International Journal of Organic Chemistry
Vol.06 No.01(2016), Article ID:64681,13 pages
10.4236/ijoc.2016.61004
Synthesis, Spectroscopy and X-Ray Characterization, of Novel Derivatives of Substituted 2-(Benzothiazol-2’-ylthio)acetohydrazide
Fatima Al-Omran*, Adel Abou El-Khair
Department of Chemistry, Faculty of Science, Kuwait University, Safat, Kuwait

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 13 January 2016; accepted 14 March 2016; published 17 March 2016
ABSTRACT
Treatment of 2-(benzo[d]thiazol-2’-ylthio)acetohydrazide (3) with acetylacetone afforded N-(4- oxopentan-2-ylidine) acetohydrazide derivative 5. The acetohyrazide derivatives 3 and 5 were utilized as a key intermediate for the synthesis of a novel heterocyclic compounds. The synthesis of a novel series of condensation and substituted derivatives of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide in good yield has been reported. The newly synthesized compounds were characterized by elemental analysis 1H-NMR, 13C-NMR spectra and X-ray crystallographic investigations. The reported crystal structures of these novel 2-(benzo[d] thiazol-2’-ylthio)acetohydrazide derivatives are expected to be a remarkable contribution to the crystallographic database of heterocyclic compounds.
Keywords:
Acetohydrazide, Benzothiazole, Pyrazole, 1,2,4-Triazolopyridine, N’-Acetylcinnamohydrazide

