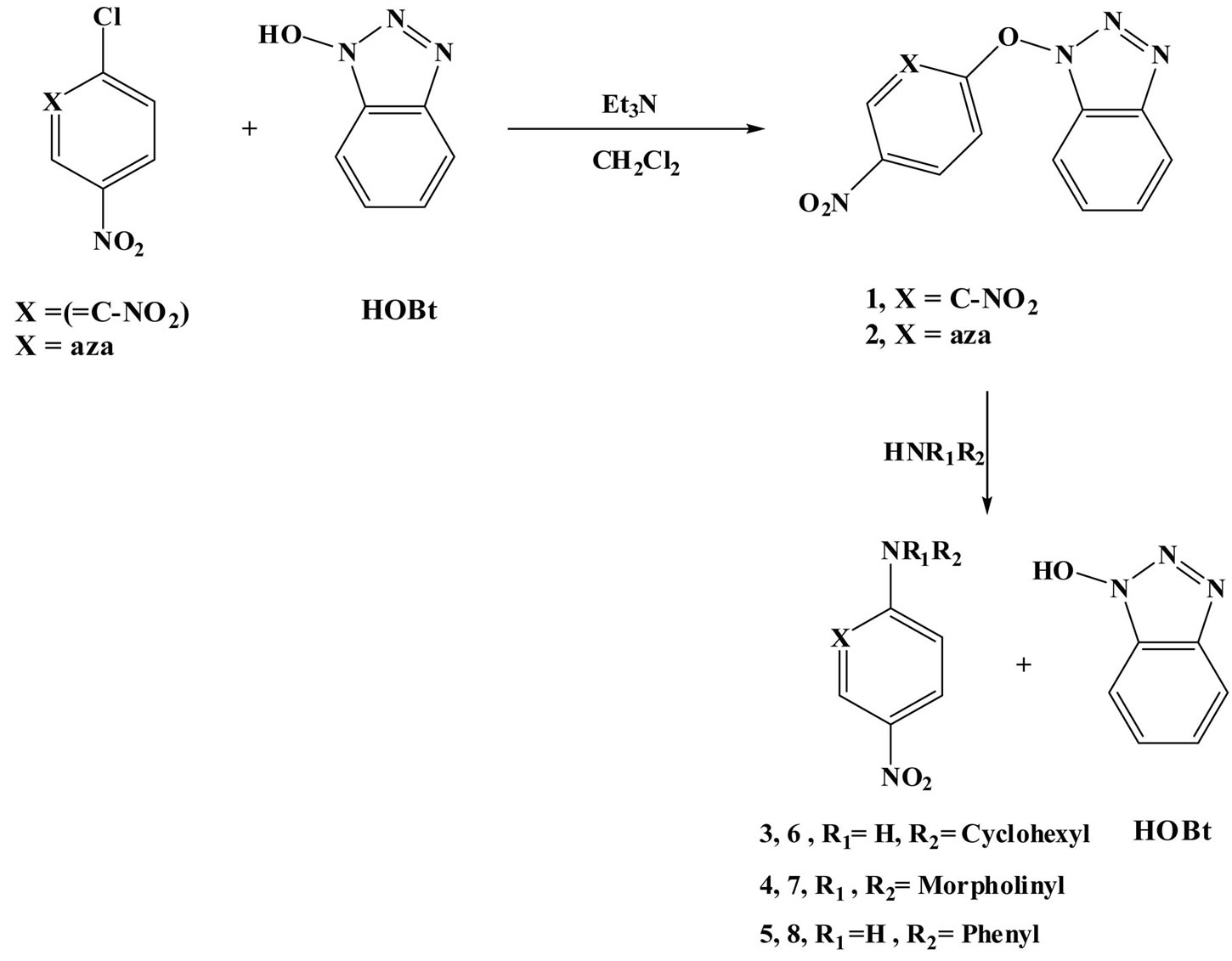
Scheme 1. Synthesis and reaction of 1 and 2 with cyclohexylamine (CHA), morpholine (Mo) and aniline (An).
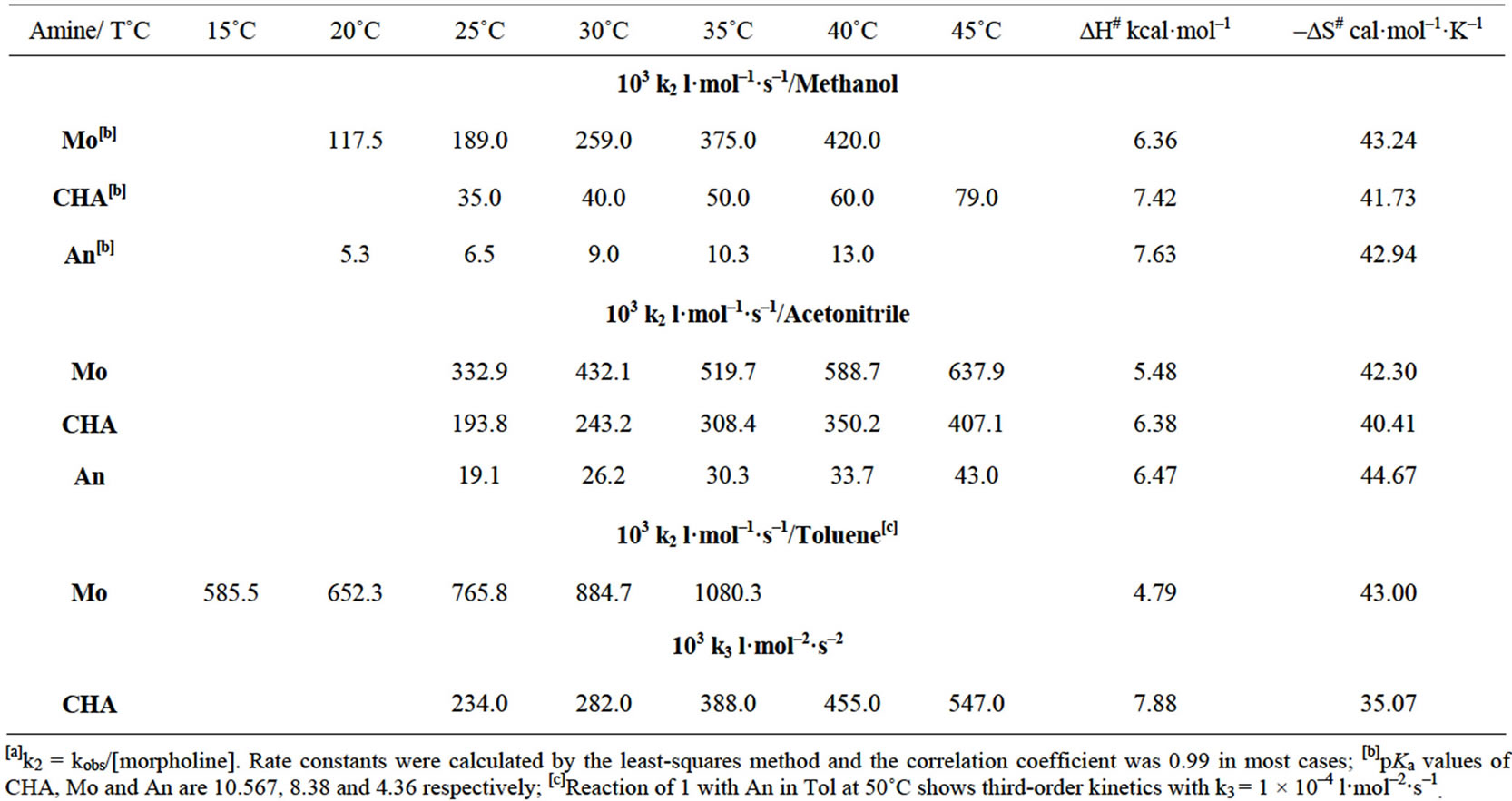
Table 1. Rate constants[a] for the reactions of 1-(1-hydroxybenzotriazolyl)-2,4-dinitrobenzene 1 with Mo, CHA and An in MeOH, AN and Tol.
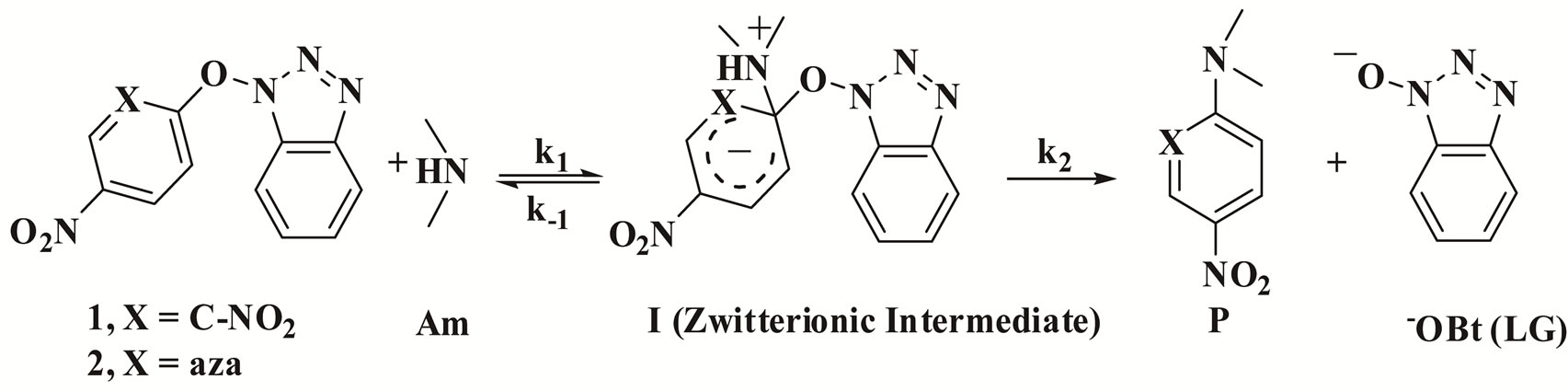
Scheme 2. The mechanism of the reaction of 1 and 2 with cyclohexylamine (CHA), morpholine (Mo) and aniline (An).
kobs = ko+ kA [Amine] (Amine = Mo, CHA, An) (1)
It shows that the rate is inconsistent with a stronger nucleophile as normally observed for a typical nucleophilic aromatic substitution reactions.
