Journal of Tuberculosis Research
Vol.1 No.2(2013), Article ID:36723,11 pages DOI:10.4236/jtr.2013.12005
Tuberculosis-related miRNAs have potential as disease biomarkers*
![]()
Beijing Key Laboratory of Drug Resistance in Tuberculosis, Beijing Chest Hospital, Capital Medical University, Beijing, China; #Corresponding Author: sunzg75@hotmail.com
Copyright © 2013 Yuhui Xu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 20 June 2013; revised 3 August 2013; accepted 23 August 2013
Keywords: miRNA; Tuberculosis; Biomarker; Differently-Expressed Genes
ABSTRACT
Background: More effective biomarkers for use in tuberculosis prevention, diagnosis, and treatment are urgently needed. The potential of miRNAs for use as biomarkers of human disease has received much attention; however, suitable miRNA biomarkers for use in tuberculosis (TB) diagnosis and treatment have not yet been identified. Methods: We used human miRNA arrays to identify miRNAs in Peripheral Blood Mononuclear Cells (PBMCs) that are differentially expressed in subjects with active disease, those with latent TB infections (LTBI) and healthy individuals. The relationship between differentially-expressed miRNAs and mRNAs was examined using TargetScanS, Pic-Tar and miRanda. The expression profiles of selected miRNAs in subjects with active disease, those with LTBI and healthy individuals were validated by qRT-PCR. Results: miRNA array analysis of PBMCs from subjects with active disease, those with LTBI and healthy individuals identified 26 differentially-expressed miRNAs. Analysis of gene expression levels in THP-1 cells using mRNA arrays identified 87 differentially-expressed genes, 80 of which were up-regulated (ratio >2) and 7 of which were down-regulated (ratio <1/2). In silico miRNA target prediction identified target mRNAs for 15 of the 26 differentially-expressed miRNAs. Differentially-expressed miRNAs were identified for 90 of the 178 differentially-expressed genes. has-miR-21* and has-miR-26b had the highest numbers of differentially-expressed target mRNAs. PCR validation of has-miR-21* and has-miR- 15b* demonstrated the fidelity of our microarray results. Conclusion: Whole-genome transcriptional profiling identified differentially-expressed mRNAs and miRNAs. Differentially-expressed miRNAs combined with predicted differentially-expressed mRNAs from the same whole-genome transcriptional profiling may be used as the new ways to better understand TB disease. This discovery of differentially-ex-pressed miRNAs and mRNAs provides a resource for further studies on the role of miRNAs in tuberculosis.
1. BACKGROUND
Tuberculosis (TB) remains a significant human health issue. It is estimated that up to two billion individuals throughout the world are currently infected with Mycobacterium tuberculosis (M. tb), 10% of which will develop active disease during their lifetime [1]. There is an urgent need to find suitable biomarkers which can differentiate between the disease, latent infections and absence of disease to improve the accuracy of tuberculosis diagnosis. A biological marker, or biomarker, is a characteristic that is objectively measured and evaluated as an indicator of a physiological or pathological process or pharmacological response(s) to a therapeutic intervention [2]. Very few biomarkers exist that fulfill these requirements [3].
Transcriptomic, proteomic and metabolomic profiling combined with broad scale immunological profiling can provide clues to the key questions in TB, which will help in the design of novel intervention strategies [4].
miRNAs, a class of small non-coding RNAs of approximately 21 nucleotides in length that are found in plants, animals, and some viruses [5], are more stable than mRNAs and are thus good candidates for use as biomarkers [6]. They modulate gene function at the posttranscriptional level and act in fine tuning various processes such as development, proliferation, cell signaling, and apoptosis. The use of miRNAs as potential bio-markers of human disease has been extensively studied and reviewed [7-11].
miRNAs that play an important role in human infectious diseases have most commonly been identified in studies on viral infections; for example, the human miRNA Hsa-miR-32 restricts the replication of retrovirus PFV-1 in human cells [12]. Viruses also use miRNAs to regulate host cellular environments. For instance, miRNA miR-K12-11 encoded by Kaposi’s sarcoma-associated herpesvirus (KSHV) down-regulates an extensive set of common host mRNAs [13]. As an intracellular bacteria, M. tuberculosis depends on the tolerance of host immune system to control its survival and replication. We hypothesize that miRNAs may also be involved in the pathology of tuberculosis.
Microarray technology permits the simultaneous measurement of the expression of hundreds of miRNAs. This technology has already been widely used and promises to become a standard tool in the near future [14,15]. The miRNA expression profiles of PBMCs among patients with active TB, subjects with latent TB infection (LTBI), and healthy controls have been compared using microbaray-based expression profiling followed by real-time quantitative PCR validation [16,17], and levels of circulating miRNAs in patients with active pulmonary tuberculosis and matched healthy controls have been compared [18]. Those reports showed some potential use of miRNA, such as hsa-miR-365, hsa-miR-223, hsa-miR- 144, hsa-miR-451, hsa-miR-424, miR-155 and miR-155* etc. Target prediction and interaction networks have been used to link miRNA and mRNA expression data [16,17, 19]. Some of these correlations need to be discovered between miRNAs and their target mRNAs in the disease of tuberculosis. Here, we investigated the miRNA expression profile in the peri-blood mononuclear cells of active TB, subjects with latent TB infection (LTBI), and healthy controls using Angilant microarrays. Three different databases were included to predict the targets between differentially-expressed miRNAs and differentially-expressed mRNAs and results indicate that there is considerable inconsistency in the sensitivity and efficiency of these databases. Two differentially-expressed miRNAs, miR-21* and mir-15b*, were quantified to validate their expression pattern using Quantitative RT-PCR.
2. MATERIALS AND METHODS
2.1. Samples
We used two independent mRNA gene expression datasets. The first dataset (Table S1) contained 31 samples obtained from the GEO database (GSE6112) [19]. The gene expression profiles obtained by microarray analysis of peripheral blood mononuclear cells from patients with active TB, LTBI and healthy donors are listed in Table S2 for TB patients, latent-infected and completely healthy subjects [20].
The second dataset (Table S2) comprised four samples obtained in our lab by infecting THP-1 cells with drug-resistant and drug-sensitive M. tb at an MOI of 10 for 18 h (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE15539), and was included to extend the relevance of our miRNA analysis to subjects who are resistant to some anti-TB drugs.
The 27 subjects used in this mRNA microarray analysis, characterized as patients with active TB, LTBI and healthy controls were college student volunteers aged 20 to 29 from the Changping District of Beijing, China. Subjects were selected randomly after performing tuberculin skin tests (TST) and X-ray examinations. Two of the 9 active TB patients were later confirmed as mutidrug resistant (MDR) TB by absolute concentration method on L-J slant and the bloods were collected within two weeks of treatment. The TST was considered positive if the diameter of the induration was >10 mm after an intradermal injection of 5 tuberculin units of PPD (Xiangrui Corp., Beijing, China). Results of the TST were read after 48 h.
PBMCs were prepared using a Ficoll-Paque PREMIUM (GE) kit according to the manufacturer’s instructions. Samples for each group consisted of 2 ml of blood from three volunteers. Three mRNA samples were isolated from each group, each sample being pooled from three subjects, to rule out the possibility that differences in gene expression were due to variation in the relative abundance of cell populations between individuals.
2.2. Total RNA Isolation
Total RNA was isolated from THP-1 cells and PBMCs using a mirVana™ RNA Isolation Kit (Applied Biosystem p/n AM1556) according to the manufacturer’s instructions. THP-1 cells and PBMCs were prepared as described below.
THP-1 cells for M. tb infection were first sub-cultured every third day to an initial density of 2 × 105 cells/ml and then differentiated into adherent, well-spread macrophages by addition of 100 nM phorbol myristate acetate (PMA). Prior to infection with M. tb at an MOI of 10, the THP-1 cells were washed twice with PBS. M. tb and THP-1 cells were then gently rocked for 0.5 h at 37˚C, 5% CO2, before removing M. tb not associated with THP-1 cells by repeated washing with PBS.