1. Introduction
Benzothiazole and its derivatives represent one of the most biologically active classes of compounds, displaying a remarkable diversity of bioactivities in the medical and the agrochemicals field [1] - [3] . Recently, several publications reported that hydrazide-hydrazones are valuable intermediates in the synthesis of heterocyclic compounds with potential pharmaceutical and biological activities [4] [5] . Based on those reports and in continuation of our interest in the synthesis of heterocycles containing a 2-(benzo[d]thiazol-2’-ylthio) moiety [6] - [8] , we reported herein the results of our study of the behavior the hitherto unreported of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide (3) towards a variety of reagents in order to synthesis of a novel 2,4-dihydropyrazole, 1,2,4-triazolopyri- dine, N’-acetyl cinnamohydrazide and 1,2,4-triazolo-3-(4H)thione bearing 2-(benzo[d]thiazol-2’-ylthio) in the hopes of obtaining compounds that have biological and pharmacological applications.
2. Results and Discussion
2.1. Synthesis
The 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide (3) proved to be a useful key intermediate in the synthesis of several heterocyclic nuclei [3] . Therefore, 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide (3) was prepared as previously described in the literature via treatment of 2-MBT (1) with ethyl chloroacetate yielded 2. The latter compound reacted with hydrazine hydrate afforded 3 in good yield [3] [9] . The structure of 3 was confirmed on the basis of elemental analysis and spectral information. Treatment of 3 with acetylacetone in refluxing ethanol, afforded N-(4-oxopentan-2-ylidene)acetohydrazide derivative 5 and not the pyrazole derivative 4 as it reported in Literature [9] . The structure of 5 is confirmed by elemental analysis and spectral data. The mass spectrum showed the peak of the molecular ion at m/z 321[M+, 10.0%] corresponding to molecular formula C14H15N3O2S2 (cf. Scheme 1). The 1H NMR spectrum of 5 revealed, in addition to the aromatic benzothiazole protons, two doublets at δ H 2.97 and 2.84 ppm., with J = 16 Hz, as required for the methylene protons of H-3 indicating that
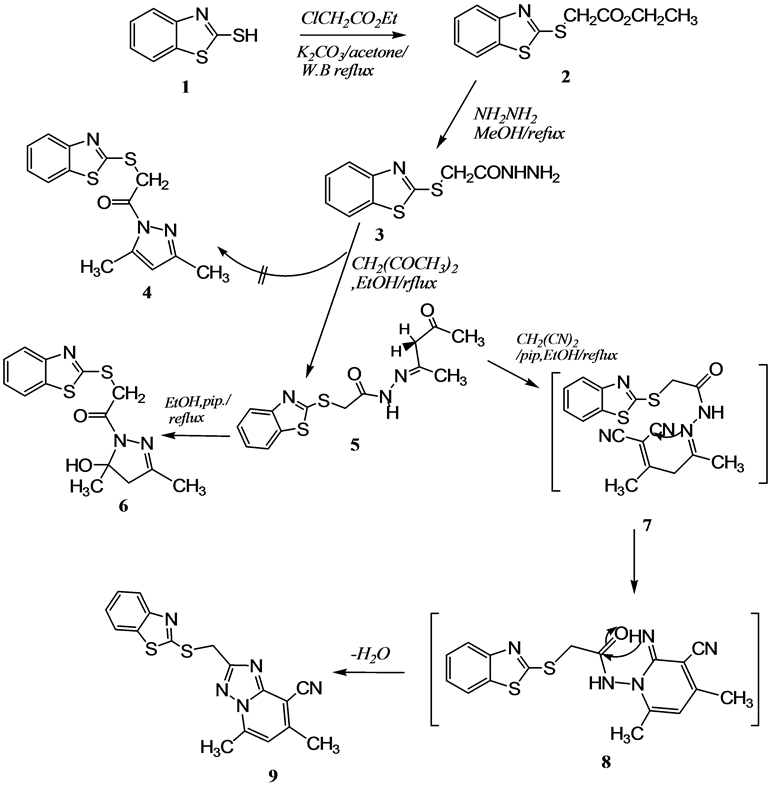
Scheme 1. Synthesis route of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide and their derivatives.
these protons are in a different environment. Likewise, the 1H NMR spectrum exhibited two singlet signals at δH 4.58 and 10.70 ppm for -SCH2-protons and NH group respectively. The assignments of 1H and 13C chemical shifts of compound 5 were registered in the Experimental Section. On the other hand, when the compound 5 heated in ethanol, containing a catalytic amount of piperidine, afforded 5-hydroxy-4, 5-dihydropyrazole derivative 6 in good yield. The mass spectrum of the obtained product revealed a molecular ion peak [M+] at m/z 321[15%] and it is compatible with the molecular formula C14H15N3O2S2. It is worth mentioning that both compounds 5 and compound 6 have the same molecular formula, but has different melting points and spectral data (cf. Scheme 1). The complete assignments of 1H and 13C chemical shift for 6 based on COSY and HSQC were recorded in the Experimental Section. The structure of compound 6 was established based on X-ray crystallographic analysis, the pyrazole ring containing the chiral center (at C 10) and the benzothiazole moiety has located almost the same plane (cf. Figure 1 and Table 1) [10] .
Moreover, treatment of acetohydrazide derivative 5 with malononitrile in refluxing ethanolic piperidine afforded the [1,2,4]-triazolo[1,5-a]pyridine derivative 9 in good yield via the intermediary 7-8 (cf. Scheme 1).
Analytical and spectroscopic data confirmed the structure of compound 9. The mass spectrum of 9 revealed the molecular ion peak (which is the base peak as well) at m/z value 351 [M+, 100%]. The complete assignments of the 1H and 13C chemical shift of the compound were recorded in the Experimental Section. It is worth noting that recently, [1,2,4]-triazolopyridine derivatives have been shown to be useful antifungal activities compare with the commercial pesticide [11] .
On the other hand, treatment of acetohydrazide derivative 3 with 2,3-butanedione in refluxing ethanolic piperidine afforded the acetohydrazide derivative 10 in good yield (cf. Scheme 2). The analytical and spectral data of the last reaction product are entirely consistent with the proposed structure. The complete assignments of 1H and 13C chemical shifts of compound 10 based on COSY and HSQC experiments were recorded in the Experimental Section. Moreover, Nuclear Overhauser Effect (NOE) experiments were carried out to establish the configuration of acetohydrazide derivatives 10 whether it is (E) or (Z)isomers. Therefore, irradiation of a signal of the NH proton at δH 11.40 ppm,we observed increasing a signal of the methyl protons at δH 1.95 ppm. While, irradiation of a methyl signal at δH 1.95 ppm, increased the a NH signal at δH 11.40 ppm and have no effect on an acetyl signal at δH 2.41 ppm. Indicate the 2-(benzo[d]thiazol-2’-ylthio)acetamide moiety and the methyl group

Figure 1. A general view and atom labeling of X-rays structure of 6.