The second order rate law points out that the rate determining step is either the formation of the zwitterion intermediate I or the uncatalysed leaving group departure step, Scheme 2. It involves the reaction of 1 with the titled amines Am (Mo, CHA, An) which is proceeding by the attack of the amine on the ipso carbon of the substrate to form the zwitterionic intermediate I, followed by departure of 1-hydroxyl benzotriazolyl anion to give the substitution product.
The differentiation between the two considerations depends greatly on the rate of leaving group departure compared to the rate of the formation of the zwitterionic intermediate I and the pKa values of the amine used. This will be achieved by changing the nature of the leaving group i.e. element-effect like and the use of amines with different pKa values.
The present kinetic results shows difference in rates with changing in the nature of the leaving group from OBt to Cl, I, O-N = C(COOEt)2, O-N = C(CN)COOEt, O-N = C(CN)2 [36,37,51-53].
This indicates that the rate constants follow the order ON = C(CN)2 > ON = C(CN)COOEt > OBt > I > Cl. As a result, one can conclude that the departure of the leaving group in the reactions of 1 with Mo, CHA and An in MeOH and AN is the rate-determining step, Scheme 2, unlike most nucleophilic aromatic substitution reactions [1-7].
The rate enhancement for the reactions of the amines with compound 1 follows the order Mo > CHA > An which is not in harmony with the order of pKa values of the amines under investigation. Also the small rate constant ratios between two amines compared to the difference between the pKa values of the corresponding amines is a further indication that the rate determining step is the departure of the –OBt group.
The thermodynamic parameters computed in Table 1 indicate that the reactions in MeOH and AN are not greatly influenced by the nature of the amines. Entropies of activation are negative as expected for bimolecular reactions. The small difference of activation parameters ΔH# and ΔS# for the reaction of 1 with Mo, CHA, An in MeOH and AN may be due to a slight solvent participation in the transition state.
2) In Toluene The kinetic of the reaction of 1 with Mo, CHA and An were studied at 15˚C - 50˚C under pseudo-first-order conditions with respect to 1. The second-order rate constants of the reaction at various concentrations of Mo are shown in Table 1. These data show that no significant acceleration in rate constants occurred with the increase in the Mo concentrations. Hence the reaction is not catalyzed by base and it can be suggested that the formation of the zwitterionic intermediate is the rate determining, Scheme 2. This suggestion is consistent with the difference in rate constants as well as the activation parameters for the reaction of 1 with the same amine in MeOH and AN. The rate constants for the reaction of 1 with Mo in Tol at 25˚C, 30˚C and 35˚C are higher than those in MeOH by 4, 3.4, 2.8 times and in AN by 2.3, 2.0, 3.5 times. Also, the enthalpy of activation in Tol (∆H# = 20.04 kJ/mol) is smaller than those in MeOH (∆H# = 26.59 kJ/mol) and AN (∆H# = 22.89 kJ/mol) reflecting a higher rate in the former solvent presumably due to the change in the rate determining step in Scheme 2.
On the other hand, the values of the second-order rate constants k2 for the CHA and An increased linearly with amine concentrations, i.e. catalyzed reaction. A plot of k2 (values kobs/[amine]) versus amine (An or CHA) concentrations showed a straight line with an intercept on the y-axis indistinguishable from zero. This indicates that the contribution of k2, the unanalyzed pathway, is negligible, Scheme 3.
In these systems, CHA is more efficient than An in catalyzing the reaction as shown from k2 values, Table 1. The dependence of the reaction on two amine molecules for the reactions of 1 with An and CHA in Tol can proceed by a dimer mechanism [28,29], specific base-general acid (SB-GA) [27], or specific base catalysis (SB) [22-26] (Scheme 3). The dimer mechanism is rejected on the ground that the plots of k2 against [amine] for the reactions of 1 with An and CHA do not exhibit a curvilinear response and the plots of quotient k2 [amine] against [amine] did not gave straight lines [28,29].
The confirmation that the reaction is subjected to SBGA mechanism is done by measuring the reaction rates in the presence of anilinium hydrochloride and cyclohexylamine hydrochloride at constant An and CHA concentrations respectively and vice versa. Actually, neither aniline hydrochloride or cyclohexylamine hydrochloride are completely soluble in toluene. This excludes catalysis by SB-GA mechanism, which as previously reported, occurs only in dipolar solvents such as acetonitrile and DMSO [54].
The general base catalysis (SB) is likely to involve rate limiting deprotonation of the formed zwitterionic intermediate I, followed by rapid elimination of the leaving group [55]. Thus by adding or omitting bases in the reaction mixture, we may exert considerable control of the relative rate of the product formation and thus on the overall rate-controlling step, Scheme 3.
The kinetic expression of SB mechanism when the amine acts as both nucleophile and catalyst base can be expressed by Equation (2).
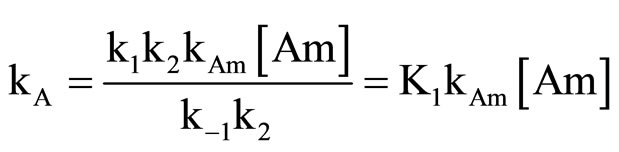 (2)
(2)
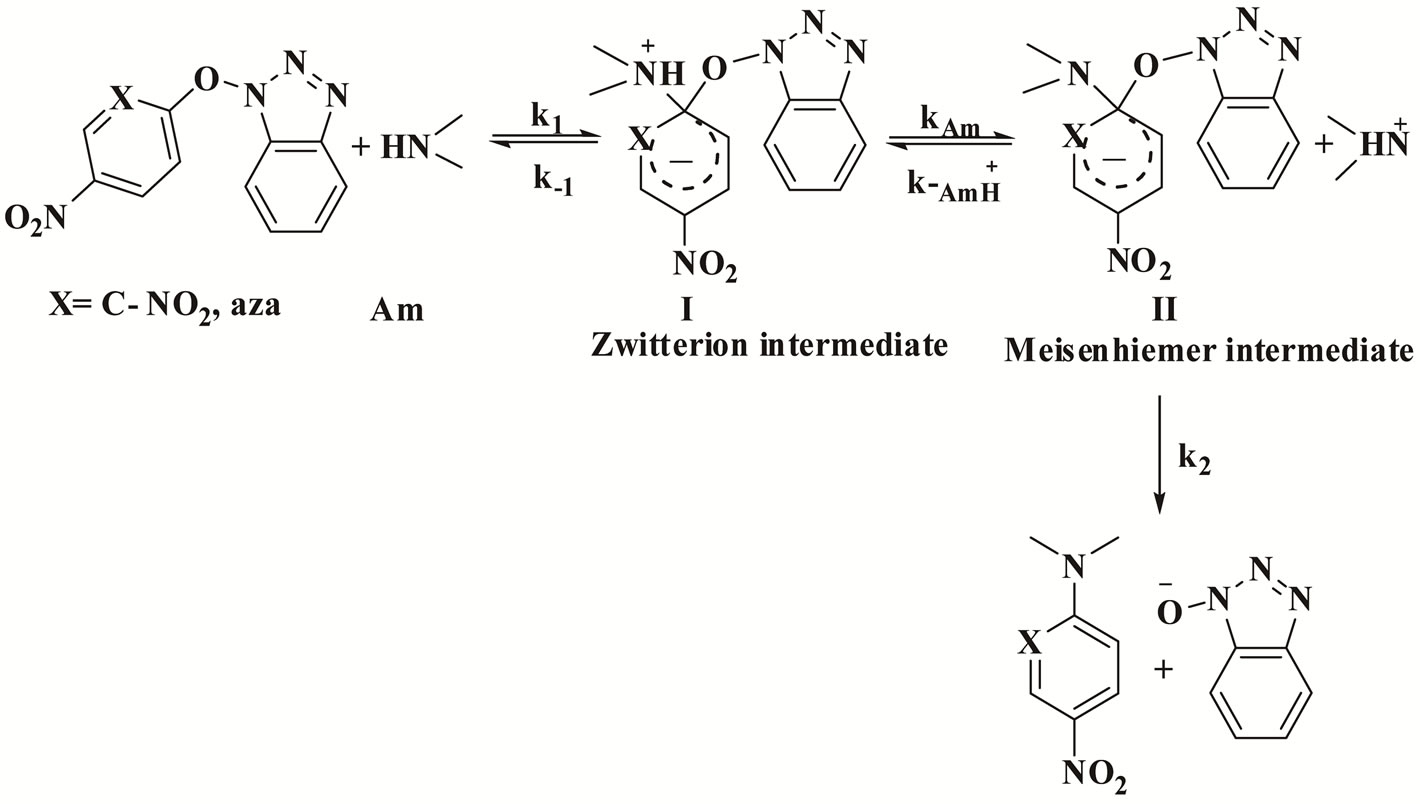
Scheme 3. The mechanism of the reaction of 1 and 2 with cyclohexylamine (CHA) and aniline (An) in Tol by the specific base catalysis mechanism (SB).
where, K1 is the apparent second-order rate constant. Where,
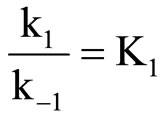
Equation (2) is consistent with the amine catalysis for the reaction of 1 with amine in Tol because a linear correlation between the kA value and [Am] values passing through the origin or intercept on the y-axis indistinguishable from zero is observed. Accordingly, the proton transfer process is the rate-determining step. This confirmed that the reaction is subjected to general base catalysis as shown in Scheme 3, where the measurements was carried out in the presence of 1,4-diazabicycle-[2,2,2] octane (DABCO). A complete study of the combined influence of amines was then outlined as follow: the reaction of 1 with CHA in Tol was run at a fixed [amine] (in the range 0.0525 mole·dm–3) in the presence of varying amounts of [DABCO] (up to 0.1049 mole·dm–3). The reactions were also measured at fixed [DABCO] (0.0525 mole·dm–3) in presence of varying [amine]. Results are set forth in Table 2. The plot of kA vs [DABCO] gives straight lines and excellent correlation coefficient. The slopes are also given in the table. The value of 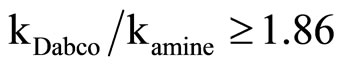 in presence of constant [DABCO] and variable [amine], where it is 2.27 in presence of constant [amine] and variable [DABCO].
in presence of constant [DABCO] and variable [amine], where it is 2.27 in presence of constant [amine] and variable [DABCO].