2.3. mRNA and miRNA Expression Profiling
A 22K Human Genome chip representing the 21,522 human ORFs was used along with a 70-mer oligonucleotide probe (CapitalBio Corp., Beijing, China) to analyze mRNA expression in THP-1 cells before and after infection. Microarray hybridization was performed according to published protocols [9,13]. Fluorescence intensities of Cy3 and Cy5 dyes at each spot on the 22K Human Genome Array were scanned and quantified using a double tunnel laser scanning LuxScan 10KA (CapitalBio). Gene expression scanning data were first analyzed using LuxScan 3.0 (CapitalBio) and then submitted for further analysis as outlined in [21]. Only genes whose expression was up-regulated at least 2.0-fold or down-regulated at least 0.5-fold were accepted as significant.
miRNA expression profiling was performed using a Human miRNA microarray v2.0, which contains 723 human and 76 human viral miRNAs, each replicated 16 times. 100 ng of each RNA sample was hybridized to an Agilent Human miRNA Microarray v2.0 (G4470B, Agilent Technologies). MiRNA labeling, hybridization and washing were carried out according to the manufacturer’s instructions. Images of hybridized microarrays were acquired with a DNA microarray scanner (Agilent G2565BA), and features were extracted using Agilent feature extraction (AFE) software version 9.5.3. image analysis tool version A.9.5.3.1 with default protocols and settings [22]. Raw data are available on the National Center for Biotechnology Information Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo/) using accession number GSE25435 [23].
2.4. Prediction of mRNA-Associated miRNAs
Three miRNA databases, TargetScan (http://www.targetscan.org), PicTar (http://pictar.mdc-berlin.de) and miRanda (http://www.microrna.org/microrna/home.do) were used to predict miRNAs and their target mRNAs in humans. We searched these databases for miRNA targets using mRNAs that were differentially-expressed in the two datasets used in this study. Only miRNAs that were differentially-expressed along with their mRNA targets were considered as potential markers.
2.5. PCR Validation
Quantitative RT-PCR validation was performed using SYBR Green Realtime PCR Master Mix (TaKaRa) and miScript Reverse Transcription Kit and primers according to the manufacturer’s instructions (Qiagen). Reactions were performed on an ABI7700 thermocycler (Applied Biosystems), and cycle threshold values were determined using the manufacturer’s software. Each group of subjects with active disease (pulmonary tuberculosis) or latent-infection and the group of healthy controls had eight members. Signals were normalized to expression levels of the U6 snRNA transcript. Fold changes were plotted as ΔCt (ΔCt = Cttarget − Ctcontrol) using GraphPad Prism 5.0.
3. RESULTS
3.1. miRNA Array
To perform a microarray-based integrated analysis of mRNA and miRNA data, we first determined the miRNA profile of patients with active TB, patients with LTBI and healthy controls. PBMCs from subjects were isolated and counted to make sure that equal numbers of cells (5 × 106) were used for the extraction of total RNA. The entire data set is accessible in the GEO public database (GSE25435).
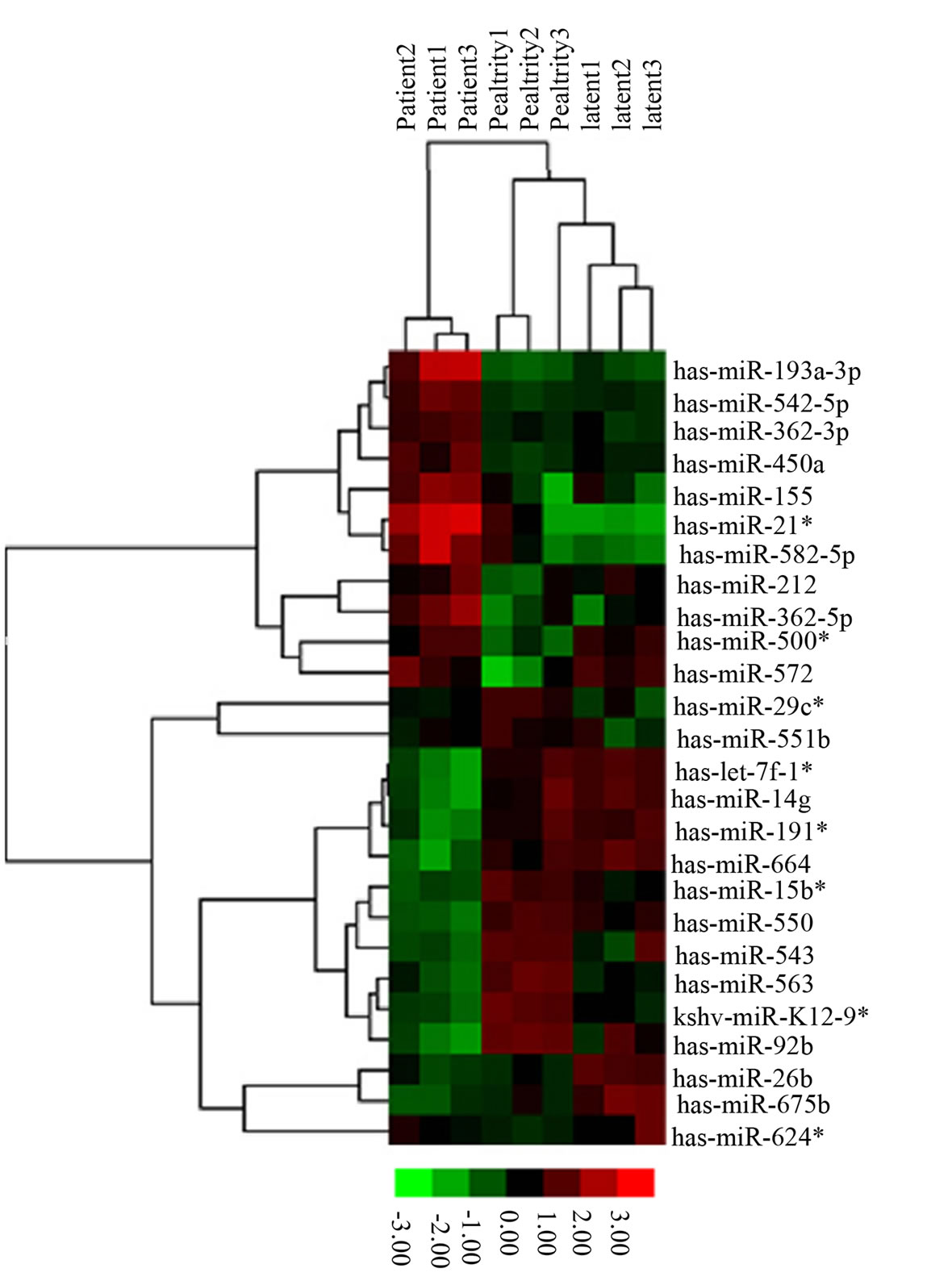
Microarray analysis (Figure 1(a)), followed by analysis of variance (ANOVA), revealed that 26 miRNAs were differentially-expressed among subjects with active disease, those with latent infection and healthy controls. Twenty-three of these miRNAs were differentially expressed between subjects with active disease and those with latent infection, and 20 miRNAs were differentially expressed between subjects with active disease and healthy controls. Only four miRNAs were differentially expressed between subjects with latent infection and healthy controls (Figure 1(b)).