Table 1. Selected bond lengths (˚A) & bond angles for compound 6.

Scheme 2. Reactions of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide with carbonyl compounds and phthalic anhydride.
are on opposite sides of the C=N, as required by an (E)-form. The crystal structure of 10 [12] depicted in Figure 2, besides to the selected bond lengths and bond angles was registered in Table 2. It observed from crystallographic image for the compound 10 that two molecules of 10 were shown in one asymmetric unit of these crystals; they are chemically same, only different in crystallographic point of view, due to nonequivalence in symmetry characteristics within the unit cell. In an attempt to obtain pyrolo[1,2-b][1,2,4]triazole derivative 11, the compound 10 treated with malononitrile in refluxing ethanolic piperidine; it was found to be abortive.
Moreover, treatment of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide 3 with phthalic anhydride in refluxing acetic acid, afforded N-(1,3-dioxoisoindolin-2-yl)acetamide derivative 13 in good yield and not 2,3-dihy- drophthalazine-1,4-dione derivative 12 as it reported in the literature [9] (Scheme 2). Complete assignments of 1H and 13C chemical shifts of compound 13 based on COSY and HSQC experiments were recorded in the Experimental Section. The crystal structure of 13 [13] depicted in Figure 3, beside to the selected bond lengths and bond angles was registered in Table 3. The crystal networks contain dioxane molecule as the space-filling solvent (crystalline solution). The phthalimide fraction is oriented nearly perpendicular to the plane of benzothiazole fraction (Figure 3). In an attempt to convert compound 13 to 1-aminoethylidine malononitrile derivatives 14 by treatment with malononitrile in refluxing ethanolic piperidine were ineffective.
On the other hand, condensation of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide (3) with 1,3-cyclohexadione yielded N’-(3-oxocylohex-1-enyl) acetohydrazide hydrate derivative (15) as depicted in Scheme 2. The structure

Figure 2. A general view and atom labeling of X-rays structure of 10.

Figure 3. A general view and atom labeling of X-rays structure of 13.
Table 2. Selected bond lengths (˚A) & bond angles for compound 10.
Table 3. Selected bond lengths (˚A) & bond angles for compound 13.
of this acetohydrazide hydrate derivative (15) deduced from its elemental analysis and spectroscopic data. The mass spectrum revealed a molecular ion peak at m/z 333[M+ -H2O], which corresponds to a molecular weight consistent with a formula of C15H15N3O2S2 (cf. Experimental Section). Furthermore, the X-ray crystallography provided an unambiguous evidence of structure 15 as proposed in Scheme 2 [14] given in Figure 4.
The single crystal of 15 also contains water molecules in its network as space filling solvent. Based on bond length (Table 4) it is indicating the presence of keto-enol transformation from N3 to O2 through C10 and C11 as follows: the bond length between O1-C9 is 1.214 (6). While bond length between O2-C12 is 1.249 (6); elongated a little due to partial enol-form. Also, bond length between N2-C9 is 1.346 (6) while bond length between N3-C10 is 1.318 (7); reduced due to the partial double bond character. Besides, the two bond length between C10-C11 and C11-C12 are same [1.426 (6) and 1.422 (8) respectively] due to resonance. The inter conversion of the two forms involves the movement of the lone pair electrons on the N3 to the most electronegative atom (cf. Figure 5).

Figure 4. A general view and atom labeling of X-rays structure of 15.