In addition, values of K1kDABCO, where kDABCO represent the kA rate constants for the reactions of 1 with An DABCO catalyzed route, were obtained from the slopes of the approximately linear plot of the data in Table 3.
It is observed that the rate constant in presence of DABCO is larger by about 5 and 2 times than in its absence, (at constant amine concentration and varying DABCO and at constant DABCO concentration and varying An concentration, respectively). Consequently, the catalysis present for the reaction of 1 with both amines in Tol is general base catalysis.
2.2.2. Reaction of 1-(1-Hydroxybenzotriazolyl)-5-nitropyridine 2 with Mo and CHA in MeOH, AN and Tol
All reactions of 2 with CHA and Mo were carried out under pseudo-first-order conditions, with various amine concentrations and obeyed the kinetic law given in Equation (3).
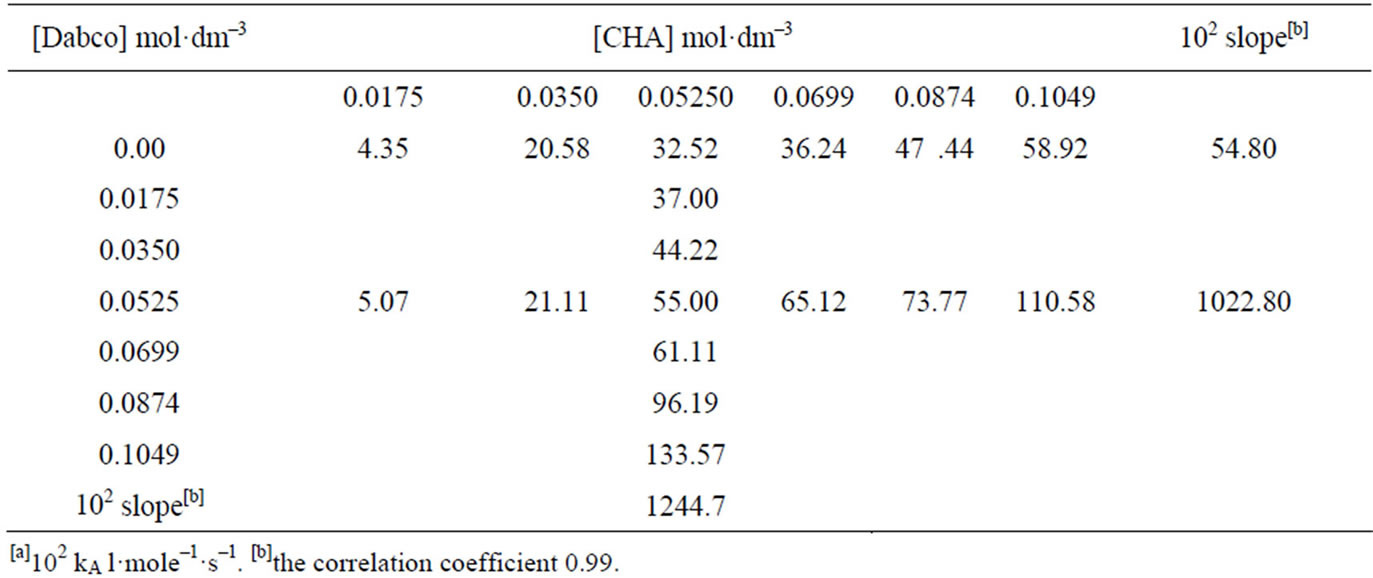
Table 2. Kinetics[a] of the reaction of 1 with CHA in the presence of DABCO in toluene at 40˚C.
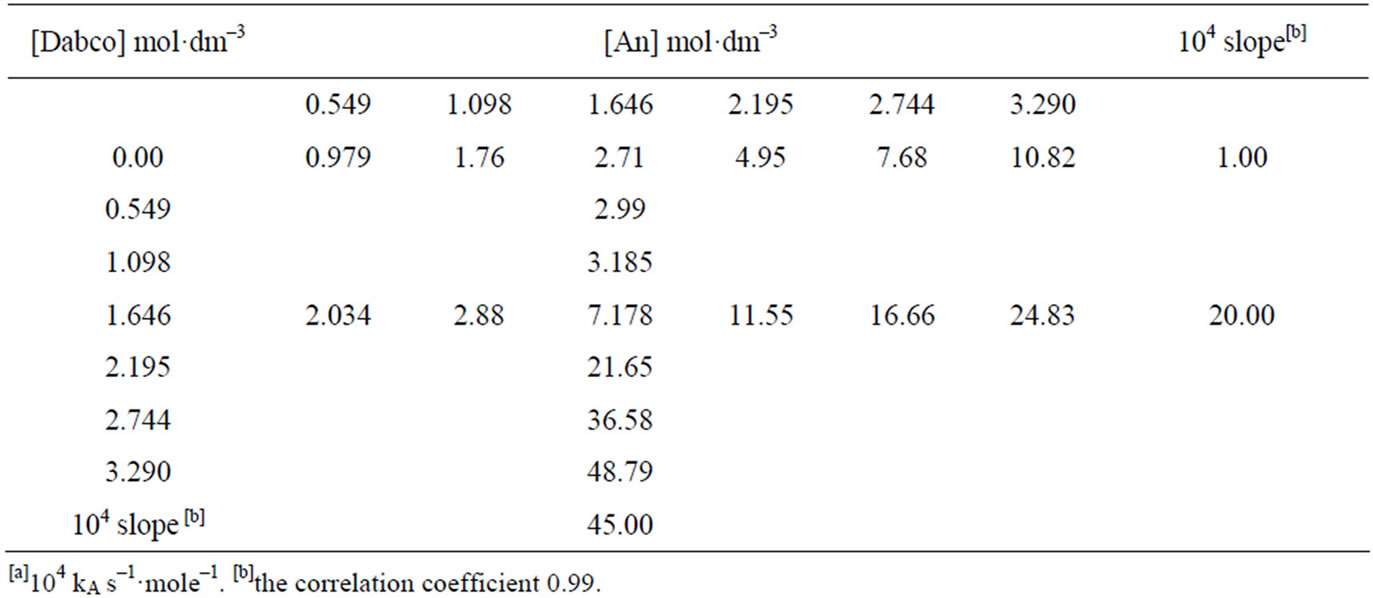
Table 3. Kinetics[a] of the reaction of 1 with An in the presence of DABCO in toluene at 50˚C.
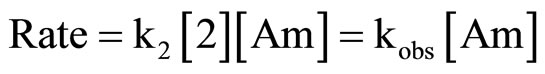 (3)
(3)
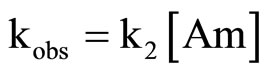 (4)
(4)
The second-order rate constants, kA, summarized in Table 2, were obtained from a straight-line plot of kobs vs. the concentration of the amine. It should be noted that compound 2 was too unreactive for satisfactory measurement for the reactions with An in all solvents even toluene due to the low reactivity of compound 2 and aniline (An). The kinetic data for the reactions of 2 with Mo and CHA in MeOH and AN are compatible with rate limiting nucleophilic attack and also with an intermediate decomposition without amine catalysis. In a similar way as discussed previously for the reaction of 1 with the same amines, the differentiation between the two considerations depends greatly on the same factors. This leads us to suggest that the rate determining step is the expulsion of the leaving group step, Scheme 2. Inspection of the activation parameters, Table 4, shows that the activation enthalpy values are slightly variable and the activation entropy values are mainly responsible for the rate difference for the reactions of 2 with Mo and CHA in MeOH and AN.
The same kinetic behavior and mechanism are observed for the reaction of Mo with compound 2 in Tol. The data show that reaction is not catalyzed by a base and the formation of the zwitterionic intermediate is the rate determining, Scheme 2. This is corroborated with the difference in rate constants as well as the activation parameters for the reaction of 2 with the same amine in MeOH and AN, Table 4.
The kinetic results for the reactions of 2, with CHA in Tol indicate a squared dependence of kobs on amine concentration reflecting the condition 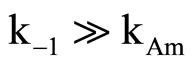 so that proton transfer is rate-limiting, Scheme 3. Plots of k2 versus CHA concentration were linear with negligible intercept indicating the uncatalyzed decomposition of the intermediate, the k2 step. The dimer and SB-GA mechanisms are rejected due to the reasons mentioned previously for the reaction of 1 with the same amine.
so that proton transfer is rate-limiting, Scheme 3. Plots of k2 versus CHA concentration were linear with negligible intercept indicating the uncatalyzed decomposition of the intermediate, the k2 step. The dimer and SB-GA mechanisms are rejected due to the reasons mentioned previously for the reaction of 1 with the same amine.
The suggested SB mechanism, Scheme 3 is supported by measurements of the reaction of 2 in the presence of added 1,4-diazabicycle [2,2,2] octane (DABCO). A complete study of the combined influence of amines was then outlined: The reactions were run at a fixed [amine] (in the range 0.525 mole·dm–3) in the presence of varying amounts of [DABCO] (up to 1.049 mole·dm–3). In addition, the reactions were also measured at fixed [DABCO] (0.525 mole·dm–3) in the presence of varying [amine]. Results are set forth in Table 5. The plot of k2 vs [DABCO] gives straight lines and excellent correlation coefficient. The value of 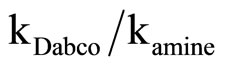 is ≥ 2.00 in presence of constant [DABCO] and variable [amine] and it is 5.00 in presence of constant [amine] and variable [DABCO]. Accordingly the kinetic results of the reaction of 2 with CHA is best analyzed in terms of Scheme 3 in which base catalysis is attributed to rate-limiting proton transfer from the zwitterionic intermediate I to base, followed by rapid expulsion of −OBt anion.
is ≥ 2.00 in presence of constant [DABCO] and variable [amine] and it is 5.00 in presence of constant [amine] and variable [DABCO]. Accordingly the kinetic results of the reaction of 2 with CHA is best analyzed in terms of Scheme 3 in which base catalysis is attributed to rate-limiting proton transfer from the zwitterionic intermediate I to base, followed by rapid expulsion of −OBt anion.