3.2. Infection Array
Bearing in mind that significant changes in mRNA expression may take place during the many steps of PBMC sample preparation, we used THP-1 cells instead of PBMCs to measure mRNA gene expression after in vitro infection. When patients with tuberculosis are diagnosed, they have usually passed the initial infection period. To ascertain the role miRNA plays during the initial course of the disease, we examined mRNA expression patterns in THP-1 cells infected with different strains of M. tb (T36 and T242; drug sensitive and extensively drug-resistant tuberculosis strains, respectively, each belonging to the Beijing genotype family [24]) at an MOI of 10 using an mRNA array. Results (Table S1) show that 87 genes were differentially-expressed, 80 of which were up-regulated (ratio >2) and seven of which were down-regulated (ratio <1/2) (Figure 2). Most of the differentially-expressed genes were chemokines/cytokines and their receptors or receptor precursors (Table S1). Only four differentially-expressed genes (encoding AREG, CXCL2, CXCL3 and CXCL1) were present in
 (a)
(a)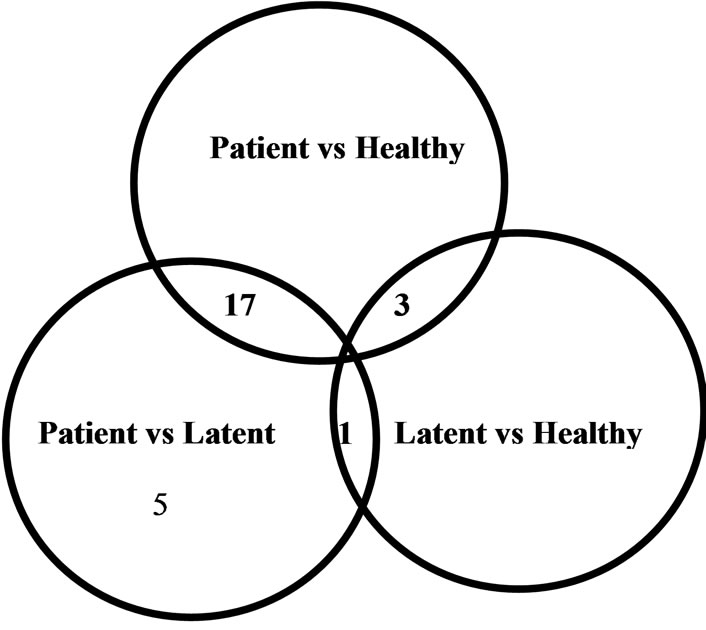 (b)
(b)
Figure 1. The 26 differentially-expressed miRNAs identified in this study. (a) Unsupervised hierarchical clustering of the expression of the 26 miRNAs. The tree was generated by unsupervised hierarchical clustering of PBMC miRNA profiles for patients with active TB, subjects with LTBI, and healthy controls. miRNAs with 2-fold upor down-regulated expression in any two types of enrolled subjects were selected for unsupervised analysis (n = 26). Sample clusters can be compared using the clinical parameters given at the top of each profile. (b) Venn diagram showing the distribution of the 26 miRNAs identified in this study by comparison between subjects. Patient, patients with active TB; Latent, subjects with latent TB infection; Healthy, healthy controls.
reported data 1 (Table S1) and our data (Table S2), indicating that they were shared during the disease process and early infection of macrophages.
3.3. In Silico miRNA Target Prediction
We hypothesized that miRNAs are involved in tuberculosis disease progression. To verify this we examined the relationship between the miRNA array results (Figure 1) and their target mRNA array results (Figure 2) using PicTar, MirTar and TargetScan miRNA target prediction algorithms. Target mRNAs that were also differentially-expressed during M. tb infection were found for 15 of the 26 differentially expressed miRNA in PBMCs.
These 15 miRNAs targeted 90 of the 178 differentially-expressed genes (Table 1). Many miRNAs were predicted to target the same mRNA (Table S2), indicating that they may be involved in the same pathways during tuberculosis disease progression. Both has-miR-21* and has-miR-26b* had six target mRNAs, while has-miR- 543 and has-miR-21* all targeted CXCL1, IGFBP3, PDE4 and E2F5 (Table 1). We hypothesize that differential expression of miRNAs results in differences in the expression of their target mRNAs among patients with active TB, subjects with LTBI, and healthy controls, however, this requires further experimental verification.
The three miRNA prediction databases gave quite different results (Table 1). They have differing abilities to
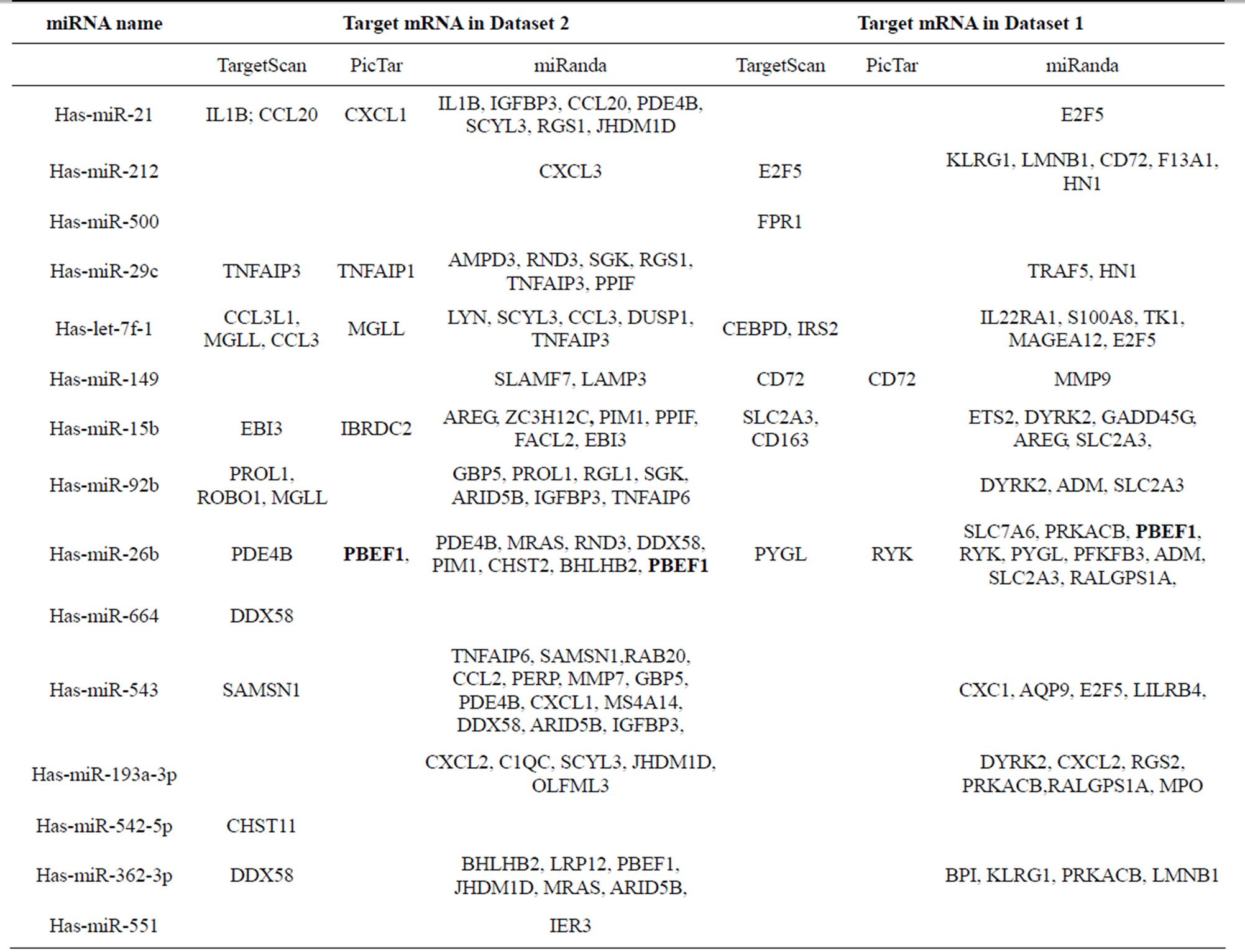
Table 1. Target mRNAs in datasets 1 and 2 for miRNAs identified in this study predicted using online databases.
determine the relationship between differentially-expressed miRNAs and mRNAs. Target mRNAs of differentially-expressed miRNAs were readily found when prediction was performed using miRanda, while PicTar gave the fewest predictions. Of the 178 differentiallyexpressed genes, only 16 mRNAs targeted by 9 miRNAs were common to any two of the prediction databases, and none of them were predicted simultaneously by all three prediction databases.