Figure 5. The keto-enol tautomeric forms of compound 15.
Table 4. Selected bond lengths (˚A) & bond angles for compound 15.
In continuation of our efforts to generate new routes to the different functional thiophenes [6] , we have used Gewald’s reaction [6] by treatment of compound 15 with malononitrile in refluxing ethanol containing an elemental sulfur and a catalytic amount of piperidine. Nevertheless, this procedure was found to be unsuccessful to obtain the expected 2-amino-3-cyanothiophene derivative 16. Moreover, condensation of acetohydrazide derivative 3 with thiophene-2-carbaldehyde afforded a mixture of both configurations E/Z acetohydrazide derivative 17 in a ratio of 3:2 respectively, based on the 1H-NMR spectrum. The 1H-NMR spectrum, revealed, in addition to the expected aromatic signals, four downfield signals, two corresponding to the N=CH at δH 8.25 and 8.47 ppm. The other two signals corresponding to NH at δH 11.74 and 11.82 ppm. These protons assigned to the E and Z-configuration, respectively. The latter two signals underwent facile hydrogen, deuterium exchange upon addition of deuterium oxide. Also, the 1H-NMR spectrum exhibited two singlet signals for methylene protons of two isomer E and Z-configuration at δH 4.63 and 4.23 ppm respectively. In an attempt to synthesize pyrazole derivative 18 by a one-pot reaction of acetohydrazide derivative 3 with thiophene-2-carbaldehyde in the refluxing methanol, followed by treatment with malononitrile, however, it was unsuccessful (cf. Scheme 2).
On the other hand, treatment of acetohydrazide derivative 3 with triethyl orthoformate, in refluxing acetic anhydride afforded N’-(acetyl) acetohydrazide derivative 19. In a similar manner, upon treatment of acetohydrazide derivative 3 with chloroacetyl chloride in dimethylformamide (DMF), afforded the N’-(2-chloroacetyl)- acetohydrazide derivative 20 in good yield. The structure of 20 also confirmed by its spectral data. It should remark at this point that many publications have recently reported the broad spectrum of biological usefulness exhibited by N, N-diacylhydrazine derivatives such as anti-HIV, herbicidal and antifungal activities [15] . In contrast to the above-observed chemistry, the acetohydrazide derivative also reacted with cinnamoyl chloride in 1,4-dioxane and the presence of triethylamine afforded of E-configuration of N’-(acetyl cinnamohydrazide derivative 21. Treatment of acetohydrazide derivative 3 with phenyl isothiocyanate in refluxing ethanolic sodium hydroxide solution, afforded the [1,2,4]-triazole-3(4H)-thione derivative 22. The reaction of acetohydrazide derivative 3 with dimethyl acetylene dicarboxylate (DMAD) provided a mixture of both configuration (E/Z ) acetyl hydrazono succinate derivative (23) in a ratio of 3:1 respectively (based on the 1H-NMR spectrum). The chemical shift of protons 23 was assigned using COSY and HSQC measurements were recorded in the Experimental Section (cf. Scheme 3).

Scheme 3. Reactions of compound 3 with HC(OEt)3/Ac2O, ClCH2COCl, cinnamoly chloride, PhNCS and DMAD.
Moreover, the 1H-NMR spectrum showed in addition to the expected aromatic signals, two downfield signals at δH 11.74 and 11.44 ppm and two upfield signals at δH 4.71 and 4.45 ppm assignable to the E and Z-configu- ration for NH and methylene protons respectively. The crystal structure, bond lengths and bond angles data of 23 [16] depicted in Figure 6 and Table 5. From single crystal X-ray crystallography of dimethyl 2-[2-(- 2(benzo[d]thiazol-2’-ylthio)acetyl)hydrazono] succinate (23), it is indicated that the two acetate groups are present as tail units of the molecule. Out of which, one of the acetate group is oriented in the molecular plane of the benzothiazole, while the other acetate tail is situated perpendicular to this plane. Furthermore, the distortionless enhancement by polarization transfer (DEPT) experiment was too utilized for separate the carbon signals for the compound 23. The DEPT spectrum of the aforementioned compound showed four positive signals δ126.8, 125.0, 122.3 and 121.6 for the aromatic benzothiazole carbon (CH) and two positive signals δ53.1 and 52.6 for methyl carbons , while two negative signals at δ35.8 and 32.6 for the methylene carbon. Also an attempt to prepare the dihydropyridine 3,6-dione 24 via condensation of 23 with hydrazine hydrate was unsuccessful and the product identified as 2-(benzo[d]thiazol-2'-ylthio) acetohydrazide 3 (cf. Scheme 3).
3. Experimental Section
3.1. General Procedures
Melting points are reported uncorrected and determined on a Gallenkamp apparatus. The Infrared spectra recorded on a Jasco FT/IR-6300 FT-IR uses KBr disks. 1H-NMR and 13C-NMR spectra were measured on a Bruker DPX 400 MHz and Bruker AVANCE ΙΙ 600 MHz spectrometers, with DMSO-d6 or CDCl3 as solvent using TMS as an internal standard. The methods used for the purpose of NMR assignment were COSY, HSQC, DEPT and NOE. The chemical shifts expressed as a δ unit in parts per million (ppm) and TMS = 0.00 ppm. The following abbreviation is used: s = singlet, d = doublet, t = triplet; q = quartet; m = multiple; br. = broad. Mass spectra measured on GC/MS DFS, THERMO instrument. Microanalyses performed on a CHNS-Vario.
Micro Cube Analyzer; Single crystal X-ray crystallography was performed using Rigaku Rapid ΙΙ and Bruker X8 Prospector diffractometers. The starting materials ethyl-2-(benzo[d]thiazol-2’-ylthio) acetate 2, mp 59˚C (lit.