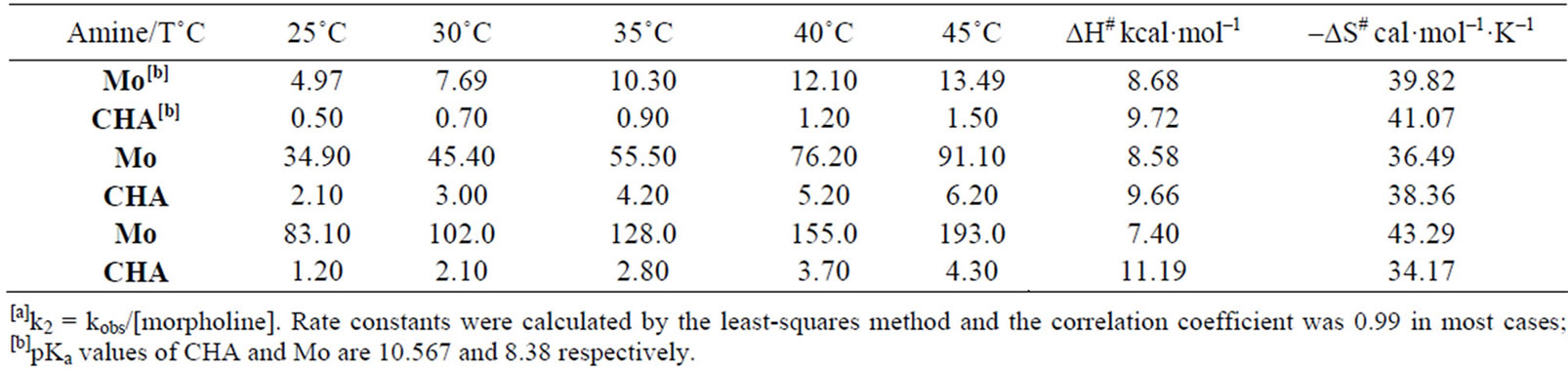
Table 4. Rate constants[a] l·mol−1·s−1 for the reactions of 2-(1-hydroxybenzotriazol-yl)5-nitropyridine 2 with Mo and CHA in MeOH, AN and Tol.
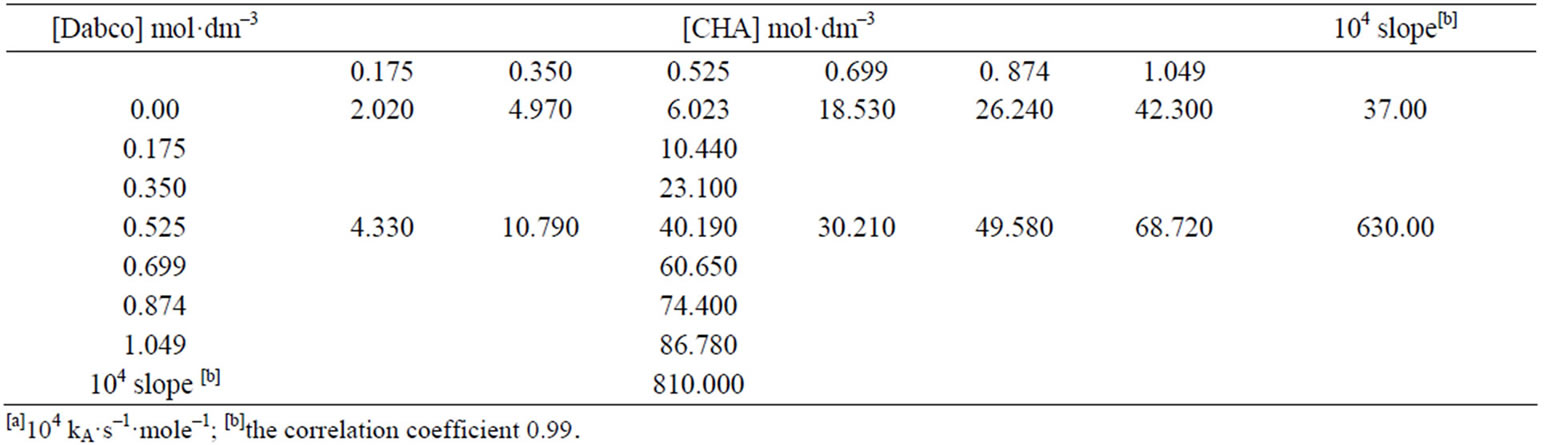
Table 5. Kinetics[a] of the reaction of 2 with CHA in the presence of Dabco in toluene at 40˚C.
2.3. Comparison between the Reactions of 1 and 2 with Mo, CHA and An in MeOH, AN and Tol
2.3.1. Relative Activation of a Ring-Nitrogen and 2-Nitro of Substrates 1 and 2
The bulk of the 2-substituent as well as its activating effect clearly have to be considered. The effect of changing the nature of the ring substituent on the rate constants kA is given in Tables 1 and 4. Kinetic data for the reactions of Mo, CHA and An with 1 and Mo, CHA with 2 in MeOH allow an assumption of the relative activation of a ringnitrogen and a 2-nitro group expressed in terms of (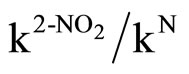 ) ratio. Values for MeOH/Mo is 33 - 38; AN/Mo is ≈ 7 - 9 and Tol/Mo is 8 - 9. The results show that the activation effects of the ortho substituents is in the order NO2 > ring aza. Hence, in the absence of steric encumbrance [37,56], the activating power of a 2-nitro group is greater than that of a ring-nitrogen in SNAr. The low difference in rates can be attributed to the ground state stabilization [24,57] i.e. the mesomeric effect of the 4-nitro group, Figure 1; (b) the more efficient delocalization of the negative charge with a nitro group than with a ring-nitrogen in the transition state [58].
) ratio. Values for MeOH/Mo is 33 - 38; AN/Mo is ≈ 7 - 9 and Tol/Mo is 8 - 9. The results show that the activation effects of the ortho substituents is in the order NO2 > ring aza. Hence, in the absence of steric encumbrance [37,56], the activating power of a 2-nitro group is greater than that of a ring-nitrogen in SNAr. The low difference in rates can be attributed to the ground state stabilization [24,57] i.e. the mesomeric effect of the 4-nitro group, Figure 1; (b) the more efficient delocalization of the negative charge with a nitro group than with a ring-nitrogen in the transition state [58].
2.3.2. Effect of Solvent on Kinetic Results and Mechanism
It has long been recognized that the rate of reactions involving the formation of extensively ionic transition state from uncharged molecules are strongly solvent dependent. Methanol is much better than acetonitrile and greater than toluene at solvating charged polarizable species such as the zwitterions. In methanol, hydrogen bond is relatively weak and the intermediates, because they are zwitterionic in character, are extensively solvated. Toluene represents an ideal medium to promote the need for base catalysis for the decomposition of the intermediate. Therefore, aromatic nucleophilic substitution reactions are more prone to base catalysis in aprotic solvents of low relative permittivity than in polar and dipolar solvents [58].
It is observed that the reactions of 1 and 2 with Mo in the three solvents and with CHA and An in MeOH and AN proceeded by uncatalysed nucleophilic aromatic substitution, Tables 1 and 4. Therefore, the rate of the reaction is greatly depended on the effect of solvent on the stability of the zwitterionic intermediate.
Therefore, the increase in stability of the zwitterionic intermediate led to an increase in the rate constants, when the formation of intermediate is the rate limiting step and causes a decrease in rate when the departure of the leaving group is the controlling step. This stabilization arises from the intramolecular hydrogen bonding between the ammonium hydrogen and the ortho-nitro or the aza ring and the extensive solvation of the intermediate through intermolecular hydrogen bond. We do not neglect the solvation of amine.
Actually, the rate constants in MeOH showed lower values than that in AN which in turn showed different kinetic order in Tol; for the reactions of 1 with Mo, CHA and An and for the reactions of 2 with Mo and CHA. Compound 1 with Mo (Tol:AN:MeOH, 4:1.7:1), with CHA (AN:MeOH, 5:1), with An (AN:MeOH, 2-2.9:1); reaction of compound 2 with Mo (Tol:AN:MeOH 16-14:7-6:1) and with CHA (Tol:AN; 4:1).
Our results postulate that 1) methanol molecules extensively solvate the intermediate I and the amine and accordingly weaken the intramolecular hydrogen bonding in the intermediate I; 2) the dipolar solvent AN slightly solvates the amine and timely solvates the intermediate I by intermolecular hydrogen bond; 3) The higher rate constants for the reactions of compounds 1 and 2 with Mo in toluene compared to those in methanol and AN at all temperatures is due to the destabilization of the zwitterion intermediate which overcomes the maximization of intramolecular hydrogen bonding between the ammonium hydrogen and ortho-nitro or aza groups in the intermediate [37,40,41,59]. The higher rate constants of 1 and 2 with Mo and CHA in AN than in MeOH can be explained by the stabilization of the intermediate I by MeOH molecules through intermolecular hydrogen bonding interaction.
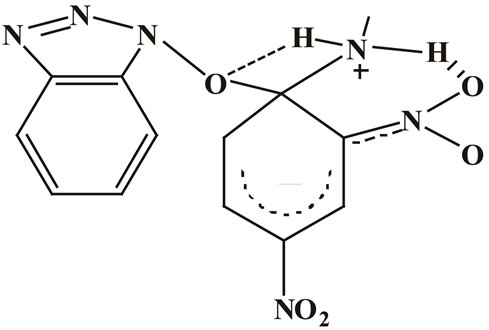
The relatively low ΔH# values and the absence of amine catalysis in MeOH and AN, Table 1, suggest a fast proton transfer, Scheme 2. While the large negative ΔS# in both solvents perhaps indicates intermolecular (in MeOH, Figure 2) and intra as well as intermolecular hydrogen-bond in AN, Figure 3 (cyclic transition state) and Figure 4 respectively.