3.4. PCR Validation
PCR assays quantify miRNA precursors (primary precursor and precursor) and not the active, mature miRNA. Here we examined the expression of two of the differentially-expressed miRNAs identified in this study, miR- 21* and mir-15b*, in subjects with active disease, those with LTBI and completely healthy subjects (Figure 3). Levels of miR-21* were significantly higher in subjects with active disease (p < 0.05), but low in those with latent infection and healthy subjects, with the increase of 1.55 fold vs. healthy subjects and 1.92 fold vs. subjects with LTBI. Levels of mir-15b* were significantly higher in healthy subjects, but not different between subjects with active disease and those with latent-infection (p > 0.05), with the increase of 1.28 fold vs. subjects with LTBI and 1.41 fold vs. active patients.
4. DISCUSSION
Routine clinical methods for diagnosing TB, involving radiography, culture of sputum and the tuberculin skin test (TST), have many shortcomings. Finding new biomarkers in tuberculosis is not only necessary for diagnosing patients with TB, but also for the staging or classification of TB, TB prognosis, and TB drug and vaccine trials [4,20]. The use of miRNAs as biomarkers for different kinds of cancers has been intensively investigated and some promising candidates have been developed as vaccines [25]. With respect to their use as biomarkers for infectious diseases, in this study we analysed miRNAs involved in tuberculosis using a microarray approach
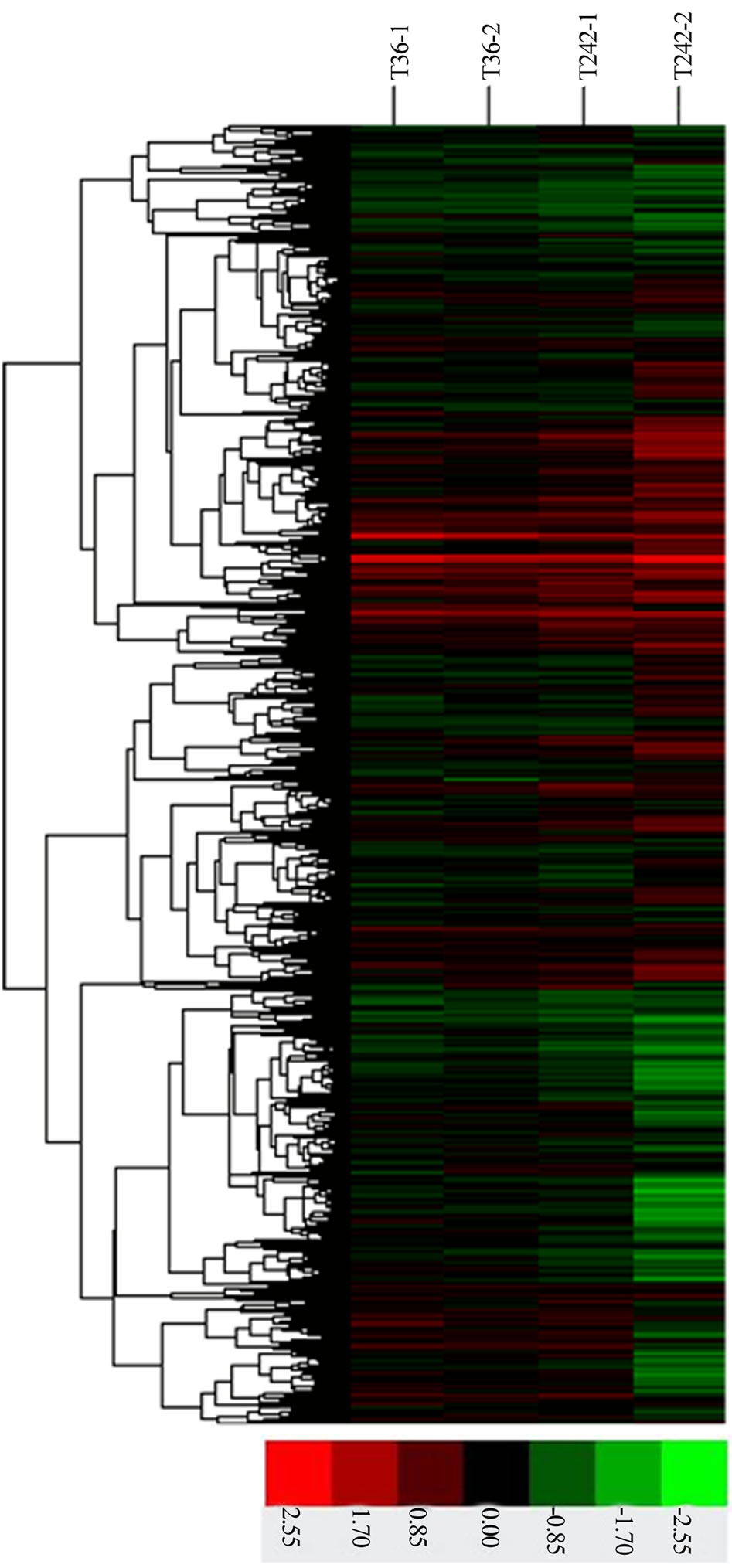
Figure 2. Cluster analysis of genes detected by 22K Human Genome microarrays (CapitalBio Corp., Beijing, China) of the macrophage-like THP-1 cell line exposed to 2 different strains of M. tb for 18 h. Colours represent log 2 ratios of the means of triplicate measurements of infected cells versus the non-infected control according to the scales shown below.
combined with the use of three online mRNA target prediction methods.
Using microarrays is an easy way to analyze changes in the expression of miRNAs during the progression of disease. Over the past year, a number of different approaches have been described for quantifying miRNAs, including cDNA arrays [21,26-28], a modified Invader
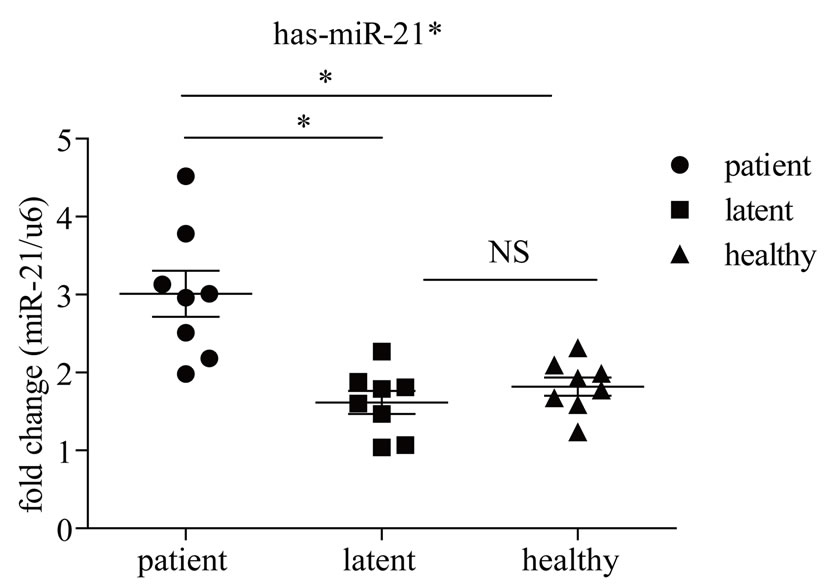
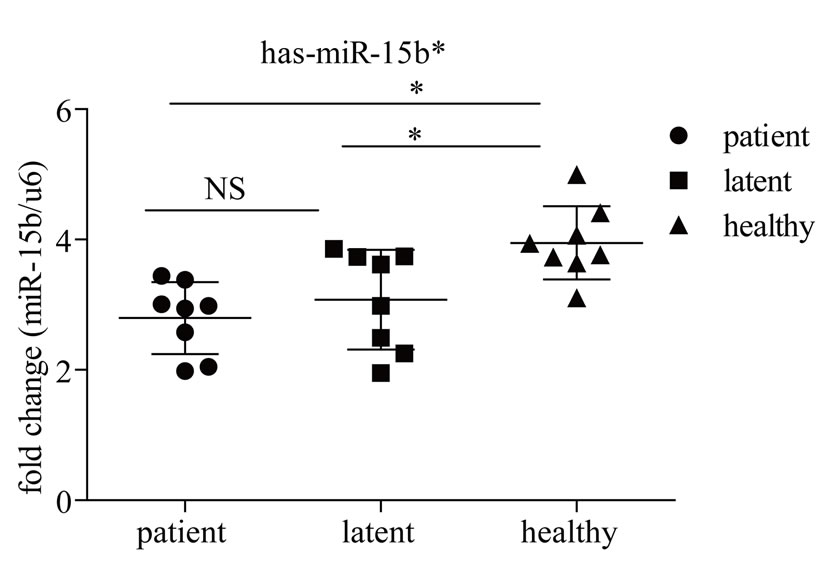
Figure 3. Quantitative analysis of miRNA expression in PBMCs from subjects with active disease, those with latent infections and completely healthy subjects. Expression levels were normalized to U6 snRNA transcript levels. miRNA expression levels are depicted as ΔCt values (Y-axis). Statistical analysis was performed using unpaired t-tests. **P < 0.01, *P < 0.05, NS: not significant. Patient, patients with active TB; Latent, subjects with latent TB infection; Healthy, healthy controls.
