Stem Cell Discovery
Vol.3 No.2(2013), Article ID:29932,18 pages DOI:10.4236/scd.2013.32015
Long-term phenotypic characterization of human bone marrow and adipose tissue derived mesenchymal stromal cells*
![]()
Department of Biomedical Engineering, Tufts University, Medford, USA; #Corresponding Author: David.Kaplan@tufts.edu
Copyright © 2013 Dean L. Glettig, David L. Kaplan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 2 February 2013; revised 2 March 2013; accepted 30 March 2013
Keywords: Stem Cells; Adipose Stem Cells; Mesenchymal Stem Cells; Flow Cytometry; Surface Markers; Human Cells; Long-Term Cell Culture
ABSTRACT
We present methods to characterize mesenchymal stromal cells (MSC) over long time periods in vitro. The methods entail passaging cells multiple times and performing differentiation studies with the cells at each passage. Using an array of surface markers and flow cytometric quantification, the data can be correlated to traditional measures of differentiation such as PCR and staining. Using these methods to quantify the amount of differentiation, we concluded that many common MSC markers do not specifically define MSCs with true stem cell properties. Additionally, adipose-derived as opposed to bone marrow-derived MSCs show long-term CD34+ labeling. The methods described can be used to help identify stem cell markers and to characterize the state of stem cells in vitro. Compiling these data from multiple laboratories would be helpful to determine source, extraction and culture methods needed to obtain high yields of useful stem cells.
1. INTRODUCTION
In the rapidly evolving field of tissue engineering and regenerative medicine, human bone marrow-derived mesenchymal stromal cells (hMSCs) have been extensively studied with respect to their differentiation potential towards adipocytes, osteoblasts and chondrocytes, among other tissues, and provide an essential source for these tissue outcomes [1]. In many cases, hMSCs are also being used due to their reported immunosuppressive functions [2,3]. More recently, studies have shown that adipose-derived stem cells (hASCs) show similar differentiation properties [4].
When comparing methods in different publications, it becomes apparent that a multitude of extraction protocols and culture conditions exist. Essentially though, most of these studies extract hMSCs by adhesion to tissue culture plastic (TCP) as this method is gentle to the cells, relatively inexpensive and easy to perform [5]. In contrast, isolation by adherence generally yields a heterogeneous population and a mixed population might not behave in the same manner as a pure isolated cell population.
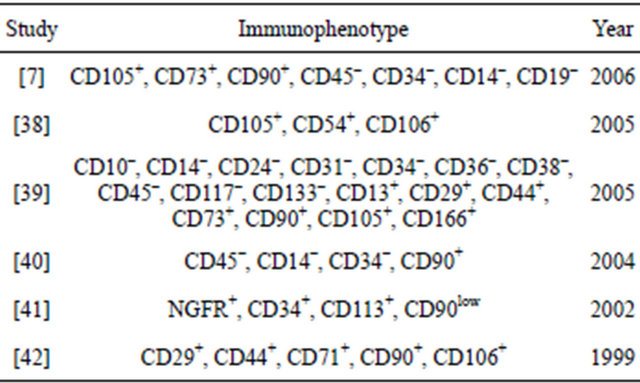
Studies using mixed cell populations can be both useful, as well as misleading, as it is often unknown which specific cell type responds to the culture conditions used [6]. The predominant use of the facilitated adhesion extraction method is also related to the fact that there is no unique phenotypic marker that defines a true hMSC. To circumvent this problem, many groups, most recently the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy [7], have listed a combination of surface marker profiles to define hMSCs. Table 1 shows both the range, but also the variety, of markers currently being used. Unfortunately, these definitions do not agree on a single distinguishable marker. This large variation in profile definition has not only led to confusion as to which profile combination defines the actual stem cell, but subsequently different groups use different combinations as their definition of hMSCs. This approach impairs comparison of results between different research groups, given the already large variance from donor variability, extraction and culTable 1. List of surface marker profiles used in the literature to define MSCs.
ture conditions [8-10]. Without clear methods to generate consistent assessments of stem cell populations, variability in clinical trial outcomes would be problematic.
Current differentiation studies with hMSCs and hASCs are limited due to the lack of true quantitative analysis. Endpoint analysis of differentiated cells is generally based on RNA quantification of cell specific genes using quantitative polymerase chain reaction (qPCR), as well as staining methods to determine the presence of tissue-specific components, e.g. calcium phosphate staining with Alizarin red for osteogenesis. These supposedly quantitative methods should be regarded as qualitative methods as they only provide information regarding the overall population but do not reflect how many cells responded to a specific treatment and therefore how many stem cells were present before and after differentiation. A more quantitative analytical method is therefore of high interest with respect to the decreasing differentiation potential of stem cells with increasing time in culture [11].
In an attempt to standardize hMSC definition, we have developed methods to correlate the quantitative analytical strength of flow cytometry to the qualitative outcomes of functional assays. For this we followed the procedure as outlined in Figure 1. At first cells were extracted from a respective cell source and expanded as P0 cells. These cells were passaged and expanded several times, up to a maximum passage number of 6. At each passage a subset of the cells were not passaged, but instead differentiated towards either adipocytes or osteoblasts. At each passage, differentiated as well as undifferentiated cells were analyzed with both qualitative and quantitative analytical tools such as flow cytometry, qPCR and staining.
If this relatively inexpensive and simple method was utilized for MSCs in research, a large database could be assembled that would allow researchers to compare results, improve stem cell extraction, characterization, and streamline outcomes.
2. MATERIALS AND METHODS
2.1. Bone Marrow Derived Human MSC Extraction
hMSCs were extracted according to a common procedure [12-14] from commercially obtained fresh human bone marrow aspirates (Lonza) from two different donors (MSC1 and MSC2). Aspirate donors were male, under 25 years of age and free of HIV, hepatitis B and hepatitis C, and had a cell count of at least 15 × 106 leukocytes/mL (as measured by a Coulter counter). Briefly, an aspirate volume of 25 mL was diluted 10-fold with MSC expansion medium consisting of DMEM:F12 basal medium (DMEM:F12) supplemented with 10% fetal bovine serum (FBS), antibiotics-antimycotics (100 U/mL penicillin, 100 µg/mL streptomycin, 0.25 µg/mL fungizone), 0.1 mM nonessential amino acids and 1 ng/mL basic fibroblast growth factor (bFGF) (Invitrogen). This diluted bone marrow was plated on tissue culture treated plastic (TCP) T-185 flasks at an average seeding density of 350,000 leukocytes/cm2. After 10 days the nonadherent cells were removed and the adherent cells were kept in expansion medium to reach confluence.
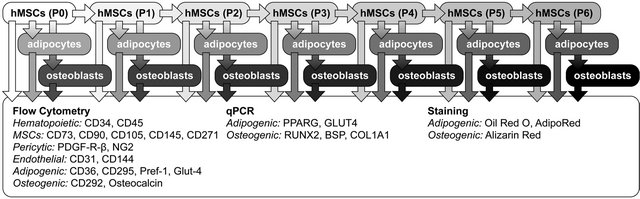
Figure 1. Outline of the procedure.
2.2. Adipose Tissue Derived MSC Extraction
hASCs were extracted from two different donors (ASC1 and ASC2) following a standard procedure using general anesthesia from human subcutaneous adipose tissue from elective abdominoplasty surgeries under approved protocols [15]. Briefly, primary isolation of the hASCs was conducted by first homogenizing a minimum of 200 mL of the extracted adipose tissue to obtain smaller tissue masses. These masses were then washed three times with phosphate buffered saline (PBS) to remove excess blood cells as well as non-adipogenic tissue. After the washes the adipogenic tissue was digested with equal volumes of 1 mg/mL collagenase type I (Invitrogen) in pre-warmed PBS for one hour at 37˚C on a shaker plate. This dissociated the tissue, releasing the hASCs from the adipocytes. Centrifugation at 300 g for 10 min allowed the hASCs to pellet, which were then washed twice in PBS and defined as SVF. These cells were then plated on TCP at a density of 0.16 mL original tissue volume per cm2. hASCs were then expanded to confluence using DMEM:F12 medium with 10% FBS and 1% antibiotics-antimycotics.
2.3. Osteoblasts
Human osteoblasts derived from healthy adults (NHO st) were purchased commercially (Lonza). These were used as controls to measure as standards in comparison to MSC2 cells differentiated to osteoblasts.