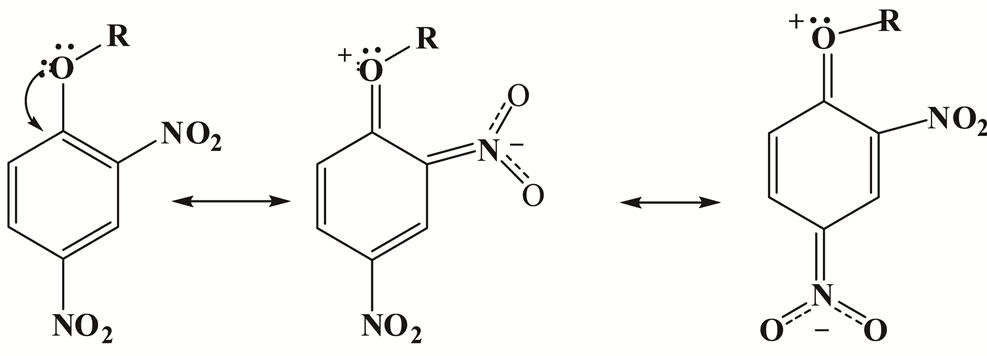
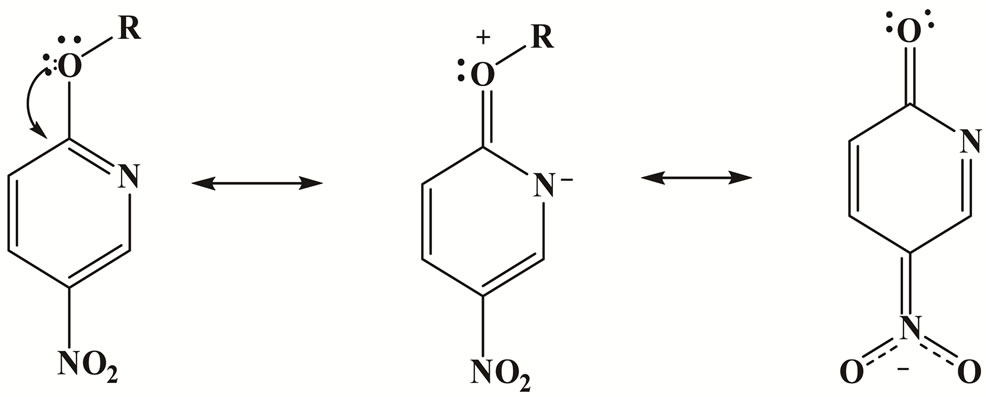
Figure 1. The ground state resonance structures of 1 and 2.
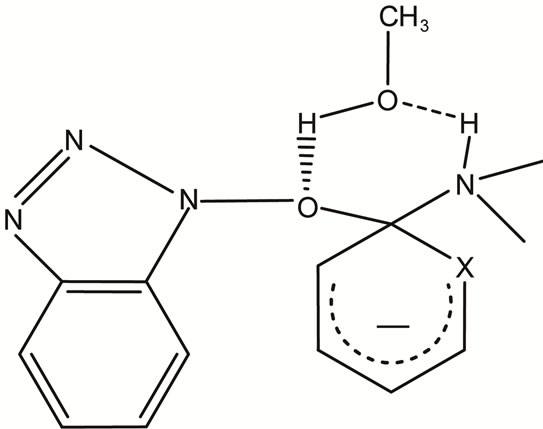
Figure 2. Intermolecular hydrogen bond of compounds 1 and 2 with MeOH.
 (a)
(a)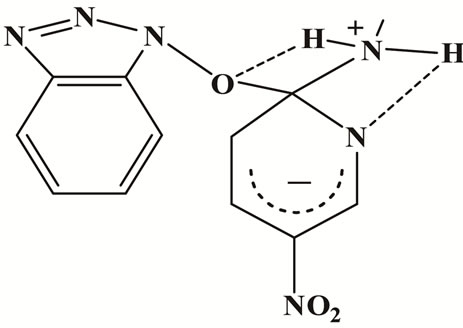 (b)
(b)
Figure 3. (a) Intramolecular hydrogen-bond of compound 1 in AN; 3(b) Intramolecular hydrogen-bond of compound 2 in AN.
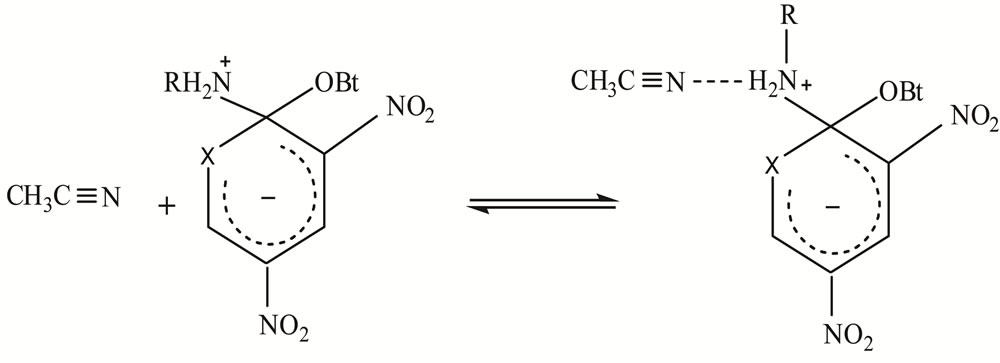
Figure 4. Intermolecular hydrogen-bond of compound 1 in AN.
2.3.3. The Effect of the Nature of Amines (Mo, CHA, An)
In general, base catalysis is more observed in less polar solvents. Unusually, when amines of the same nature and under the same experimental conditions react by different mechanism, there is considerable difference in their basicities which has been ascribed to an increase in the value of k–1 with decreasing basicity of the nucleophile [57,59,60]. pKa values, measured in MeOH, for the conjugate acids of the amines are given in Table 1.
Generally, the rate constants in MeOH showed lower values than that in AN which in turn showed less rate constant values than that in Tol; for the reactions of 1 with Mo, CHA and An; and for the reactions of 2 with Mo and CHA, Table 1 and Table 6. These results indicate that for the uncatalysed reaction, the amine with less pKa value (Mo) reacts with 1 and 2 faster than the reaction of CHA, that has higher pKa value, with the same substrates. This is consistent with Scheme 3 in which the departure of the leaving group is the rate determining step.
2.3.4. The Activation Parameters ΔS# and ΔH#
It seems that the reactions of (1, 2) with Mo, CHA and An proceed via very similar transition states, Figure 5. This involves much greater charge separation than exists in the reactants. The more polar nature of the transition state (than of the reactants) means that the change from reactants to transition state will be accompanied by considerable loss of freedom of the solvent and consequent decrease in entropy, Tables 1 and 4.
The low activation enthalpies ΔH# and the highly negative activation entropies ΔS#, Table 1, are in accordance with the proposed transition state T2# for the aza compound 2, whereas compound 1 exhibit T1# (hydrogen bond to the o-nitro group). Such hydrogen bonding facilitates the attack of the amine (Mo, CHA, An) to form zwitterionic intermediate I. Actually, the energy of formation of the N-H…..O bond is ~2000 cal and that of N-H…..N is ~1930 cal [37]. The expulsion of OBt− anion in the rate-determining step is assisted by the intramolecular hydrogen-bonding with the ammonio hydrogen present in T#1,2, which accordingly will lower the ΔH# value, whereas the structured and rigid transition state reflects the observed large negative ΔS# value.
Table 6. Comparison between the amines: Mo, CHA and An.
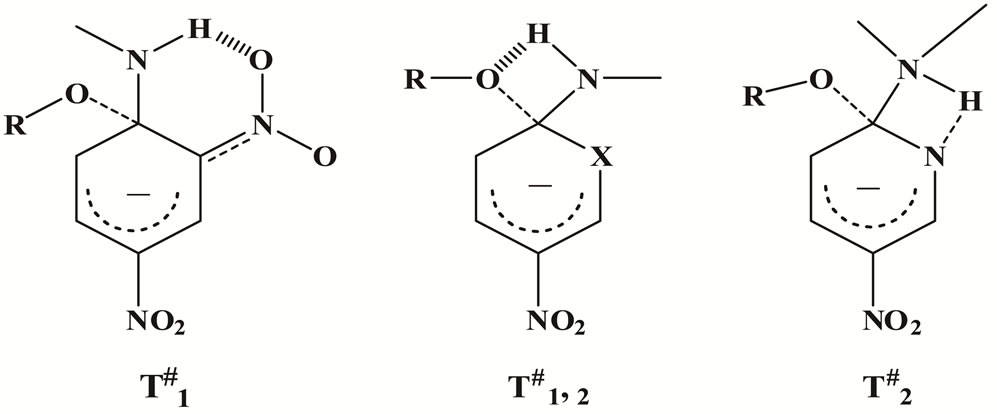
Figure 5. Possible transition states for the reaction of 1 and 2 with amines.
3. Conclusions
The kinetic data for the reactions of compounds 1 and 2 with Mo, An and CHA in MeOH and AN proceeded by uncatalysed mechanism in which the rate limiting step is the leaving group departure, whereas their reactions with Mo in toluene proceeded also by uncatalysed process, in which the formation of zwitterionic intermediate is the rate determining step. On the other hand, the reaction of 1 with CHA and An and the reaction of 2 with CHA in Tol proceeded by SB mechanism in which the rate determining step is the proton transfer process.
The rate for the reactions of (1, 2) with Mo, An and CHA depends on 1) the more efficient delocalization of the negative charge with a nitro group than with a ring-nitrogen in the transition state; 2) the stabilization of the formed activated complex by intramolecular hydrogen bonding in AN and Tol and by intermolecular hydrogen bonding in MeOH and AN; 3) the nucleophilicity of Mo, An and CHA.