assay [29] and real-time PCR to measure miRNA precursors [30-31]. However, unlike cancers, the use of miRNAs as markers has not yet been verified for bacterial infectious diseases. Recently several research groups have reported studies on miRNAs involved in TB [16- 18]. Some of the miRNAs which have been described as potential markers in TB diagnosis, such as the miR-155 [16,17], miR-21*, miR-500*, miR-550 [17] were also detected in our study. Other miRNAs shown in this study to be related to TB need further validation.
TargetScanS, PicTar and miRanda seem to be the best methods for predicting miRNA targets, with sensitivity values ranging between 65% and 68%. The overlap between the target gene sets we obtained for differentiallyexpressed miRNAs was not strong. Sethupathy et al. suggest some answers which address this problem. These three programs inevitably have different features in terms of specific base-pairing rules, such as sequence match rules and thermodynamics, and cross-species conservation requirements [32]. In agreement with the analysis conducted by Sethupathy et al., miRanda was more sensitive here than TargetScan, however, PicTar was not as sensitive in our analysis as in their report [32]. TargetScan features an efficient reduction in the false-positive rate, however the required strict complementarity in the seed region is more likely to lead to the loss of loosely conserved targets and those containing wobble pairings, including 3’-compensatory sites [33]. Although miRanda is more sensitive than the other two programs, it seems to be slightly less effective in terms of specificity, although this issue will be tackled in the next versions to be released. PicTar has the lowest sensitivity of the three programs, but the experimental validation of seven out of 13 tested predicted targets, and the validation of eight of nine previously known targets, demonstrates the efficiency of the algorithm [33]. As a result, we combined the prediction results from all three programs and obtained the target mRNAs of 15 miRNAs (Table 1).
We validated the differentially-expressed miRNAs by qRT-PCR. miRNA-21* had potential as a biomarker for active disease, while miRNA-15b* was able to discriminate between healthy subjects and subjects infected with the M. tb bacterium. It was not, however, able to discriminate between subjects with active disease and those with latent infection. Real-time PCR has unparalleled sensitivity and specificity; however, it quantifies the miRNA precursor and not the active, mature miRNA. Moreover, clinical specimens show great variation in disease progression and genetic diversity.
5. CONCLUSION
Whole-genome transcriptional profiling uncovered some potential signatures in the development of the disease [4,20]. Transcripts identified by the microarray methods included not only mRNA, but also non-coding RNAs, such as miRNA. We report changes in miRNA expression profiles associated with disease process in individuals with active TB, latent TB and healthy controls. The putative miRNA targets identified suggest that differentially-expressed miRNAs combined with differentially-expressed mRNAs from the same whole-genome transcriptional profiling experiments may be used as the new ways to better understand the TB disease. Further testing and validation are needed to determine whether the differentially-expressed miRNA-mRNA pairs identified here can be used as biomarkers for monitoring TB infection, disease progression or treatment outcomes.
6. ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (30901283), Beijing Municipal Natural Science Foundation (7103169) and the Beijing Talent training program “215” (2011- 3-069).
REFERENCES
- Kaufmann, S.H. and McMichael, A.J. (2005) Annulling a dangerous liaison: Vaccination strategies against AIDS and tuberculosis. Nature Medicine, 11, S33-S44. doi:10.1038/nm1221
- Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clinical Pharmacology, 69, 89- 95.
- Braunwald, E. (2008) Biomarkers in heart failure. The New England Journal of Medicine, 358, 2148-2159. doi:10.1056/NEJMra0800239
- Parida, S.K. and Kaufmann, S.H. (2010) The quest for biomarkers in tuberculosis. Drug Discovery Today, 15, 148- 157. doi:10.1016/j.drudis.2009.10.005
- Ambros, V. (2004) The functions of animal microRNAs. Nature, 431, 350-355. doi:10.1038/nature02871
- Liu, A., Tetzlaff, M.T., Vanbelle, P., Elder, D., Feldman, M., Tobias, J.W., Sepulveda, A.R. and Xu, X. (2009) MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. International Journal of Clinical and Experimental Pathology, 2, 519-527.
- Weiler, J., Hunziker, J. and Hall, J. (2006) Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs implicated in human disease? Gene Therapy, 13, 496- 502. doi:10.1038/sj.gt.3302654
- Calin, G.A. and Croce, C.M. (2006) MicroRNA signatures in human cancers. Nature Reviews Cancer, 6, 857-866. doi:10.1038/nrc1997
- Alvarez-Garcia, I. and Miska, E.A. (2005) MicroRNA functions in animal development and human disease. Development, 132, 4653-4662. doi:10.1242/dev.02073
- Jackson, A. and Linsley, P.S. (2010) The therapeutic potential of microRNA modulation. Discovery Medicine, 9, 311-318.