2.4. Expansion and Passaging
Upon reaching confluence, the expanded cells from each flask were passaged using 0.25% trypsin-EDTA (Invitrogen) and expanded in 4 new T-185 flasks. An average of 5 × 106 cells per flask at confluence yielded a passaging seeding density of 6750 cells/cm2, suggesting a 2-fold cell doubling to reach confluence. This expansion was carried out continuously for up to 6 passages (Figure 1). Continuous expansion (P1 - P6) was performed using the same expansion medium, although without bFGF as this has been reported to play a negative role on hMSCs [10] and should therefore be avoided in cultures that are intended for tissue implants.
2.5. Differentiation
Additional flasks were differentiated towards adipocytes and in the case of hMSCs also towards osteoblasts (Figure 1). Though an hMSC is defined as a stem cell with the capability to differentiate towards all three lineages (adipocytes, osteoblasts and chondrocytes), differentiation was commenced towards just one or two cell types as a measure of stemness. Lack of differentiation towards at least one cell type rules out an hMSC being a true stem cell. Therefore not requiring differentiation towards all three cell types allows a reduction of cells needed for analysis.
To commence differentiation, the medium of the confluent flasks was exchanged with a differentiation medium. In the case of adipogenic differentiation the medium consisted of DMEM:F12 with 3% FBS, 1% antibiotics-antimycotics (100 U/mL penicillin, 100 µg/mL streptomycin, 0.25 µg/mL fungizone) (Invitrogen) supplemented with 33 µM biotin, 17 µM D-pantothenic acid hemicalcium salt, 1 µM human insulin, 1 µM dexamethasone, 50 mM 3-isobutyl-1-methylxanthine and 5 µM 2,4-thiazolidinedione (Sigma). For osteogenic differentiation the medium was DMEM:F12 with 10% FBS, 1% antibiotics-antimycotics (100 U/mL penicillin, 100 µg/mL streptomycin, 0.25 µg/mL fungizone) (Invitrogen) supplemented with 10 mM glycerol-2-phosphate disodium salt hydrate, 400 µM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 10 nM 1α, 25-dihydroxyvitamin D3 and 10 nM dexamethasone (Sigma). Medium was fully replenished biweekly and the cells of each passage were kept in the respective differentiation medium for 3 weeks to allow full differentiation.
2.6. Microscope Images and Staining
Changes in cell morphology preand post-differentiation were observed using an inverted microscope. To determine successful differentiation, cells were also stained with Oil Red O (Sigma) in the case of adipogenic differentiation or Alizarin Red (Sigma) for osteogenic differentiation. Additionally, AdipoRed (Lonza) staining was used after hASC differentiation as it is faster and more quantifiable due to fluorescence measurements.
2.7. RNA Isolation, Purification, and qPCR
RNA was isolated from 6 biological replicates of cells both before and after differentiation using the Qiagen RNEasy Mini kit (Qiagen). To synthesize cDNA, reverse transcription was performed on 1 µg of purified RNA using the High Capacity cDNA Archive kit (Applied Biosystems). Osteoblast differentiation markers include runt-related transcription factor 2 (RUNX2), bone sialoprotein (BSP) and collagen type I (COL1A1). Adipogenic differentiation markers include perixosome proliferator-activated receptor γ (PPARG) and insulin-responsive glucose transporter 4 (GLUT4). Primers and probes for the bone-related and adipose-related genes above were obtained from TaqMan® Gene Expression Assay kits (Applied Biosystems). Transcript expression levels were quantified with a Stratagene Mx3000P QPCR System (Stratagene). Expression levels were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and reported relative to values of P0 undifferentiated cells. We have previously reported PCR reaction conditions and primers [12,16].
2.8. Flow Cytometry@NolistTemp#2.8.1. Phenotypic Markers
Flow cytometric analysis was performed on cells of each passage, both after reaching confluence and after 3 weeks of differentiation. Cells were labeled with antibodies pre-conjugated with either fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorphyll protein (PerCP) or allophycocyanin (APC). Phenotypic cell markers were selected for each of the following cell types: hematopoietic stem cells (CD34, clone: 581, BD Biosciences; CD45, clone: MEM-28, Abcam), adipocytes (CD36, clone: 255606, R&D Systems; CD295, clone: 52263, R&D Systems; Pref-1, clone: 211309, R&D Systems, Glut-4, clone EP930(2)AY), osteoblasts (CD292, R&D Systems; osteocalcin, clone: 190125, R&D Systems), endothelial cells (CD31, clone: WM59, Abcam; CD144, clone 16B1, eBioscience) and pericytes (PDGFR-β, clone: PR7212, R&D Systems; NG2, clone: LHM-2, R&D Systems). Additionally 3 commonly cited MSC markers (CD73, clone: AD2, BD Biosciences; CD90, clone: 5E10, Biolegend; CD105, clone: 166707, R&D Systems) as well as 2 more novel MSC markers (CD146, clone: 128018, R&D Systems; CD271, clone: ME20.4-1.H4) were included. Hematopoietic cell markers were chosen based on their high abundance in the extracted tissues. Endothelial cell markers were chosen as it has been reported that endothelial cell progenitor cells are also present in bone marrow [17] and can be co-extracted with hMSCs. Especially since many of the MSC markers are proteins expressed for cell adhesion (Table S1) it is important to be certain that the extracted cells do not include endothelial cells. Also for future studies where endothelial cells may be included in co-cultures with hMSCs for regenerative medicine, distinguishing the survival of these respective cell populations would be useful. Pericyte markers we included due to recent suggestions that both hMSCs and hASCs are located in proximity to blood vessels in vivo, where they exert the function of pericytes by supporting blood vessel structure and wound repair [18]. Due to a lack of commonly used adipogenic or osteogenic markers for flow cytometry, a few selected markers were chosen for each cell type.
2.8.2. Cell Extraction
For flow cytometric analysis, growth medium was aspirated from 3 confluent T-185 flasks (an average of 15 × 106 cells). After washing with PBS, 5 mL of 0.25% trypsin/EDTA (Invitrogen) was added to each flask and kept in an incubator for 10 min at 37˚C. Trypsinization was halted by adding 10 mL serum containing medium and the cell suspension was collected into a 50 mL conical tube. The conical tube was centrifuged for 10 min at 450 g and 4˚C. After removing the supernatant, the pellet was resuspended in 2 mL of FACS-buffer, consisting of PBS supplemented with 0.5% FBS (Invitrogen) and 5 mM EDTA (Sigma).
2.8.3. Intracellular Staining-Osteocalcin Sample
A 200 µL aliquot of the original 2 mL suspension (an average of 1.5 × 106 cells) was separated and fixed in 10% formalin solution for 10 min. The fixed cells were centrifuged for 10 min at 450 g and 4˚C. Following centrifugation, the supernatant was removed and the cells were re-suspended in 2 mL PBS. After another centrifugation step, the supernatant was removed and the cell pellet was re-suspended in 2 mL 0.1% Triton-X in PBS solution, to perforate the lipid bilayer, allowing the stain to enter the cell. Subsequent washes always included Triton-X to maintain lipid bilayer perforation.
2.8.4. Sample Labeling and Measurement
The cell suspension was split into samples in 2 mL Eppendorf tubes. Then 10 µg of human IgG (Sigma) was added to each of the samples to block any nonspecific receptors before adding the respective antibody to each sample. Unstained samples were included for autofluorescence background reduction. Each sample was incubated at 4˚C for 30 min in the dark. After incubation 1 mL of FACS-buffer was added to each sample and centrifuged for 10 min at 450 g and 4˚C. The supernatant was removed and the pellets were re-suspended in 1 mL FACS-buffer. After a second centrifugation at the same settings, the samples were re-suspended in 250 µL FACS-buffer, transferred into their respective FACStubes and kept on ice until measurement. Flow cytometric analysis was performed on a FACSCalibur (Becton Dickinson). A minimum of 50,000 events was recorded for each sample. Analysis was performed using FlowJo (Tree Star) on dot plots to obtain a more accurate quantification as compared to histogram analysis.
2.9. Statistics
Statistically significant differences between two conditions in qPCR were determined by performing a twotailed Student’s t-test. Differences were considered significant if P < 0.05, unless otherwise noted.