4. Experimental Section
4.1. Materials
Methanol, Toluene and acetonitrile were obtained from PROLABO, BDH and were used without further purification. Aniline, cyclohexylamine and morpholine were obtained from Aldrich and were distilled before use. Melting points were determined with a Mel-Temp apparatus and are uncorrected. The UV spectra were carried out on a 160-A UV-VIS recording spectrophotometer Shimadzu. Infrared spectra (IR) were recorded on a Perkin-Elmer 1600 series Fourier Transform instrument as KBr pellets. Proton NMR spectra (1H-NMR) were recorded on a JEOL 500 MHz spectrometers at ambient temperature. Tetramethylsilane (TMS) was used as an internal reference for all 1H-NMR spectra with chemical shifts reported as ppm relative to TMS. The elemental analyses were performed at the Micro analytical Unit, Cairo University, Cairo, Egypt.
4.2. Synthesis of the Starting Materials
4.2.1. Preparation of 1-Hydroxybenzotriazole: (HOBt)
1-Chloro-2-nitrobenzene (0.675 g, 5 mmol) and (1 mL, 15 mmol) of hydrazine hydrate were dissolved in 30 mL ethanol. The reaction mixture was refluxed for 24 hours, after removing the solvent in vacuum, the residue was dissolved in aqueous Na2CO3 (10%) solution. The solution was washed with ether to remove the starting material and acidified with concentrated HCl to precipitate the product. The product was washed with water and recrystallized from ether [61].
4.2.2. Preparation of 1-(2,4-Dinitrophenoxy)-1H-benzo[d][1,2,3] Triazole 1
1-Chloro-2,4-dinitrobenzene (1) (2.025 g, 10 mmol) was added to a mixture of 1-hydroxy benzotriazole (1.35 g, 10 mmol) and Et3N (1.4 mL, 10 mmol) in CH2Cl2 (5 mL). Then the reaction mixture was stirred for 3 h at room temperature and then was refluxed on water bath for 2 hours. The crude product was filtered, washed with cold water and recrystallized from ethanol. The purity was checked by TLC (1:4, methanol:chloroform). Yield: 1.5 g (78%); m.p.: 141˚C. UV (methanol) λ nm: λmax = 250 nm (ε = 22210); UV (toluene) λ nm: λmax = 291 (ε = 6080); UV (acetonitrile) λ nm: λmax = 297 (ε = 2830) and λ = 236 (ε = 16990). IR (KBr): 1535, 1345, (NO2 asym and sym. respectively), 1073 (C-O ether) cm−1. 1H NMR [(CDCl3)]: δ 6.98 (d, 1H, J = 9.2 Hz, Ar-H), 7.56 (t, 1H, J =7.7 Hz, Ar-H), 7.60 - 7.67 (m, 2H, Ar-H), 8.16 (d, 1H, J =8.4 Hz, Ar-H), 8.39 (dd, 1H, J = 9.2, 2.3 Hz, Ar-H), 9.02 (d, 1H, J =2.3 Hz, Ar-H). C12H7N5O5: Requires: C, 49.37; H, 3.19; N, 22.15%. Found: C, 46.07; H, 2.98; N, 22.47%.
4.2.3. Preparation of 1-(5-Nitropyridin-2-yloxy)-1H-Benzo[d][1,2,3]Triazole 2
2-Chloro-5-Nitro pyridine (1.585 g, 10 mmol) was added to a mixture of 1-hydroxy benzotriazole (1.35 g, 10 mmol) and Et3N (1.4 mL, 10 mmol) in CH2Cl2 (5 mL). Then the reaction mixture was stirred for 3 h at room temperature and then was refluxed on water bath for 2 hours. The crude product was filtered, washed with cold water and recrystallized from ethanol. The purity was checked by TLC (2:8 Ethyl acetate: n-Hexane). Yield: 1.2 g, 80%; m.p.: 130˚C. UV (methanol) λ nm: λmax = 254.5 nm (ε = 18990); UV (toluene) λ nm: λmax = 289.5 (ε = 9580); UV (acetonitrile) λ nm: λmax = 284 (ε = 6230) and λ = 240 (ε = 17190). IR (KBr):1560 (C=N), 1522, 1359 (NO2 asym and sym. respectively), 1207 (C-O ether) cm–1. 1H NMR [(CD3Cl)]: δ 7.26 (d, 1H, J = 7.6 Hz, Py-H3), 7.38 (m, 2H, Ar-HOBt), 7.46 (d, 1H, J = 6.9 Hz, Ar-HOBt), 8.10 (d, 1H, J = 6.9 Hz, Py-H4), 8.65 (t, 1H, J = 6.1 Hz, Ar-HOBt), 8.90 (d, 1H, J = 2.3 Hz, Py-H6). C11H7N5O3: Requires: C, 54.54; H, 4.23; N, 24.46, O, 16.77%. Found: C, 54.78; H, 4.6; N, 24.11, O, 16.37%.
4.3. Synthesis of the Reaction Products
4.3.1. Preparation of N-cyclohexyl-2,4-Dinitroaniline 3
The reaction of 2,4-dinitrochlorobenzene (0.5 g, 2.5 mmol), with cyclohexylamine (1.5 mL, 13 mmol) in absolute methanol (10 mL) was refluxed for five hours. Work up afforded a yellow precipitate which was filtered and crystallized from benzene-petroleum ether as yellow crystals (0.52 g, 79.42%), m.p. = 212˚C - 213˚C (Lit [46] m.p. 214.1˚C). The purity was checked by TLC (1:9 ethyacetate: n-hexane). UV (methanol) λ nm: λmax = 346 (ε = 14210) and λ = 212 (ε = 14399); UV (toluene) λ nm: λmax = 346 (ε = 17690); UV (acetonitrile) λ nm: λmax = 368 (ε = 14332) and λ = 218 (ε = 14822).
4.3.2. Preparation of 4-(2,4-Dinitrophenyl)morpholine 4
The reaction of 2,4-dinitrochlorobenzene (0.5 g, 2.5 mmol), with morpholine (1.25 mL, 13 mmol) in absolute methanol (10 mL) was refluxed for five hours. Work up afforded a yellow precipitate which was filtered and crystallized from benzene-petroleum ether as yellow crystals (0.6 g, 70%), m.p. = 220˚C - 221˚C (Lit [47] m.p. 221.9˚C). The purity was checked by TLC (1:9 ethyacetate: n-hexane). UV (acetonitrile): λmax = 375 nm (ε = 14160); UV (methanol): λmax = 372 nm (ε = 15980); UV (toluene): λmax = 372 nm (ε = 15170).
4.3.3. Preparation of 2,4-Dinitro-N-phenylaniline 5
The reaction of 2,4-dinitrochlorobenzene (0.5 g, 2.5 mmol) and aniline (1.2 mL, 10.5 mmol) in absolute methanol (10 mL) was refluxed for five hours. Work up afforded a dark yellow precipitate which was filtered and crystallized from benzene-petroleum ether as a dark yellow crystal (0.49 g, 73.4%), m.p. = 158˚C - 160˚C. (Lit [44] m.p. = 159˚C - 161˚C). The purity was checked by TLC (1:9 ethyacetate: n-hexane). UV (acetonitrile): λmax = 367 (ε = 1410) and λ = 346 (ε = 17470); UV (methanol): λmax = 365 (ε = 12340) and λ = 346 (ε = 15760); UV (toluene): λmax = 367.5 nm (ε = 14990).
4.3.4. Preparation of N-cyclohexyl-5-nitropyridin-2-amine 6
The reaction of 2-chloro-5-nitropyridine (0.5 g, 2.0 mmol), with cyclohexylamine (1.5 mL, 13 mmol) in absolute methanol (10 mL) was refluxed for five hours. Work up afforded a yellow precipitate which was filtered and crystallized from benzene-petroleum ether as yellow crystals (0.7 g, 71.42%), m.p. = 184˚C - 185˚C. (Lit [49] m.p. = 186˚C). The purity was checked by TLC (1:9 ethyacetate: n-hexane). UV (acetonitrile): λmax = 370 (ε = 11490), UV (methanol): λmax = 370 (ε = 14120). λ = 346 (ε = 14710); UV (toluene): λmax = 367.5 nm (ε = 6710).
4.3.5. Preparation of 4-(5-Nitropyridin-2-yl) Morpholine 7
The reaction of 2-chloro-5-nitropyridine (0.5 g, 2.0 mmol) with morpholine (1.25 mL, 13 mmol) in absolute methanol (10 mL) was refluxed for five hours. Work up afforded a yellow precipitate which was filtered and crystallized from benzene-petroleum ether as yellow crystals (0.6 g, 70%), m.p: 137˚C - 138˚C (Lit [50] m.p. 140˚C - 142˚C). The purity was checked by TLC (1:9 ethyacetate: n-hexane). UV (acetonitrile): λmax = 375 nm (ε = 24720); UV (methanol): λmax = 375nm (ε = 20200); UV (toluene): λmax = 370 nm (ε = 22380).
4.4. Kinetic Measurements
The kinetics of 1-(1-hydroxybenzotriazolyl)-2,4-dinitrobenzene 1,2-(1-hydroxy-benzotriazolyl) 5-nitropyridine 2 with cyclohexylamine, morpholine and aniline in methanol, acetonitrile and toluene was measured spectrophotometrically using a Shimadzu (UV-160A) spectrophotometer in conjunction with a Shimadzu thermo bath (TB-85). Temperature control (±0.1˚C) was attained by circulating water through cell compartments. The kinetic runs were carried out at five temperatures (15˚C - 50˚C) in the same solvents and the absorbance variation with time were recorded at wave length λ = 370 nm, λ = 346 nm, λ = 370 nm for the product of the reaction depending on the nature of the amine. All reactions were carried out under pseudo-first-order conditions, with various concentrations of amine ranges from 1 to 10–3 M and final concentration of substrates 1, 2 (1 × 10–4 mol·dm–3) were used. The pseudo-first-order rate constants kobs were estimated by applying Equation (5).