- Ferracin, M., Veronese, A. and Negrini, M. (2010) Micromarkers: miRNAs in cancer diagnosis and prognosis. Expert Review of Molecular Diagnostics, 10, 297-308. doi:10.1586/erm.10.11
- Lecellier, C.H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., Saïb, A. and Voinnet, O. (2005) A cellular microRNA mediates antiviral defense in human cells. Science, 308, 557-560. doi:10.1126/science.1108784
- Gottwein, E., Mukherjee, N., Sachse, C., Frenzel, C., Majoros, W.H., Chi, J.T., Braich, R., Manoharan, M., Soutschek, J., Ohler, U. and Cullen, B.R. (2007) A viral microRNA functions as an orthologue of cellular miR-155. Nature, 450, 1096-1099. doi:10.1038/nature05992
- Sah, S., McCall, M.N., Eveleigh, D., Wilson, M. and Irizarry, R.A. (2010) Performance evaluation of commercial miRNA expression array platforms. BMC Research Notes, 18, 80. doi:10.1186/1756-0500-3-80
- Mack, G.S. (2007) MicroRNA gets down to business. Nature Biotechnology, 25, 631-638. doi:10.1038/nbt0607-631
- Wang, C., Yang, S., Sun, G., Tang, X., Lu, S., Neyrolles, O. and Gao, Q. (2011) Comparative miRNA expression profiles in individuals with latent and active tuberculosis. PLoS One, 6, e25832. doi:10.1371/journal.pone.0025832
- Wu, J., Lu, C., Diao, N., Zhang, S., Wang, S., Wang, F., Gao, Y., Chen, J., Shao, L., Lu, J., Zhang, X., Weng, X., Wang, H., Zhang, W. and Huang, Y. (2012) Analysis of microRNA expression profiling identifies miR-155 and miR-155* as potential diagnostic markers for active tuberculosis: A preliminary study. Human Immunology, 73, 31-37. doi:10.1016/j.humimm.2011.10.003
- Fu, Y., Yi, Z., Wu, X., Li, J. and Xu, F. (2011) Circulating microRNAs in patients with active pulmonary tuberculosis. Journal of Clinical Microbiology, 49, 4246-4251. doi:10.1128/JCM.05459-11
- Sharbati, J., Lewin, A., Kutz-Lohroff, B., Kamal, E., Einspanier, R. and Sharbati, S. (2011) Integrated microRNAmRNA-analysis of human monocyte derived macrophages upon Mycobacterium avium subsp. hominissuis infection. PLoS One, 6, e20258. doi:10.1371/journal.pone.0020258