3. RESULTS
hMSCs extracted from the two different donors were labeled MSC1 and MSC2, respectively. Likewise the hASCs extracted from two different donors were labeled ASC1 and ASC2, respectively. Due to the limited amount of cells obtained directly after extraction, the stromal vascular fraction (SVF) of ASC1 was used entirely for flow cytometric analysis. Likewise insufficient cells were available at P0 of the ASC2 cells, such that full analysis commenced at P1.
3.1. Expansion
Following the procedure outlined in Figure 1, the experimental setup allowed the same seeding density at each passage, and confluence was reached in most cases 3 weeks post expansion. The cumulative time of culture for the cells in vitro is listed in Table S2.
3.2. Microscopic Analysis
Microscopic analysis of the MSC1 cells (Figure 2) showed that both adipogenic and osteogenic differentiation was the highest at P0. Despite treating cells at each passage for the same period of time with differentiation medium, different levels of matrix deposition were observed. Figure 2 also shows that in comparison to P0, the P1 cells had a lower amount of differentiated cells. This drop in differentiation potential decreased gradually with each passage. This is especially visible in adipogenic differentiation, where at P0 adipocytes were highly abundant with large lipid vacuoles compared to P5 where adipocytes were not only scarce but also showed very
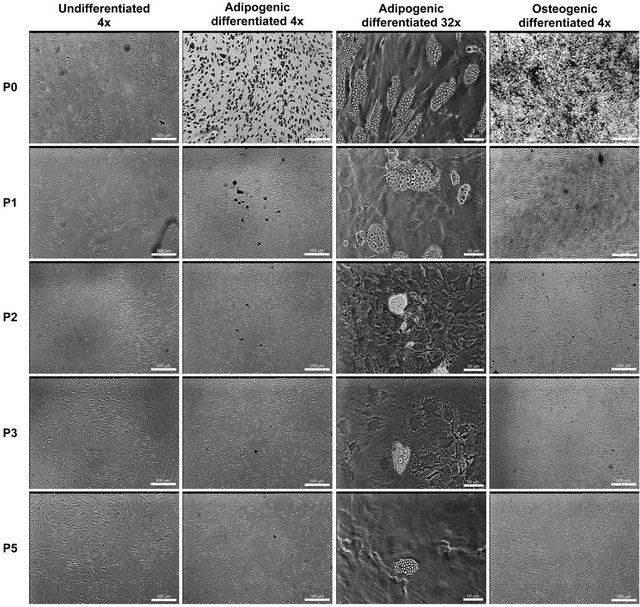
Figure 2. Microscopic images of MSC1 cells at each passage preand post-differentiation. Rows are images of cells from the same passage (P0 - P5) shortly before extraction for flow cytometric and qPCR analysis. Columns are images of undifferentiated, adipogenic and osteogenic differentiated cells measured at 4× and 32× magnification for a population overview or detailed cell morphology respectively. Scale bars in all 4× images are equal to 500 µm. In 32× images, scale bars are equal 50 µm.
small lipid vacuoles. The same decrease in differentiation was observed in both MSC2 and ASC2 cells (Figure S1).
3.3. qPCR
Adipogenic differentiation was confirmed with qPCR analysis with an increase of perixosome proliferatoractivated receptor γ (PPARG) at all passages (Figures 3 and S2). Additionally, the PPARG levels of undifferentiated hMSCs gradually decreased with each passage. An increase in PPARG was also observed at all passages after osteogenic differentiation. Glucose transporter 4 (GLUT4) expression followed no distinct trend. With regard to osteogenic differentiation, the same gradual baseline decrease of all markers was measured with the undifferentiated cells at each passage. In each case runt-related transcription factor 2 (RUNX2) levels and bone sialoprotein (BSP) levels increased after 3 weeks of differentiation, however collagen type I (COL1A1) levels showed no clear trend.
3.4. Flow Cytometric Analysis
The two hematopoietic markers CD34 and CD45 showed very low positive labeling at all passages of hMSCs (Figures 4(a) and S3(a)). In comparison, hASCs showed a significantly higher CD34+ labeling. This labeling decreased in undifferentiated cells after 2 passages (Figure S3(c)). Contrarily the CD34+ percentages increased to over 33% after adipogenic differentiation, which was not observed in hMSC cells.
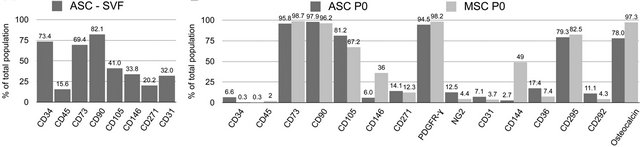
The higher CD34+ labeling on ASCs was initially observed with the SVF of the ASC1 cells (Figure 5(a)): These cells showed slightly lower labeling percentages of CD73, CD90 and CD105, slightly higher labeling of CD45 and CD31 and a significantly higher labeling of CD34 cells as compared to hMSCs. Subsequent measurement of the ASC2 cells showed similar surface marker profiles for P0 cells as compared to MSC1 cells (Figure 5(b)).
Further analyzing the common MSC markers, CD73 and CD90 labeling was close to 100% over all passages in all hMSC and hASC sources. This labeling remained high even after adipogenic and osteogenic differentiation. CD105 labeling remained at a relatively high rate on undifferentiated MSC1 cells. In contrast MSC2 and ASC2 cells showed as similar trend, where following an initially low labeling the percentages increased gradually to around 80% at P3 - P5 and dropped at P6 again. After differentiation the average labeling decreased in general to the CD105+ level of early passages.
The novel hMSC markers CD146 and CD271 showed very different labeling patterns when comparing cell sources. In general, CD146 percentages were lower than the CD105 percentages and CD271 was lower than
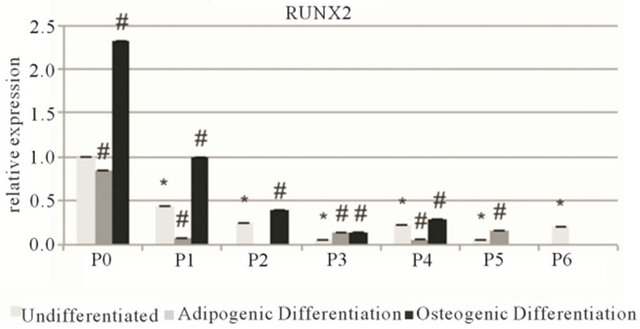
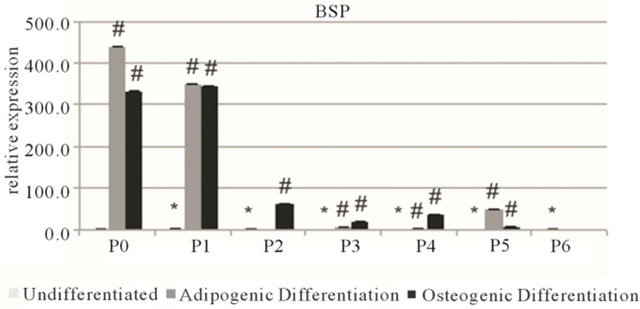

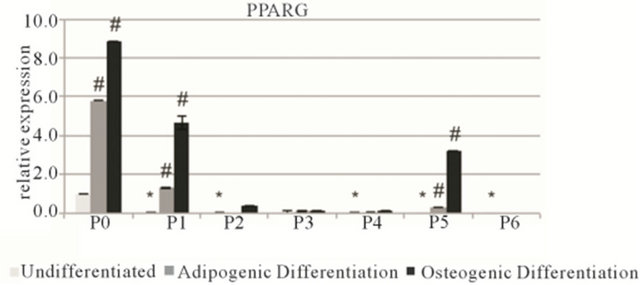
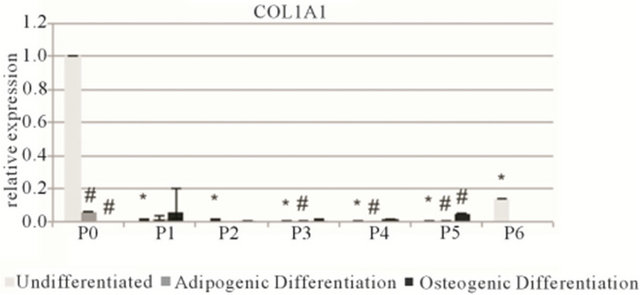
Figure 3. Relative gene expression of MSC1 cells. Top row: osteogenic markers runt-related transcription factor 2 (RUNX2), bone sialoprotein (BSP) and collagen type I (COL1A1). Bottom row: adipogenic markers: perixosome proliferator-activated receptor γ (PPARG) and glucose transporter 4 (GLUT4). Data is grouped by passage number (P0 - P6) showing data from undifferentiated, adipogenic and osteogenic differentiated cells. Data points are mean relative expression ± standard deviation (n = 6). Marked samples are statistically different, *relative to undifferentiated P1 samples (P < 0.05), #relative to undifferentiated samples of same passage number (P < 0.05).