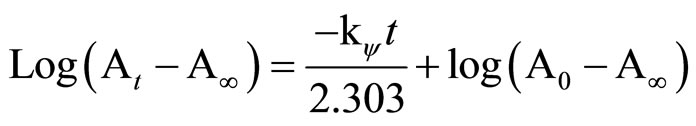 (5)
(5)
where A0, At and A¥ are the values of absorbance at zero time, time t and at the end of the reaction, respectively. The A¥ for each run was taken as the experimentally determined values and kY is the pseudo-first-order rate constant.
Plots of kobs values verses at least six amine concentration gave straight lines passing through the origin with slope equal to the second-order rate constants (k2) for the reaction in acetonitrile, methanol and toluene. Except for 1 and 2 with CHA in toluene and 1 with An in toluene the plots of kobs versus square amine concentrations gave straight lines passing through the origin with slope equal to the third-order rate constants (k3). Whereas, plots of kA values verses amines concentration gave parabolic curve for the reaction of 1 and 2 with CHA in toluene.
REFERENCES
- J. Miller, “Aromatic Nucleophilic Substitution,” Elsevier, New York, 1969, p. 18.
- F. Terrier, “Nucleophilic Aromatic Displacement,” The Influence of the Nitro Group, VCH, New York, 1991.
- E. Buncel, M. R. Crampton, M. J. Sttrauss and F. Terrier, “Electron Deficient Aromatic and Anionic Sigma Complexes,” Elsevier, Amsterdam, 1984.
- M. N. Glukhovtsev, R. D. Bach and S. Laiter, “SingleStep and Multistep Mechanisms of Aromatic Nucleophilic Substitution of Halobenzenes and Halonitrobenzenes with Halide Anions: Ab Initio Computational Study,” Journal of Organic Chemistry, Vol. 62, No. 12, 1997, pp. 4036-4046. doi:10.1021/jo962096e
- A. Patrícia Bento, M. Solà and F. Matthias Bickelhaupt, “Ab Initio and DFT Benchmark Study for Nucleophilic Substitution at Carbon (SN2@C) and Silicon (SN2@Si),” Journal of Computational Chemistry, Vol. 26, No. 14, 2005, pp. 1497-1504. doi:10.1002/jcc.20261
- N. S. Nudelman, “The Chemistry of Amino,” Nitroso, Nitro and Related Groups, Wiley & Sons, London, 1996.
- S. Park and S. Lee, “Effects of Ion and Protic Solvent on Nucleophilic Aromatic Substitution (SNAr) Reactions,” Bulletin of the Korean Chemical Society, Vol. 31, No. 9, 2010, pp. 2571-2573. doi:10.5012/bkcs.2010.31.9.2571
- N. Cheron, L. El Kaim, L. Grimaud and P. Fleurat-lessard, “Evidences for the Key of Hydrogen Bonds in Nucleophilic Aromatic Substitution Reactions,” Chemistry: A European Journal, Vol. 17, No. 52, 2011, pp. 14929-14934. doi:10.1002/chem.201102463
- J. Burdon and I. W. Parsons, “A Recent Attempt to Apply Frontier Orbital Theory to Nucleophilic Aromatic Substitution,” Journal of the American Chemical Society, Vol. 99, No. 23, 1977, pp. 7445-7447. doi:10.1021/ja00465a007
- I. Fernández, G Frenking and E. Uggerud, “Rate-Determining Factors in Nucleophilic Aromatic Substitution Reactions,” Journal of Organic Chemistry, Vol. 75, No. 9, 2010, pp. 2971-2980. doi:10.1021/jo100195w
- S. L. Edward and M. I. Joan, “The Reactions of Diazonium Salts with Nucleophiles. X. A Tracer Demonstration of the Reversible Step in Diazonium Ion Hydrolysis,” Journal of the American Chemical Society, Vol. 86, No. 1, 1964, pp. 32-34. doi:10.1021/ja01055a009
- J. F. Bunnett, “Mechanism and Reactivity in Aromatic Nucleophilic Substitution Reactions,” Quarterly Review of the Chemical Society, Vol. 12, No. 1, 1958, pp. 1-16. doi:10.1039/qr9581200001
- F. Pietra, “Mechanisms for Nucleophilic and Photonucleophilic Aromatic Substitution Reactions,” Quarterly Review of the Chemical Society, Vol. 23, No. 4, 1969, pp. 504-521. doi:10.1039/qr9692300504
- M. K. Stern, F. D. Hileman and J. K. Bashkin, “Amination of Nitrobenzene via Nucleophilic Aromatic-Substitution for Hydrogen-Direct Coupling of Aniline and Nitrobenzene,” Journal of the American Chemical Society, Vol. 114, No. 23, 1992, pp. 9237-9238. doi:10.1021/ja00049a095
- N. B. Chapman, J. Shorter and K. J. Toyne, “High Coupled Orbitals,” Journal of the American Chemical Society, Vol. 98, 1976, pp. 6702-6710.
- G. Guanti, G. Petrillo and S. Thea, “Aromatic Nucleophilic Substitution Reactions in the Naphthalene Series. A Kinetic Study of the Reaction of 2,3-Dinitronaphthalene with Piperidine in Benzene,” Tetrahedron, Vol. 38, No. 4, 1982, pp. 505-511. doi:10.1016/0040-4020(82)80094-X
- D. Spinelli and G. Consiglio, “Linear Free Energy Relationships in the Thiophene series. Part I. Leaving Group Effect in Piperidino-Substitution in Methanol of some 2-L-3-Nitro-5-Thiophene,” Journal of the Chemical Society, Perkin Transactions 2, No. 10, 1975, pp. 989-993. doi:10.1039/p29750000989
- J. F. Bunnett and R. E. Zahler, “Aromatic Nucleophilic Substitution Reactions,” Chemical Reviews, Vol. 49, No. 2, 1951, pp. 273-412. doi:10.1021/cr60153a002
- A. J. Parker, “Protic-Dipolar Aprotic Solvent Effects on Rates of Bimolecular Reactions,” Chemical Reviews, Vol. 69, No. 1, 1969, pp. 1-32. doi:10.1021/cr60257a001
- L. Forlani, C. Boga and M. Forconi, “Kinetics and Mechanism of Reactions between 2,4,6-Trinitrofluorobenzene and Alcohols,” Journal of the Chemical Society, Perkin Transactions 2, No. 7, 1999, pp. 1455-1458. doi:10.1039/a900436j
- S. K. Dotterer and R. L. Harris, “Study of Nucleophilic Aromatic-Substitution,” Journal of Organic Chemistry, Vol. 53, No. 4, 1988, pp. 777-779. doi:10.1021/jo00239a015
- A. C. Arvanites and D. W. Boerth, “Modeling of the Mechanism of Nucleophilic Aromatic Substitution of Fungicide Chlorothalonil by Glutathione,” Journal of Molecular Modeling, Vol. 7, No. 7, 2001, pp. 245-256. doi:10.1007/s008940100032
- S. Tascioglu and M. B. Gurdere, “Elucidation of the Mechanism of an Aromatic Substitution Reaction by the Utilization of Micelles as Mechanistic Probes,” Colloids and Surfaces A, Vol. 173, 2000, pp. 101-107.
- M. R. Crampton, T. A. Emokpae, C. Isanbor, A. S. Batsanov, J. A. K. Howard and R. Mondal, “Effects of Orthoand Para-Ring Activation on the Kinetics of SNAr Reactions of 1-Chloro-2-nitro and 1-Penoxy-2-nitrobenzenes with Aliphatic Amines in Acetonotrile,” European Journal of Organic Chemistry, Vol. 2006, No. 5, 2006, pp. 1222-1230. doi:10.1002/ejoc.200500774
- C. Isnabor and T. A. Emokpae, “Anilinoysis of NitroSubstituted Diphenyl Ethers in Acetonitrile: The Effect of Some Ortho-Substituents on the Mechanism of SNAr Reactions,” International Journal of Chemical Kinetics, Vol. 42, 2009, pp. 37-49.
- R. A. Akpojivi, T. A. Emopkae and J. Hirst, “The Origin of the Dichotomy of Amine Effects in Aromatic Nucleophilic Substitution Reactions,” Journal of the Chemical Society, Perkin Transactions 2, No. 3, 1994, pp. 443-449.
- J. F. Bunnett and G. Davis, “Kinetics of Reactions of 2,4-Dinitrochlorobenzene with Some Familiar Nucleophilic Reagents,” Journal of the American Chemical Society, Vol. 76, No. 11, 1954, pp. 3011-3015. doi:10.1021/ja01640a044
- C. E. S. Alvar and N. S. Nudelman, “Unusual Solvent Effects in the Reactions of 1-Halo-2,4-dinitrobenzenes and Aniline in Aprotic and Dipolar-Aprotic Solvents. Effects of Aggregates,” ARKIVOC, Vol. X, 2003, pp. 95- 106.
- C. E. S. Alvaro, M. C. Savini, V. Nicotra, J. S. Yankelevich and N. S. Nudelman, “Reaction of 2,4-Dinitrochlorobenzene with Aromatic Amines in Toluene: Effect of Nucleophile Structure,” Molecules, Vol. 5, No. 3, 2000, pp. 401-402. doi:10.3390/50300401
- M. F. Fathalla, M. F. Ibrahim and E. A. Hamed, “Kinetics of Alkaline Hydrolysis of 2-Thioaryl-3,5-dinitropyridine Derivatives in 50% v/v DMSO-Water,” Journal of Chemical Research, Vol. 2004, No. 2, 2004, pp. 150-151.
- H. O. H. Al-Howsaway, M. F. Fathalla, A. A. El. Bardan and E. A. Hamed, “Reaction of 4-Chloro-3,5-dinitrobenzotrifluoride with Aniline Derivatives. Substituent Effects,” Journal of Chemical Research, Vol. 2007, No. 9, 2007, pp. 509-512.
- M. F. Fathalla, T. S. Kassem and E. A. Hamed, “Kinetics of the Reaction between Methyl-2,4-dichloro-3,5-dinitro Benzoate and Piperidine: Solvent Effect,” Indian Journal of Chemistry, Vol. 47A, 2008, pp. 1348-1354.