- Jacobsen, M., Repsilber, D., Gutschmidt, A., Neher, A., Feldmann, K., Mollenkopf, H.J., Ziegler, A. and Kaufmann, S.H. (2007) Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. Journal of Molecular Medicine, 85, 613-621. doi:10.1007/s00109-007-0157-6
- Krichevsky, A.M., King, K.S., Donahue, C.P., Khrapko, K. and Kosik, K.S. (2003) A microRNA array reveals extensive regulation of microRNAs during brain development. RNA, 9, 1274-1281. doi:10.1261/rna.5980303
- Agilent technologies: Agilent feature extraction reference guide. 2007.
- López-Romero, P., González, M.A., Callejas, S., Dopazo, A. and Irizarry, R.A. (2010) Processing of agilent microRNA array data. BMC Research Notes, 3, 18. doi:10.1186/1756-0500-3-18
- Sun, Z., Zhang, J., Song, H., Zhang, X., Li, Y., Tian, M., Liu, Y., Zhao, Y. and Li, C. (2010) Concomitant increases in spectrum and level of drug resistance in Mycobacterium tuberculosis isolates. The International Journal of Tuberculosis and Lung Disease, 14, 1436-1441.
- Cawood, R., Chen, H.H., Carroll, F., Bazan-Peregrino, M., van Rooijen, N. and Seymour, L.W. (2009) Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathogens, 5, e1000440. doi:10.1371/journal.ppat.1000440
- Babak, T., Zhang, W., Morris, Q., Blencowe, B.J. and Hughes, T.R. (2004) Probing microRNAs with microarrays: tissue specificity and functional inference. RNA, 10, 1813-1819. doi:10.1261/rna.7119904
- Sun, Y., Koo, S., White, N., Peralta, E., Esau, C., Dean, N.M. and Perera, R.J. (2004) Development of a microarray to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Research, 32e188.
- Liu, C.G., Calin, G.A., Meloon, B., Gamliel, N., Sevignani, C., Ferracin, .M., Dumitru, C.D., Shimizu, M., Zupo, S., Dono, M., Alder, H., Bullrich, F., Negrini, M. and Croce, C.M. (2004) An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proceedings of the National Academy of Sciences, 101, 9740-9744.