 (a)
(a)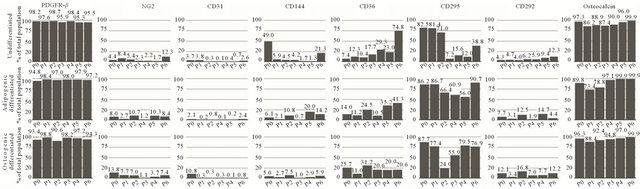 (b)
(b)
Figure 4. Flow cytometric analysis of MSC1 cells. A minimum of 50,000 events was recorded for each sample. Data is given in percentages of total population of undifferentiated (top row), adipogenic differentiated (middle row) and osteogenic differentiated cells (bottom row). Data is ordered by passage number (P0 - P6) and grouped in each column by marker type. (a) Analysis of MSC markers with CD34 and CD45 as common negative, CD73, CD90 and CD105 as common positive and CD146 and CD271 as novel positive MSC markers; (b) Analysis of pericyte (PDGFR-β and NG2), endothelial cell (CD31 and CD144), adipocyte (CD36 and CD295) and osteoblast (CD292 and osteocalcin) markers.
 (a) (b)
(a) (b)
Figure 5. Flow cytometric profile of undifferentiated cells given in percentages of total population. (a) Common MSC markers of ASC1 cells derived from the SVF; (b) Comparison of MSC, pericyte, endothelial cell, adipocyte and osteoblast markers on unpassaged (P0) cells of ASC2 and MSC1 cells. A minimum of 50,000 events was recorded for each sample.
CD146.
Of the pericyte markers, PDGFR-β showed very high labeling preand post-differentiation on MSC1 cells (Figure 4(b)). In comparison only the undifferentiated MSC2 cells had the same high labeling percentage (Figure S3(b)). After differentiation the PDGFR-β values were low at early passages however gradually reached high percentages proportional to increasing passage number. On the other hand NG2 levels were very low in all passages, differentiation conditions and sources.
Endothelial markers CD31 and CD144 were present at a very low level with an outlier at P0 of undifferentiated MSC1 cells.
The adipogenic marker CD36 showed very low labeling at early passages with a gradual increase proportional to passage number. This was the only marker that exhibited this pattern consistently over all cell sources and differentiation condition. CD295 showed no apparent correlation with respect to passage, differentiation or cell source and the same observation was made on the surface marker staining of Pref-1 and Glut-4 on ASC2 cells (Figure S3(d)).
With respect to the osteogenic markers, CD292 labeling was very low and only gradually increased with each passage. Osteocalcin labeling on the other hand was comparably high in all passages of undifferentiated hMSCs, however the levels of MSC1 and MSC2 varied greatly post-differentiation.
Comparison of P0 MSC2 and NHOst cells (Figure S3) showed very little difference in marker labeling, except for a lower PDGFR-β and a higher osteocalcin staining.
Another observation derived from flow cytometric analysis was a gradual increase of autofluorescence with increasing passage (Figures 6 and S4). After an initial increase in fluorescence when excited by a 488 nm laser and detected at 530/30 nm (FL1-H), fluorescence gradually also increased when excited by a 635 nm laser and detected at 661/16 nm (FL4-H).
4. DISCUSSION
An initial reaction to this method may be the need for a lot of cells, predominantly for flow cytometric analysis, such that e.g. insufficient cells were available for full analysis of early passages of hASCs. This concern can be put in perspective in that we used up to 15 markers to cover a broad spectrum of phenotypes. Using the same doubling rates, we calculated that only 0.5 × 106 cells are needed at initial seeding of each cell source to investigate 1 surface marker, perform osteogenic and adipogenic differentiation and commence continuous passaging. Thus, the majority of stem cell sources should be readily amenable to the methods described in the present paper.
The lack of using bFGF for this study was visible in the relatively long times it took to reach confluence at each passage. Interestingly, there was generally no change in the expansion rate of the cells even after several months in vitro. Considering the rapid drop in differentiation potential after P1, one can therefore conclude that expansion rate is not linked to differentiation potential. This adds to the debate regarding the use of bFGF in regenerative medicine [10]. The rapid drop in differentiation potential was very evident and should be greatly taken into consideration when determining the optimal balance between retaining stem cell properties and passaging cells.
The low increase in early gene markers (PPARG, RUNX) and large increase of a late marker (BSP), largely overlaps with the microscope data showing again that differentiation occurs predominantly only in early passages. Additionally, we are not aware of any study that shows the gradual decrease in the baseline of gene markers pre-differentiation. This might be an interesting aspect of the results with regard to determining the state and differentiation potential of hMSCs. However herein also lies the limitation of qPCR data: it is unclear if differentiation has been fully completed, or whether individual stem cells are still undifferentiated in culture.
The lack of hematopoietic and endothelial cell markers (CD45, CD31, CD144) in passaged cultures is explainable with the fact that the culture medium used was not targeted towards these cell types, and that they died off or were washed out during the extended culture period
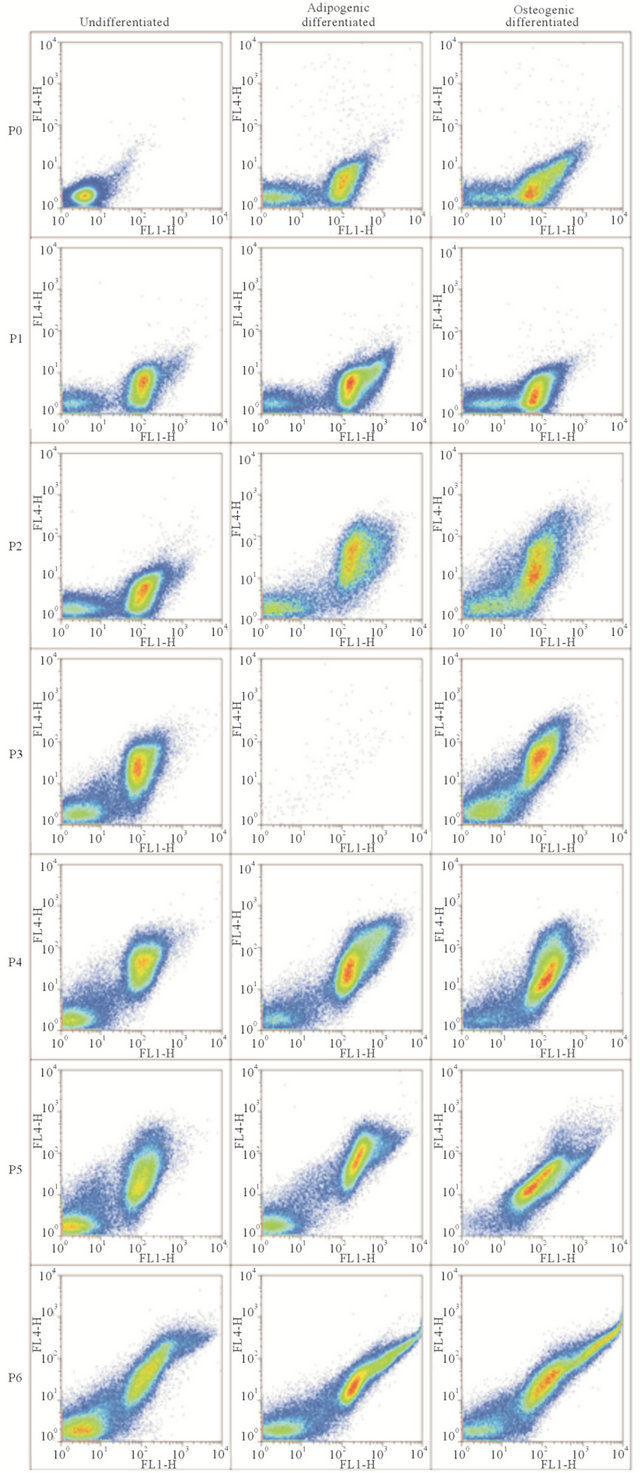
Figure 6. Autofluorescence of MSC1 cells. Rows are logscale dot plots of cells from the same passage (P0 - P6), and columns list undifferentiated, adipogenic differentiated and osteogenic differentiated conditions respectively. X-axis: FL1-H (ex: 488 nm; em: 530/30 nm). Y-axis: FL4-H (ex: 635 nm; em: 661/16 nm).