- N. M. El-Mallah, S. A. Senior, G. M. Nabil, M. Sh. Ramadan and E. A. Hamed, “Effect of Acetonitrile-Water Mixtures on the Reaction of Dinitrochlorobenzene and Dinitrochlorobenzotriflouride with Hydroxide Ion,” International Journal of Chemical Kinetics, Vol. 42, 2010, pp. 453-463.
- M. A. H., Kharaba, A. M. G. Nassar, A. A. Youssef and H. Faid-Allah, “A Comparative Kinetic Investigation of Some Nucleophilic Aromatic Displacements,” Polish Journal of Chemistry, Vol. 65, 1991, pp. 1029-1034.
- E. A. Hamed, “Nucleophilic Substitution at the Pyridine Ring. Kinetics of the Reaction of 2-Chloro-3,5-dinitropyridine with Arylthiolates in Methanol,” International Journal of Chemical Kinetics, Vol. 29, No. 7, 1997, pp. 515-521. doi:10.1002/(SICI)1097-4601(1997)29:7<515::AID-KIN5>3.0.CO;2-X
- E. A. Hamed, A. A. ElBardan, E. F. Saad, G. A. Gohar and G. M. Hassan, “Nucleophilic Substitution at the Pyridine Ring. Conformational Preference of the Products and Kinetics of the Reactions of 2-Chloro-3-Nitro and 2-Chloro-5-nitropyridines with Arenethiolates,” Journal of the Chemical Society, Perkin Transactions 2, No. 11, 1997, pp. 2415-2421. doi:10.1039/a701902e
- E. A. Hamed, “Nucleophilic Substitutions at the Pyridine Ring: Kinetics of the Reaction of 2-Chloro-3-nitro and 2-Chloro-5-nitropyridines with Piperidine and Morpholine in Methanol and Benzene,” International Journal of Chemical Kinetics, Vol. 29, 1997, pp. 599-605. doi:10.1002/(SICI)1097-4601(1997)29:8<599::AID-KIN5>3.0.CO;2-P
- N. M. El-Mallah, E. A. Hamed and A. A. El-Bardan, “Kinetics of the Reaction of 2,4-Dinitrophenyl-4'-substituted Sulfones with Morpholine in Methanol,” Egyptian Journal of Chemistry, Vol. 38, 1995, pp. 319-328.
- E. A. Hamed, A. A. El-Bardan, E. F. Saad and M. F. Fathalla, “Kinetics of the Reaction of 2,4-Dinitrophenyl aryl Sulphides Sulphoxides and Sulphones with Hydroxide Ion in 80%(v/v) Ethanol-Water,” Egyptian Journal of Chemistry, Vol. 43, 2000, pp. 177-185.
- A. A. El-Bardan, “Kinetics of the Reaction of 2-Chloro-3-nitroand 2-Chloro-5-nitropyridines with Aryloxide Ions in Methanol,” Journal of Physical Organic Chemistry, Vol. 12, No. 4, 1999, pp. 347-353. doi:10.1002/(SICI)1099-1395(199904)12:4<347::AID-POC134>3.0.CO;2-G
- F. El. M. El. Hegazy, S. Z. Abdel Fathah, E. A. Hamed and S. M. Sharaf, “Kinetics of the Reaction of 2-Chloro- 3,5-dinitropyridine with Metaand Para-Substituted Anilines in Methanol,” Journal of Physical Organic Chemistry, Vol. 13, No. 9, 2000, pp. 549-554. doi:10.1002/1099-1395(200009)13:9<549::AID-POC297>3.0.CO;2-5
- M. F., Fathalla and E. A. Hamed, “Kinetics of the Reaction between Methyl-2,4-dichloro-3,5-dinitrobenzoate and Piperidine, Piprazine, Morpholine and Thiomorpholine in Methanol and Benzene,” Journal of Chemical Research, Vol. 7, 2006, pp. 413-416.
- Sh. N. Khattab, S. Y. Hassan, E. A. Hamed and A. El-Faham, “Synthesis and Morpholinolysis of N,N-Diethyl Carbamate Derivatives of 4-HOAt, 7-HOAt and HOBt,” Journal of Chemical Research, Vol. 4, 2007, pp. 247-251.
- B. H. M. Asghar, M. F. Fathalla and E. A. Hamed, “Solvent and Substituent Effects on the Reaction of 2- and 4-Chloro-3,5-dinitrobenzotrifluorides with Substituted Anilines,” International Journal of Chemical Kinetics, Vol. 41, No. 12, 2009, pp. 777-786. doi:10.1002/kin.20455
- Sh. N. Khattab, S. Y. Hassan, E. A. Hamed, F. Albericio and A. El-Faham, “Synthesis and Aminolysis of N,N-Diethyl Carbamic Ester of HOBt Derivatives,” Bulletin of the Korean Chemical Society, Vol. 31, No. 1, 2010, pp. 75-81. doi:10.5012/bkcs.2010.31.01.075
- J. J. Blanksma and G. F. Wilmink, “Action of Cyclohexylamine on Aromatic Halogeno-nitro-compounds,” Recueil des Travaux Chimiques des Pays-Bas, Vol. 66, No. 7, 1947, pp. 445-453. doi:10.1002/recl.19470660710
- C. F. Bernasconi and P. Schmid, “Base Catalysis of the Reaction of Morpholine with 2,4-Dinitrophenyl Phenyl Ether in 10 Percent Dioxane-90 Percent Water,” Journal of Organic Chemistry, Vol. 32, No. 10, 1967, pp. 2953-2956. doi:10.1021/jo01285a003
- R. H. de. Rossi, A. B. Pierini and R. A. Rossi, “Kinetics of the Reaction of Weakly Basic Amines with Activated Aromatic Substrates. Reaction of Imidazole and Aniline with 1-Fluoro-2,4-dinitrobenzene,” Journal of Organic Chemistry, Vol. 43, No. 15, 1978, pp. 2982-2986. doi:10.1021/jo00409a010
- N. S. Nudelman and S. B. Cerdeira, “1H and 13C NMR Studies of Substituted Nitropyridines and Nitrobenzenes,” Magnetic Resonance in Chemistry, Vol. 24, 1986, pp. 507-511. doi:10.1002/mrc.1260240607
- Sh. N. Khattab, E. A. Hamed, F. Albericio and A. ElFaham, “Synthesis and Aminolysis of 2,4-Dinitrophenyl and 5-Nitropyridine N-Hydroxy Oxime Derivatives,” Bulletin of the Chemical Society Japan, Vol. 84, No. 6, 2011, pp. 633-639. doi:10.1246/bcsj.20110015
- A. A. El-Bardan, G. M. El-subruiti, F. M. El-Hegazy and E. A. Hamed, “Kinetics and Reactivity of Substituted Anilines with 2-Chloro-5-nitropyridine in Dimethyl Sulfoxide and Dimethylformamide,” International Journal of Chemical Kinetics, Vol. 34, 2002, pp. 654-650. doi:10.1002/kin.10053
- M. F. Fathalla and Sh. N. Khattab, “Spectrophotometric Determination of pKa’s of 1-Hydroxybenzotriazole and Oxime Derivatives in 95% Acetonitrile-Water,” Journal of the Chemical Society of Pakistan, Vol. 33, 2011, pp. 324-332.
- C. F. Bernasconi and P. Schmid, “The Second-Order Coefficient for Formation of 2,4-Dinitrophenol from N-(2,4-Dinitrophenyl)morpholine,” Elwood and Gates, Vol. 32, 1967, pp. 2956-2959.
- R. A. Chamberlain and M. R. Crampton, “Mechanism of Base Catalysis in the Reactions of Phenyl Aryl Ethers with Aliphatic Amines in Dimethyl Sulfoxide,” Journal of the Chemical Society, Perkin Transactions 2, No. 10, 1995, pp. 1831-1838. doi:10.1039/p29950001831
- R. A. Chamberlain, M. R. Crampton and I. A. Robotham, “Kinetic and Equilibrium Studies of σ-Adduct Formation and Nucleophilic Substitution in the Reactions of Morpholine with 1,3,5-Trinitrobenzene and Some Phenyl Aryl Ethers in Dimethyl Sulphoxide,” Journal of Physical Organic Chemistry, Vol. 9, No. 3, 1996, pp. 152-158. doi:10.1002/(SICI)1099-1395(199603)9:3<152::AID-POC769>3.0.CO;2-4
- M. F. Fathalla, M. F. Ibrahim and E. A. Hamed, “Kinetics of Alkaline Hydrolysis of 2-Thioaryl-3,5-dinitropyridine Derivatives in 50% v/v DMSO-Water,” Journal of Chemical Research, Vol. 2004, No. 2, 2004, pp. 150-151.
- T. A. Emokpae, P. U. Uwakwe and J. Hirst, “The Reactions of Aniline, N-Methylaniline, N-Butylamine and Piperidine with Some Nitroaryl Phenyl Ethers in Methanol,” Journal of the Chemical Society, Perkin Transactions 2, No. 12, 1990, pp. 2191-2195. doi:10.1039/p29900002191
- W. Eggiman, P. Schmid and H. Zollinger, “Steric Effects in Nucleophilic Aromatic Substitution Reactions with Aromatic Amines on Nucleophilic Aromatic Substitution,” Helvetica Chimica Acta, Vol. IV, 1975, pp. 257-268. doi:10.1002/hlca.19750580132
- J. F. Bunnett and G. Davis, “The Mechanism of Aminolysis of Esters,” Journal of the American Chemical Society, Vol. 82, No. 3, 1960, pp. 665-674. doi:10.1021/ja01488a043
- J. Hirst and T. O. Bankole, “Kinetics of Some of the Reactions of 2-Fluoro-and 2-Chloro-5-nitropyridine and 1-Fluoroand 1-Chloro-2,4-dinitrobenzenes with Aniline, Piperidine in Acetone and Methanol,” Journal of the Chemical Society B, No. 0, 1969, pp. 848-952.
- W. König and R. Geiger, “N-Hydroxyverbindungen Als Katalysatoren für die Aminolyse Aktivierter Ester,” Chemische Berichte, Vol. 106, 1973, pp. 3626-3635.