- Allawi, H.T., Dahlberg, J.E., Olson, S., Lund, E., Olson, M., Ma, W.P., Takova, T., Neri, B.P. and Lyamichev, V.I. (2004) Quantitation of microRNAs using a modified Invader assay. RNA, 10, 1153-1161. doi:10.1261/rna.5250604
- Takamizawa, J., Konishi, H., Yanagisawa, K., Tomida, S., Osada, H., Endoh, H., Harano, T., Yatabe, Y., Nagino, M., Nimura, Y., Mitsudomi, T. and Takahashi, T. (2004) Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Research, 64, 3753-3756. doi:10.1158/0008-5472.CAN-04-0637
- Schmittgen, T.D., Jiang, J., Liu, Q. and Yang, L. (2004) A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Research, 32E43.
- Sethupathy, P., Megraw, M. and Hatzigeorgiou, A.G. (2006) A guide through present computational approaches for the identification of mammalian microRNA targets. Nature Methods, 3, 881-886. doi:10.1038/nmeth954
- Mazière, P. and Enright, A.J. (2007) Prediction of the miRNA targets. Drug Discovery Today, 12, 452-458. doi:10.1016/j.drudis.2007.04.002
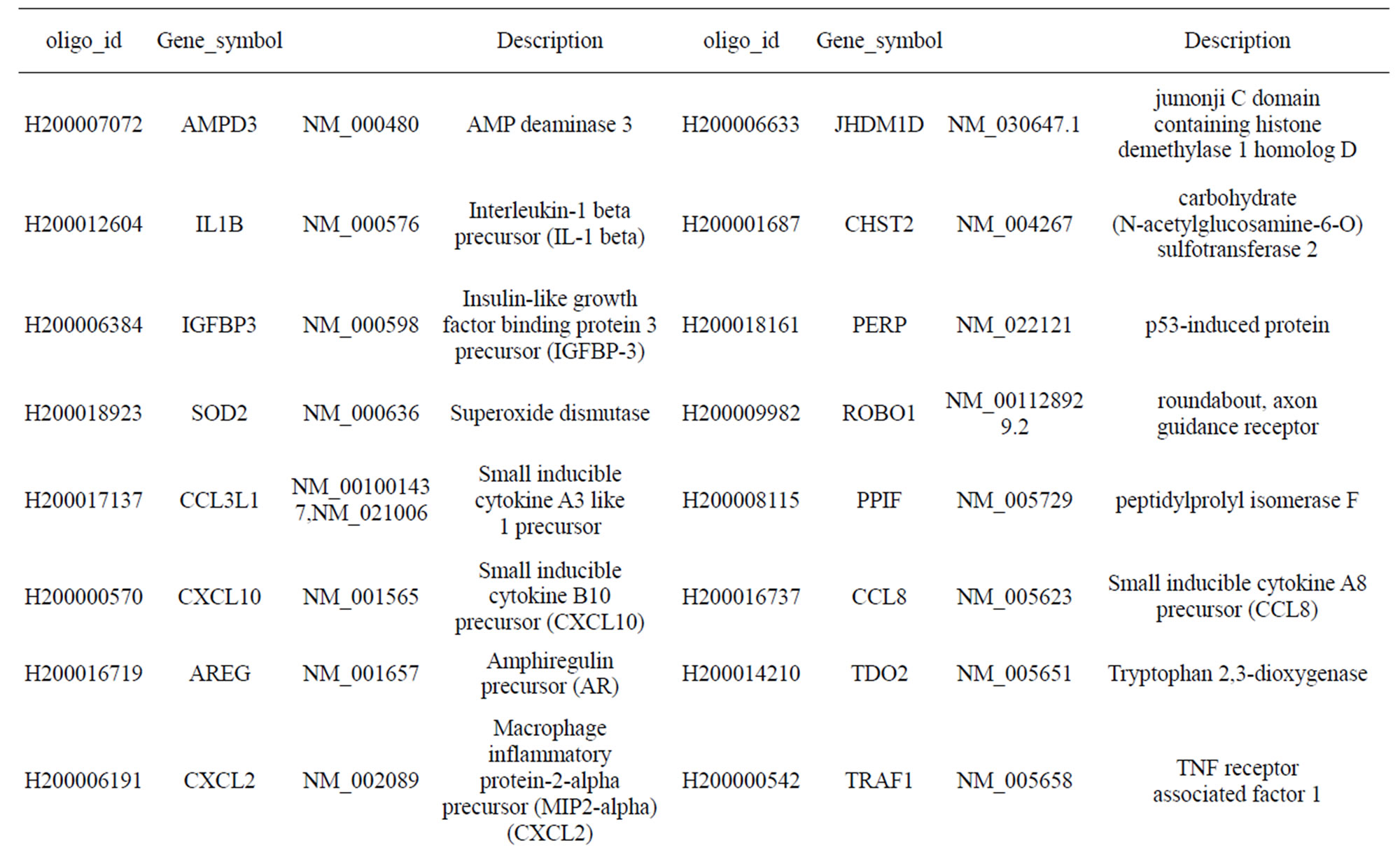
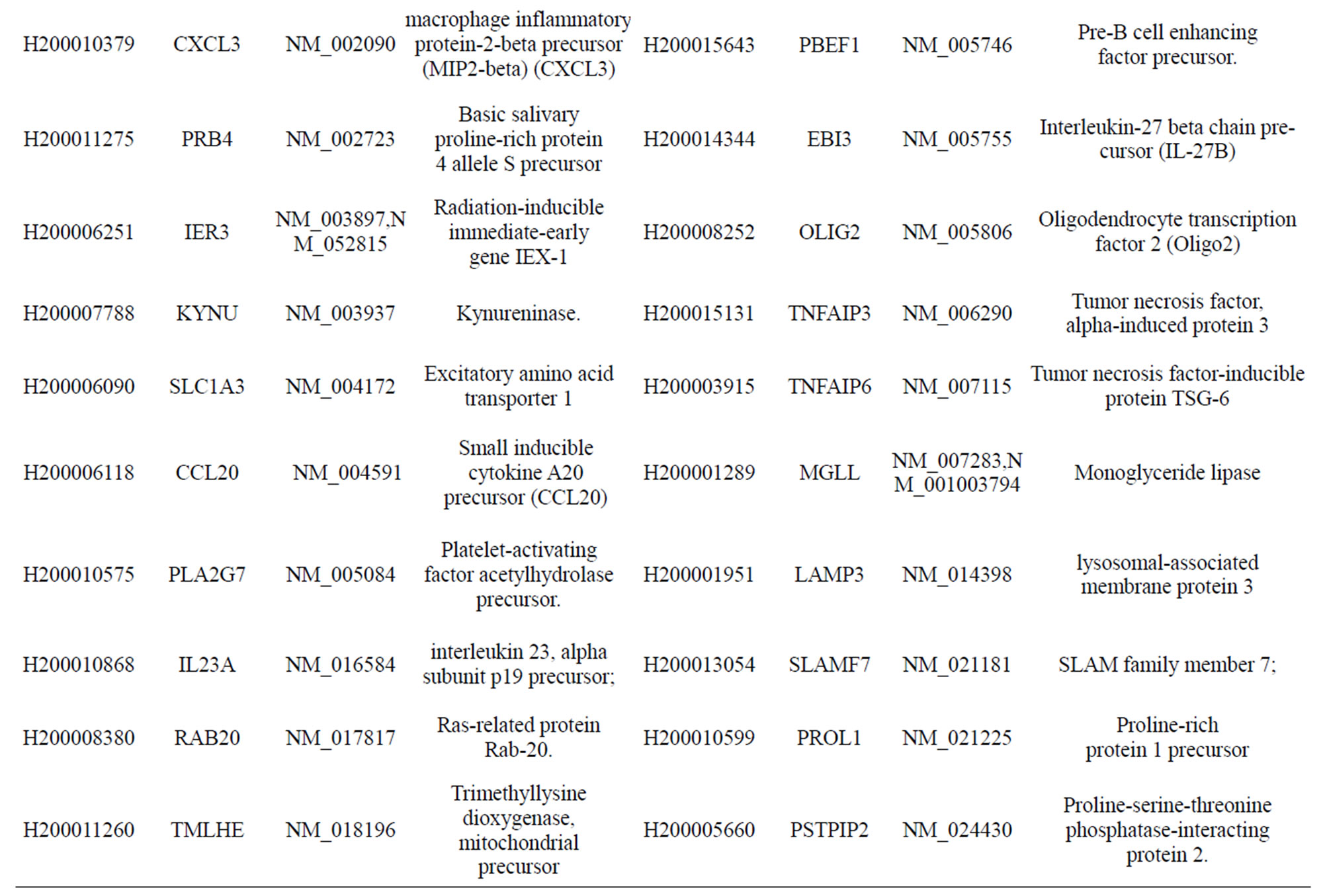
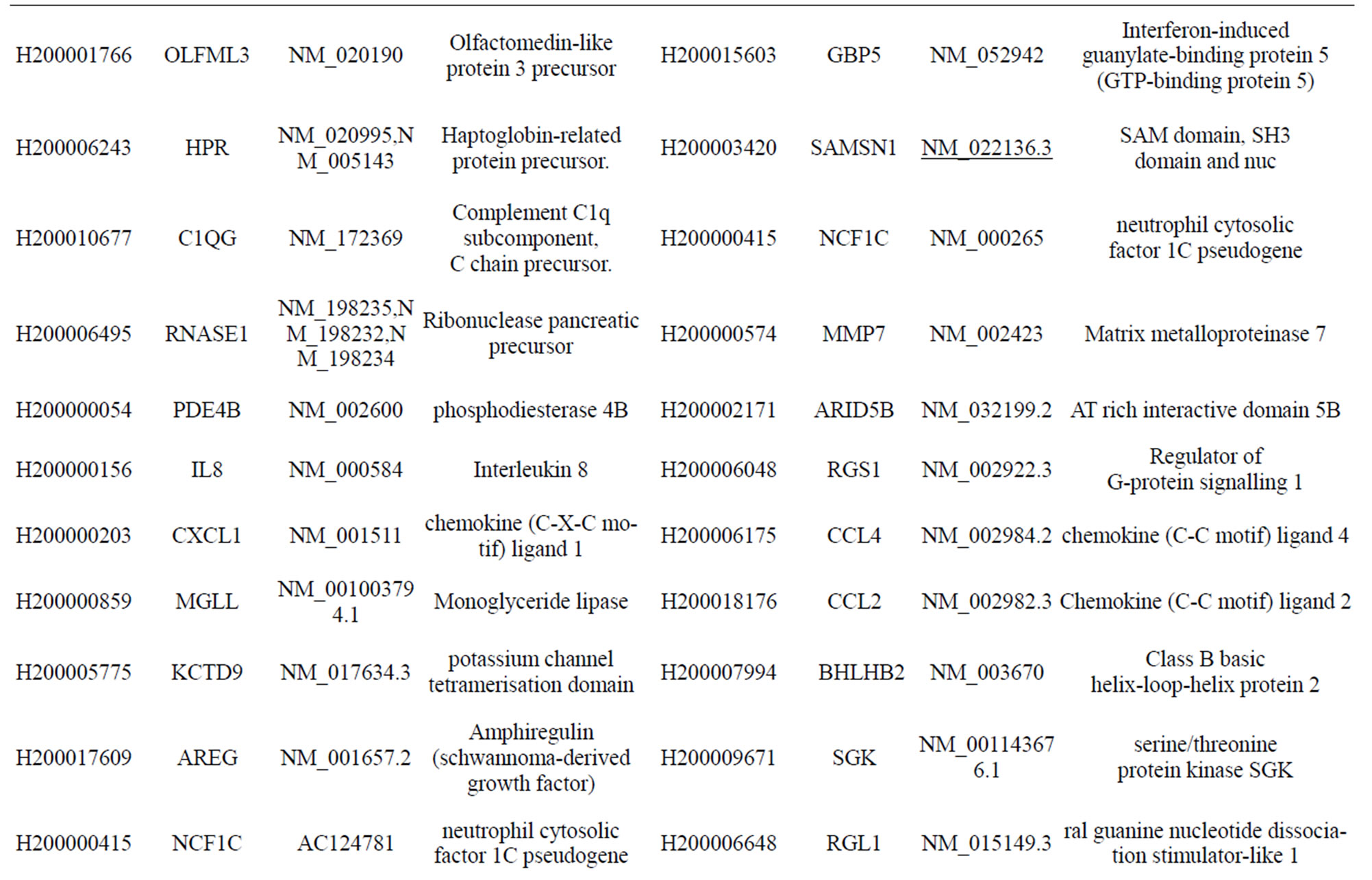
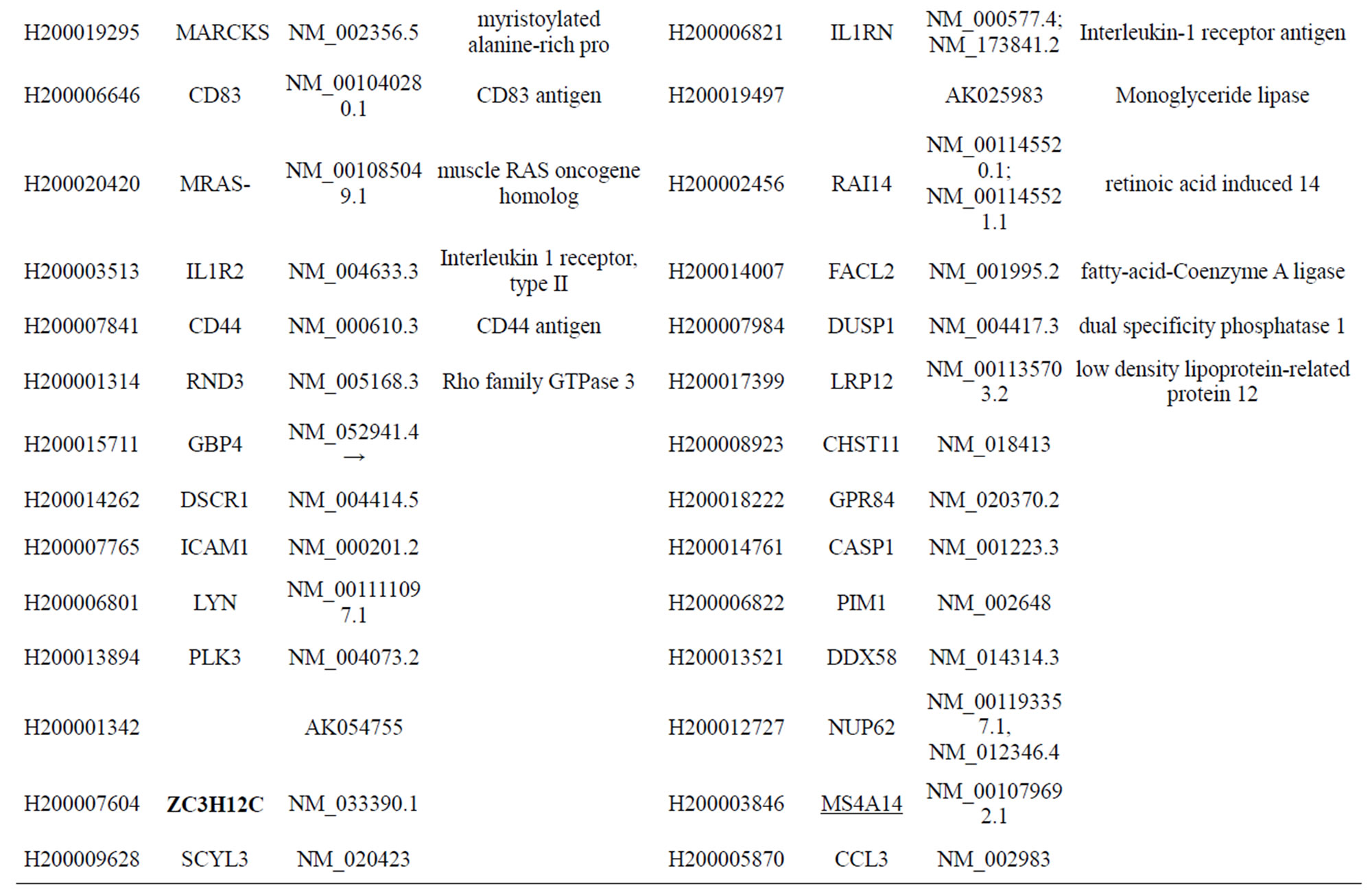
Table S1. Differentially-expressed genes with known functions that were activated by infection of MTB for 18 h at a moi of 10.
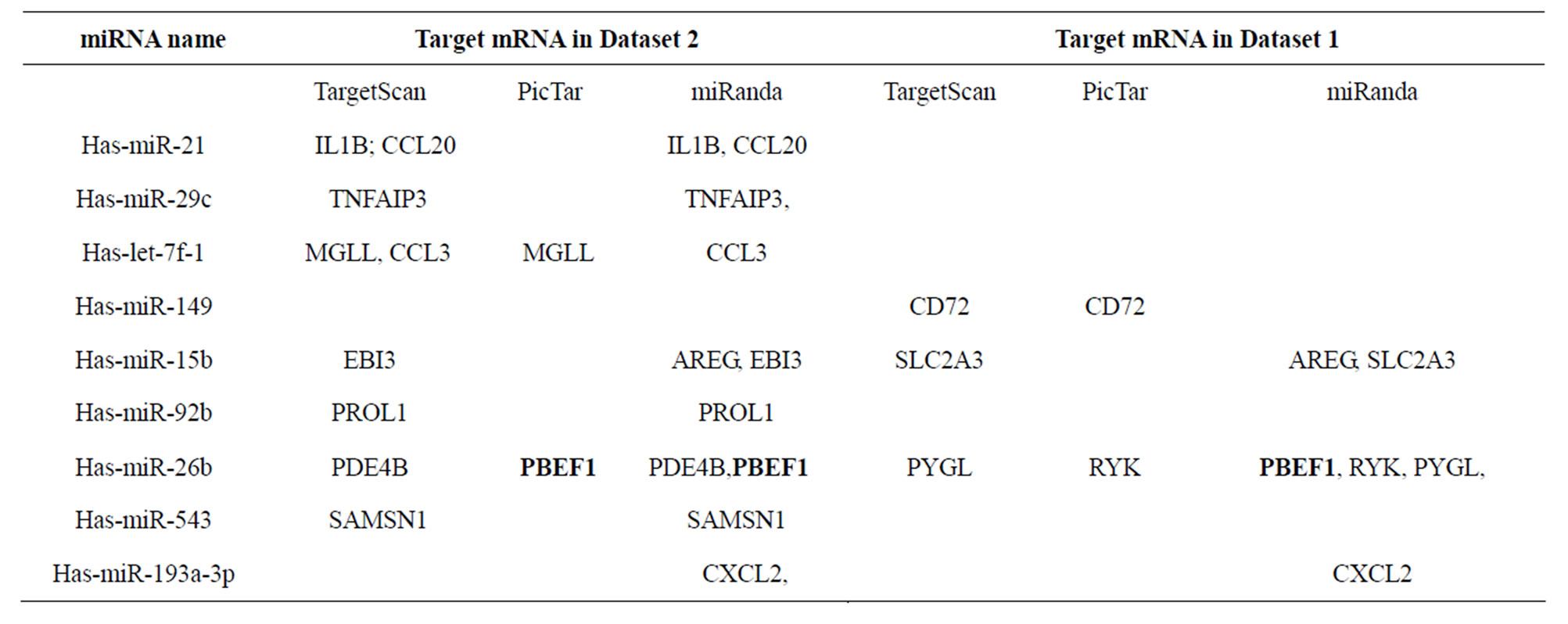
Table S2. Target mRNAs in datasets 1 and 2 for miRNAs identified in this study predicted using online databases.
NOTES
*Competing interests: The authors declare that they have no competing interests.
*Author contributions: YX, HZ, LB, CL and ZS participated in the conception and design of the experiments. ZS, NC and YL carried out the in silico prediction and analysis of miRNAs. ZS, LZ, WR, MT, XZ and CJ performed the experiments and data analyses. All authors contributed to writing this manuscript and all have read and approved the final manuscript.