This is especially obvious in the ASC cultures where a relatively high percentage of cells labeled positive for hematopoietic and endothelial cell markers in the SVF that decreased in subsequent passages. Because the SVF did not undergo extensive expansion, there was no selection for adherent MSCs, yielding a greater mixture of different cell types.
With regard to CD34 labeling, this still proves to be a negative marker for hMSCs, which is especially important as it is a positive marker for hematopoietic stem cells (HSC) which are known to be present in the bone marrow. However the presence of CD34-positive cells in the hASC population raises an interesting question: Hematopoiesis is predominantly localized within the bone marrow, therefore one would not expect a high percentage of CD34+ HSCs in adipose tissue. Though HSCs are present in bone marrow aspirates during extraction, they survive only for short periods in vitro. This makes the presence of CD34+ cells in long-term hASC cultures a potential positive marker, of which it’s function is still being discussed [19,20]. It is also very interesting to see the increase of CD34+ cells in hASC cultures after adipogenic differentiation. Though further studies will be necessary, this adds fuel to the discussion of the relationship between adipocytes and HSCs [21].
With regards to traditional MSC markers, it is striking that CD73 and CD90 labeled almost all cells, not only in both hASC and hMSC cultures, but also over all passages and differentiation conditions. Based on the qPCR and microscope data, one can assume that only very few cells still possess full differentiation potency at later passages, it must be concluded that these two markers are not very useful to define true MSCs. Especially considering the percentage of labeled cells did not decrease at all after 3 weeks of differentiation makes the use of these markers even less applicable to define a stem cell. In contrast CD105 showed a slightly lower labeling percentage. With respect to the commonly used definition of an hMSC being CD34–, CD45–, CD73+, CD90+ and CD105+, one can rather safely rely on CD105 as single marker as it is the least common denominator. However we still have strong doubts that CD105 is a valuable stem cell marker, as it again did not follow the differentiation pattern observed under the microscope. Given these observations, one must conclude that these common MSC markers do not specifically define a stem cell, but given their origin, rather just an adherent stromal cell. Based on the amount of cells visibly undergoing differentiation, we would suggest relying rather on more novel hMSC markers such as CD146 and CD271. This could be verified by sorting out these cells individually using FACS and performing differentiation studies on the isolated cell type. Additionally, further analysis using in vivo differentiation assays could provide insight in the in vivo differentiation potential of these specific markers as these can differ greatly from in vitro differentiation [22].
The high frequencies of PDGFR-β likely has origins from the fact that it is not specific for pericytes but instead is reported to be present on a wide range of cells of mesenchymal origin [23], e.g. also visible in the NHOst. On the other hand NG2 is believed to be more specific for pericytes. The low amount of labeling should not necessarily refute the suggestion of hMSCs being pericyte-like. Especially considering the observed low amount of differentiating cells, one might derive this from a low percent of stem cells.
The labeling percentages of adipogenic markers CD36, CD295, Pref-1 and Glut-4 provided no apparent correlation between the differentiation levels observed in the microscopic images. CD36 was the only marker that consistently increased with passage and may prove to be a potential marker of stem cell aging. Though the expression of the selected adipogenic proteins on the surface membrane is required for activation of important metabolic pathways of adipocytes, they do carry out many different functions [24-29]. The diversity of the metabolic pathways may be a possible reason behind the lack of a clear pattern, as the cells might be in very different stages of differentiation. In a recent publication, high levels of CD295 were correlated to the aging and dying of MSCs [30]. This could not be verified in our results. Since the chosen adipocyte markers did not lead to any conclusive results, more research needs to be done to discover adipogenic markers for flow cytometry. Possible new markers include AdipoRed staining and measuring the ratio of intracellular vs. surface expression of Glut-4 [31]. Here it should be mentioned that the lack of flow cytometric data in publications on adipocytes is due to the fact that they are difficult to measure in a flow cytometer. The strong shear forces can easily rupture the fragile lipid filled cells, though it has been shown in the past though that this can be counteracted by fixation [32]. This again leads to the importance of finding a true stem cell marker. If this could be found, flow cytometric analysis of differentiated cells would still allow accurate quantification of differentiation by counting the amount of remaining stem cells post differentiation.
Similar to the suggestions for CD36, CD292 could be a potential tool for determining the state of a stem cell. The lack of high expression after osteogenic differentiation may be understood in that it is not a requirement for differentiation and is only necessary for extracellular matrix (ECM) deposition of osteoblasts [33]. If osteoblasts secrete large amounts of ECM they may be difficult to label with flow cytometric markers. This might explain the inversely correlated relationship in differentiated MSC2 cells as well.
The use of osteocalcin, as the only intracellular marker, circumvented this issue and showed a very high, gradually increasing labeling in hMSCs reaching levels of NHOst. Future experiments should include other osteogenic markers, most notably CD106, which has recently been suggested to quantify osteogenic differentiation [34].
Finally the increase in autofluorescence may be due to accumulation of flavoproteins and lipofuscin [35,36]. Accumulation of these compounds is suggested to be related to oxidative stress and aging of cells cultured in vitro and therefore provides an additional marker for the state of stem cells. The hypothesis here would be that the higher the autofluorescence the lower the differentiation potential. This would be a relatively simple analytical tool to prescreen hMSCs for a more generalized assessment of differentiation potential.
5. CONCLUSIONSOur method of correlating quantitative flow cytometric data with current standard qualitative assays displays a straightforward and relatively inexpensive approach for detailed characterization of a batch of stem cells. To our knowledge this is the first collection of such detailed analysis of hMSCs and hASCs in long-term in vitro culture and will be helpful to aid other researchers in planning future experiments. It is important to note here that we are not postulating that our cell sources, extraction methods, or culture conditions are the gold standard for MSCs (this is obviously not the case give the low rate of differentiation at higher passages). Instead we encourage other groups to utilize these methods to analyze their stem cells with their culture conditions, providing the field with a large set of data. Using this collection of data one could back-correlate the rate of differentiation to the different parameters of cell source (e.g., gender, age, site) and also better determine the influence of different culture conditions on cell outcomes. This approach has direct implications for studies in regenerative medicine as well as for clinical therapies that involve the use of stem cells.
Using our data set, we were not only able to disqualify certain makers for true stem cell characterization (CD73 and CD90), but also highlight a marker with significant difference between hMSCs and hASCs (CD34). Since no stem cell marker followed the differentiation pattern observed under the microscope, the discovery of a unique stem cell marker remains to be identified. A true stem cell marker would not only label the same amount of cells that undergo differentiation, but also no longer be present after differentiation. Additionally, since most non-MSC markers labeled the cell populations at very different levels, it is clear that standard isolation methods yield heterogeneous populations that also change during extended culture.
Therefore hMSCs remain an undefined cell type. Though the data presented here did not directly provide the discovery of a unique stem cell marker, by using these methods we were able to disqualify the reliability of certain markers with regard to stemness. Performing this analysis on other markers and cell sources will allow one to validate the use of other phenotypes and zone in on the profile of a true stem cell. This approach should also allow one to define different stages of stemness as it is known with HSCs [37].
6. ACKNOWLEDGEMENTS
We thank Kristina Papa, Emily Shaw and Christina Thomas for assistance with cell culture, Sarah Sundelacruz and Tessa DesRochers for support with qPCR and Allen Parmelee for support with flow cytometric analysis. We also thank Peter Rubin for providing adipose derived stem cells. This work was supported by NIH P41 Tissue Engineering Resource Center (P41 EB002520) and AFIRM. No competing financial interests exist.
REFERENCES
- Gregory, C.A., Prockop, D.J. and Spees, J.L. (2005) Nonhematopoietic bone marrow stem cells: Molecular control of expansion and differentiation. Experimental Cell Research, 306, 30-35. doi:10.1016/j.yexcr.2005.03.018
- Le Blanc, K., Tammik, C., Rosendahl, K., Zetterberg, E. and Ringden, O. (2003) HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Experimental Hematology, 31, 890-896. doi:10.1016/S0301-472X(03)00110-3
- Helmy, K.Y., Patel, S.A., Silverio, K., Pliner, L. and Rameshwar, P. (2010) Stem cells and regenerative medicine: Accomplishments to date and future promise. Therapeutic Delivery, 1, 693-705. doi:10.4155/tde.10.57
- Tapp, H., Hanley Jr., E.N., Patt, J.C. and Gruber, H.E. (2009) Adipose-derived stem cells: Characterization and current application in orthopaedic tissue repair. Experimental Biology and Medicine, 234, 1-9. doi:10.3181/0805-MR-170
- Beyer Nardi, N. and da Silva Meirelles, L. (2006) Mesenchymal stem cells: Isolation, in vitro expansion and characterization. Handbook of Experimental Pharmacology, 174, 249-282. doi:10.1007/3-540-31265-X_11
- Charbord, P. (2010) Bone marrow mesenchymal stem cells: Historical overview and concepts. Human Gene Therapy, 21, 1045-1056. doi:10.1089/hum.2010.115
- Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., Horwitz, E., et al. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy, 8, 315-317. doi:10.1080/14653240600855905
- Ho, A.D., Wagner, W. and Franke, W. (2008) Heterogeneity of mesenchymal stromal cell preparations. Cytotherapy, 10, 320-330. doi:10.1080/14653240802217011
- Vater, C., Kasten, P. and Stiehler, M. (2011) Culture media for the differentiation of mesenchymal stromal cells. Acta Biomaterialia, 7, 463-477. doi:10.1016/j.actbio.2010.07.037
- Sotiropoulou, P.A., Perez, S.A., Salagianni, M., Baxevanis, C.N. and Papamichail, M. (2006) Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells, 24, 462-471. doi:10.1634/stemcells.2004-0331
- Izadpanah, R., Kaushal, D., Kriedt, C., Tsien, F., Patel, B., Dufour, J. and Bunnell, B.A. (2008) Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Research, 68, 4229-4238. doi:10.1158/0008-5472.CAN-07-5272
- Meinel, L., Hofmann, S., Betz, O., Fajardo, R., Merkle, H.P., Langer, R., Kaplan, D.L., et al. (2006) Osteogenesis by human mesenchymal stem cells cultured on silk biomaterials: Comparison of adenovirus mediated gene transfer and protein delivery of BMP-2. Biomaterials, 27, 4993-5002. doi:10.1016/j.biomaterials.2006.05.021
- Wang, Y., Kim, U.J., Blasioli, D.J., Kim, H.J. and Kaplan, D.L. (2005) In vitro cartilage tissue engineering with 3D porous aqueous-derived silk scaffolds and mesenchymal stem cells. Biomaterials, 26, 7082-7094. doi:10.1016/j.biomaterials.2005.05.022
- Bianchi, G., Banfi, A., Mastrogiacomo, M., Notaro, R., Luzzatto, L., Cancedda, R. and Quarto, R. (2003) Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Experimental Cell Research, 287, 98- 105. doi:10.1016/S0014-4827(03)00138-1
- Dubois, S.G., Floyd, E.Z., Zvonic, S., Kilroy, G., Wu, X., Carling, S., Gimble, J.M., et al. Isolation of human adipose-derived stem cells from biopsies and liposuction specimens. Methods in Molecular Biology, 449, 69-79.
- Mauney, J.R., Nguyen, T., Gillen, K., Kirker-Head, C., Gimble, J.M. and Kaplan, D.L. (2007) Engineering adipose-like tissue in vitro and in vivo utilizing human bone marrow and adipose-derived mesenchymal stem cells with silk fibroin 3D scaffolds. Biomaterials, 28, 5280- 5290. doi:10.1016/j.biomaterials.2007.08.017
- Reyes, M., Dudek, A., Jahagirdar, B., Koodie, L., Marker, P.H. and Verfaillie, C.M. (2002) Origin of endothelial progenitors in human postnatal bone marrow. The Journal of Clinical Investigation, 109, 337-346.
- Da Silva Meirelles, L., Caplan, A.I. and Nardi, N.B. (2008) In search of the in vivo identity of mesenchymal stem cells. Stem Cells, 26, 2287-2299. doi:10.1634/stemcells.2007-1122
- Festy, F., Hoareau, L., Bes-Houtmann, S., Pequin, A.M., Gonthier, M.P., Munstun, A., Roche, R., et al. (2005) Surface protein expression between human adipose tissue-derived stromal cells and mature adipocytes. Histochemistry and Cell Biology, 124, 113-121. doi:10.1007/s00418-005-0014-z
- Suga, H., Matsumoto, D., Eto, H., Inoue, K., Aoi, N., Kato, H., Yoshimura, K., et al. (2009) Functional implications of CD34 expression in human adipose-derived stem/progenitor cells. Stem Cells and Development, 18, 1201-1210. doi:10.1089/scd.2009.0003
- Sera, Y., LaRue, A.C., Moussa, O., Mehrotra, M., Duncan, J.D., Williams, C.R., Ogawa, M., et al. (2009) Hematopoietic stem cell origin of adipocytes. Experimental Hematology, 37, 1108-1120.
- Quinn, C. and Flake, A.W. (2008) In vivo Differentiation potential of mesenchymal stem cells: Prenatal and postnatal model systems. Transfusion Medicine and Hemotherapy: Offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie, 35, 239- 247. doi:10.1159/000129129
- Andrae, J., Gallini, R. and Betsholtz, C. (2008) Role of platelet-derived growth factors in physiology and medicine. Genes & Development, 22, 1276-1312. doi:10.1101/gad.1653708
- Glatz, J.F., Luiken, J.J. and Bonen, A. (2010) Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiological Reviews, 90, 367-417. doi:10.1152/physrev.00003.2009
- Jing, K., Heo, J.Y., Song, K.S., Seo, K.S., Park, J.H., Kim, J.S., Park, J.I., et al. (1791) Expression regulation and function of Pref-1 during adipogenesis of human mesenchymal stem cells (MSCs). Biochimica et Biophysica Acta, 1791, 816-826. doi:10.1016/j.bbalip.2009.04.010
- Bianco, P. (2011) Bone and the hematopoietic niche: A tale of two stem cells. Blood, 117, 5281-5288. doi:10.1182/blood-2011-01-315069
- Fruhbeck, G. (2006) Intracellular signalling pathways activated by leptin. The Biochemical Journal, 393, 7-20.
- Huang, S. and Czech, M.P. (2007) The GLUT4 glucose transporter. Cell Metabolism, 5, 237-252. doi:10.1016/j.cmet.2007.03.006
- Sul, H.S. (2009) Minireview: Pref-1: Role in adipogenesis and mesenchymal cell fate. Molecular Endocrinology, 23, 1717-1725. doi:10.1210/me.2009-0160
- Laschober, G.T., Brunauer, R., Jamnig, A., Fehrer, C., Greiderer, B. and Lepperdinger, G. (2009) Leptin recaptor/CD295 is upregulated on primary human mesenchymal stem cells of advancing biological age and distinctly marks the subpopulation of dying cells. Experimental Gerontology, 44, 57-62. doi:10.1016/j.exger.2008.05.013
- Watson, R.T. and Pessin, J.E. (2001) Intracellular organization of insulin signaling and GLUT4 translocation. Recent Progress in Hormone Research, 56, 175-193. doi:10.1210/rp.56.1.175
- Smyth, M.J. and Wharton, W. (1992) Differentiation of A31T6 proadipocytes to adipocytes: A flow cytometric analysis. Experimental Cell Research, 199, 29-38. doi:10.1016/0014-4827(92)90458-K
- Ten Dijke, P., Korchynskyi, O., Valdimarsdottir, G. and Goumans, M.J. (2003) Controlling cell fate by bone morphogenetic protein receptors. Molecular and Cellular Endocrinology, 211, 105-113. doi:10.1016/j.mce.2003.09.016
- Liu, F., Akiyama, Y., Tai, S., Maruyama, K., Kawaguchi, Y., Muramatsu, K. and Yamaguchi, K. (2008) Changes in the expression of CD106, osteogenic genes, and transcription factors involved in the osteogenic differentiation of human bone marrow mesenchymal stem cells. Journal of Bone and Mineral Metabolism, 26, 312-320. doi:10.1007/s00774-007-0842-0
- Reyes, J.M., Fermanian, S., Yang, F., Zhou, S.Y., Herretes, S., Murphy, D.B., Chuck, R.S., et al. (2006) Metabolic changes in mesenchymal stem cells in osteogenic medium measured by autofluorescence spectroscopy. Stem Cells, 24, 1213-1217. doi:10.1634/stemcells.2004-0324
- Rice, W.L., Kaplan, D.L. and Georgakoudi, I. (2010) Two-photon microscopy for non-invasive, quantitative monitoring of stem cell differentiation. PLoS One, 5, e10075. doi:10.1371/journal.pone.0010075
- Kondo, M., Wagers, A.J., Manz, M.G., Prohaska, S.S., Scherer, D.C., Beilhack, G.F. and Weissman, I.L. (2003) Biology of hematopoietic stem cells and progenitors: Implications for clinical application. Annual Review of Immunology, 21, 759-806. doi:10.1146/annurev.immunol.21.120601.141007
- Romanov, Y.A., Darevskaya, A.N., Merzlikina, N.V. and Buravkova, L.B. (2005) Mesenchymal stem cells from human bone marrow and adipose tissue: Isolation, characterization, and differentiation potentialities. Bulletin of Experimental Biology and Medicine, 140, 138-143. doi:10.1007/s10517-005-0430-z
- Wagner, W., Wein, F., Seckinger, A., Frankhauser, M., Wirkner, U., Krause, U., Ho, A.D., et al. (2005) Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Experimental Hematology, 33, 1402-1416. doi:10.1016/j.exphem.2005.07.003
- Suva, D., Garavaglia, G., Menetrey, J., Chapuis, B., Hoffmeyer, P., Bernheim, L. and Kindler, V. (2004) Non-hematopoietic human bone marrow contains long-lasting, pluripotential mesenchymal stem cells. Journal of Cellular Physiology, 198, 110-118. doi:10.1002/jcp.10396
- Quirici, N., Soligo, D., Bossolasco, P., Servida, F., Lumini, C. and Deliliers, G.L. (2002) Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Experimental Hematology, 30, 783- 791. doi:10.1016/S0301-472X(02)00812-3
- Pittenger, M.F., Mackay, A.M., Beck, S.C., Jaiswal, R.K., Douglas, R., Mosca, J.D., Marshak, D.R., et al. (1999) Multilineage potential of adult human mesenchymal stem cells. Science, 284, 143-7. doi:10.1126/science.284.5411.143
- Pruitt, K.D., Tatusova, T., Klimke, W. and Maglott, D.R. (2009) NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Research, 37, D32- D36. doi:10.1093/nar/gkn721
- Consortium, T.U. (2011) Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Research, 39, D214-D219. doi:10.1093/nar/gkq1020
SUPPLEMENT
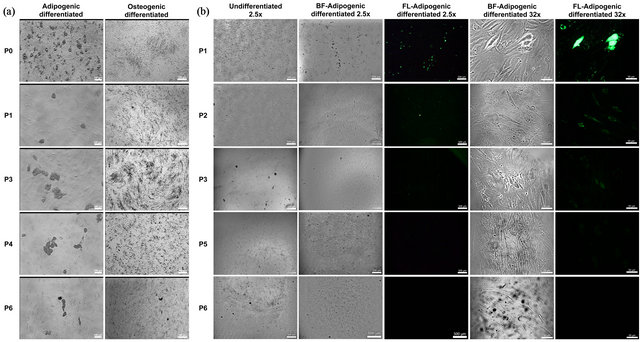
Figure S1. Microscopic images of MSC2 and ASC2 cells at each passage. (a) Rows are images of MSC2 cells from the same passage (P0 - P6) shortly before extraction for flow cytometric and qPCR analysis. Columns are images of adipogenic and osteogenic differentiated cells respectively. Scale bars in images of adipogenic cells are equal to 100 µm. In images of osteogenic cells, scale bars are equal 500 µm; (b) Rows are images of ASC2 cells from the same passage (P1 - P6) shortly before extraction for flow cytometric and qPCR analysis. Columns are either brightfield (BF) or 525 nm fluorescence (FL) images of undifferentiated and adipogenic differentiated cells measured at 2.5× and 32× magnification for a population overview or detailed cell morphology respectively. Scale bars in all 2.5× images are equal to 500 µm. In 32× images, scale bars are equal 50 µm.
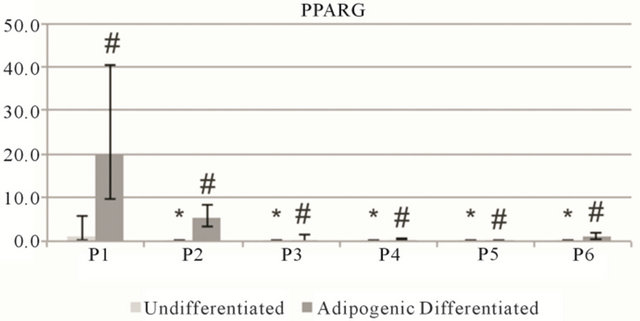
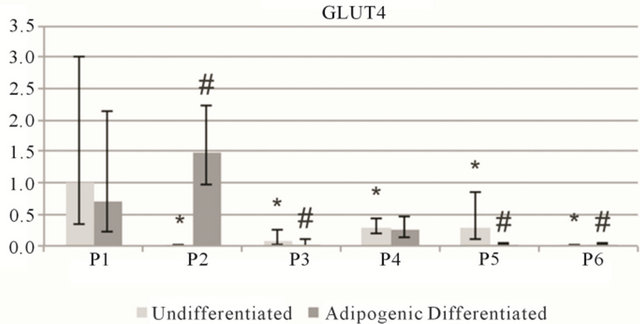
Figure S2. Relative gene expression of adipogenic markers on ASC2 cells. Perixosome proliferator-activated receptor γ (PPARG) and glucose transporter 4 (GLUT4). Data is grouped by passage number (P1 - P6) showing data from undifferentiated and adipogenic differentiated cells. Data points are mean relative expression ± standard deviation (n = 6). Marked samples are statistically different, *relative to undifferentiated P1 samples (P < 0.05), #relative to undifferentiated samples of same passage number (P < 0.05).
(a)
(b)

(c)
(d)
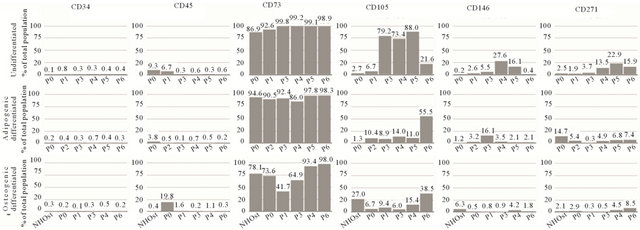
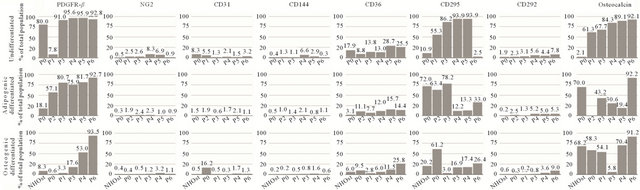
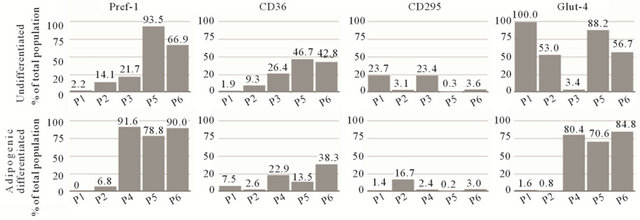
Figure S3. Flow cytometric analysis of MSC2 and NHOst as well as ASC2 cells. A minimum of 50,000 events was recorded for each sample. Data is given in percentages of total population of undifferentiated (top row), adipogenic differentiated and osteogenic differentiated cells. Data is ordered by passage number (P0 - P6) and grouped in each column by marker type. (a) Analysis of MSC2 and NHOst cells using MSC markers with CD34 and CD45 as common negative, CD73 and CD105 as common positive and CD146 and CD271 as novel positive MSC markers; (b) Analysis of MSC2 and NHOst cells using pericyte (PDGFR-β and NG2), endothelial cell (CD31 and CD144), adipocyte (CD36 and CD295) and osteoblast (CD292 and osteocalcin) markers; (c) Analysis of ASC2 cells using MSC markers with CD34 and CD45 as common negative, CD73, CD90 and CD105 as common positive and CD146 and CD271 as novel positive MSC markers; (d) Analysis of ASC2 cells using adipogenic cell markers.
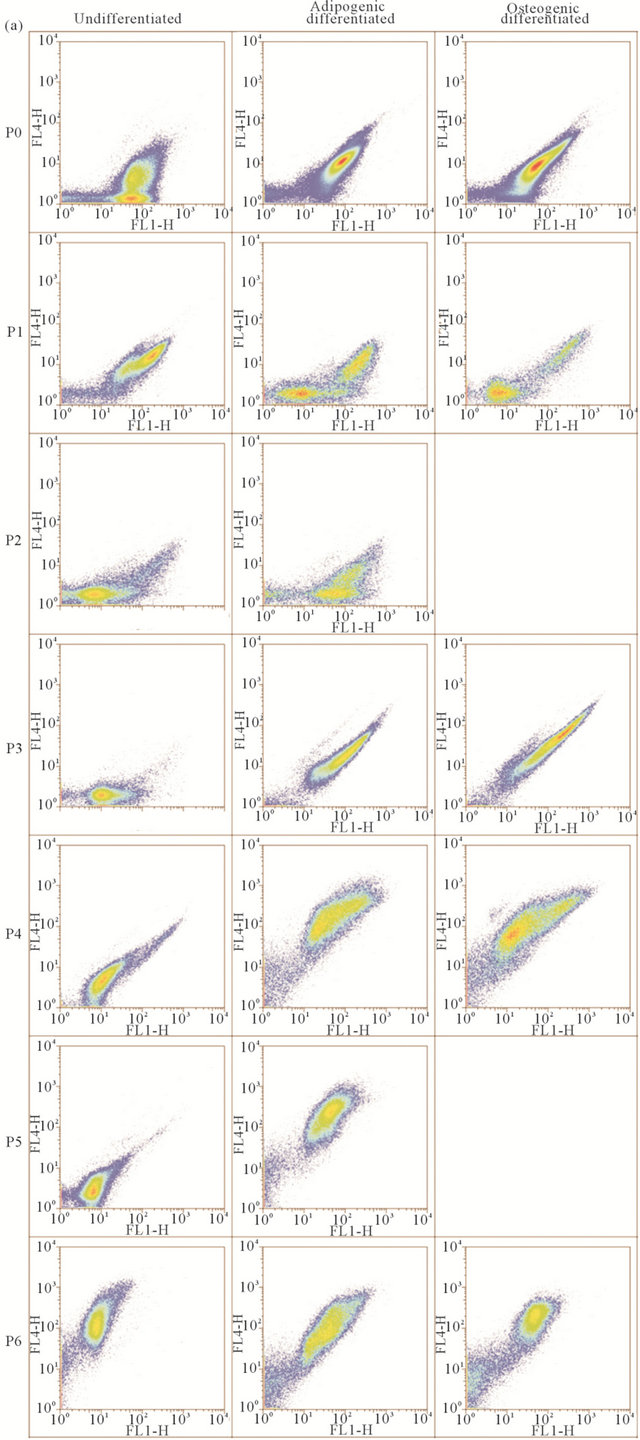
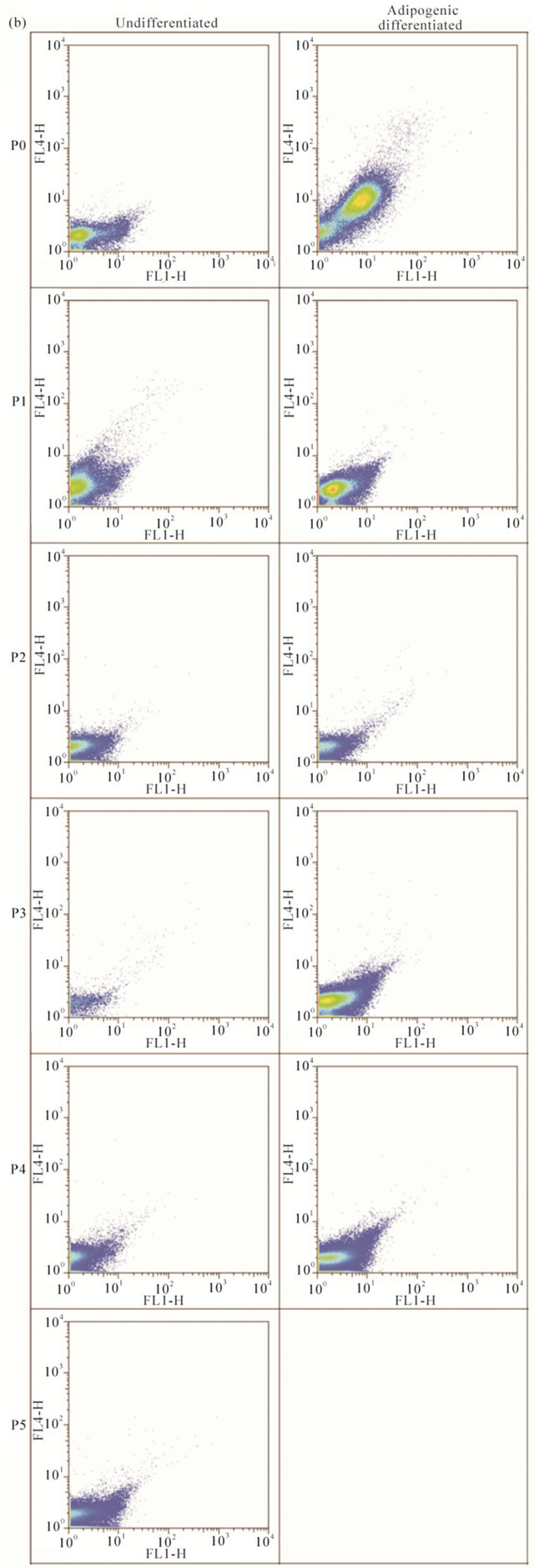
Figure S4. Autofluorescence of MSC2 and ASC2 cells. Rows are log-scale dot plots of cells from the same passage (P0 - P6), and columns list undifferentiated, adipogenic differentiated and osteogenic differentiated conditions respectively. X-axis: FL1-H (ex: 488 nm; em: 530/30 nm). Y-axis: FL4-H (ex: 635 nm; em: 661/16 nm). Autofluorescence of (a) MSC2 cells and (b) ASC2 cells.
Table S1. List of flow cytometry markers included in this study. Descriptions extracted from NCBI refSeq [43] and uniProt [44].

Table S2. Cell populations analyzed and cumulative time in culture before each passage in days. An additional 21 days should be added to determine the in vitro culture time of differentiated cells before analysis.

ABBREVIATIONS
APC—allophycocyanin bFGF—basic fibroblast growth factor BSP—bone sialoprotein CD—cluster of differentiation COL1A1—collagen type 1 DMEM—dulbecco’s modified eagle medium DNA—deoxyribonucleic acid EDTA—ethylenediamminetetraacetic acid FACS—fluorescence assisted cell sorting FBS—fetal bovine serum FITC—fluorescein isothiocyanate GAPDH—glyceraldehyde 3-phosphate dehydrogenase GLUT4—insulin-responsive glucose transporter 4 hASC—human adipose-derived stem cells hMSC—human mesenchymal stem cells IgG—immunoglobulin G NHOst—normal human osteoblasts PBS—phosphate buffered saline PE—phycoerythrin PerCP—peridinin chlorophyll protein qPCR—quantitative polymerase chain reaction RNA—ribonucleic acid RUNX2—runt-related transcription factor 2 SVF—stromal vascular fraction TCP—tissue culture plastic
NOTES
*This work was supported by NIH P41 Tissue Engineering Resource Center (P41 EB002520) and AFIRM.