Figure 6. A general view and atom labeling of X-rays structure of 23.
Table 5. Selected bond lengths (˚A) & bond angles for compound 23.
mp. 58˚C) [3] and 2-(benzo[d]thiazol-2’-ylthio)acetohydrazide (3), mp. 192˚C - 194˚C (lit mp. 193˚C) [3] were prepared according to the indicated literature method.
3.2. Procedures for the Synthesis of 2-(Benzothiazol-2’-ylthio)acetohydrazide Derivatives
3.2.1. 2-(Benzo[d]thiazol-2’-ylthio)-N’-(4-oxopentan-2-ylidene)acetohydrazide (5)
A mixture of compound 3 (2.39 gm 10.0 mmol) and 2,4-pentanedione (1.0 g, 10 mmol) in ethanol (20 mL) was refluxed for 3 h. The reaction was allowed to cool to room temperature for 24 h. The solid product formed was collected by filtration and crystallized from ethanol as white crystal. Yield: 2.40 g (75%); mp 182˚C - 184˚C; FT-IR vmax (cm−1): 3186 - 3568 (br, NH), 1690 (C=O ketone),1670(C=O amide); 1H NMR (DMSO-d6) δ ppm: 10.7 (s, 1H, NH, D2O exchangeable ), 8.02 (d, J = 8 Hz, 1H, H-4’), 7.85 (d, J = 8 Hz, 1H, H-7’), 7.46 (t, J = 8 Hz, 1H, H-6’), 7.36 (t, J = 8 Hz, 1H, H-5’), 4.58 (s, 2H, -SCH2), 2.97 (d, J = 16, 1H, H-3), 2.84 (d, J = 16, 1H, H-3a), 2.01 (s,3H, CH3), 1.77 (s, 3 H, CH3); 13C NMR (DMSO-d6) δ ppm: 168.1 (C=O), 166.4 (C=O), 165.1(C-2’), 155.9 & 152.6 (C-2 &C-3a’), 135.1 (C-7a’), 126.4 (C-6’), 124.6 (C-5’), 121.9 (C-4’), 121.2 (C-7’), 52.2 ( CH2), 37.9 (CH2), 23.0 (CH3), 14.1 (CH3). EI-MS m/z [relative intensity]: 321 [M+, 10], 264 [15%], 208 [100%], 180 [62%]. Anal. Calcdfor C14H15N3O2S2 (321.42): C, 52.32; H, 4.70; N, 13.07%. Found: C, 52.16; H, 4.53; N, 13.39%.
3.2.2. 2-(Benzo[d]thiazol-2’-ylthio)-1-(5-hydroxy-3,5-dimethyl-4,5-dihydropyrazole-1-yl) ethanone (6)
A solution of 5 (3.21 g, 10 mmol ) in ethanol (20 mL) containing a few drops of piperidine was refluxed for 3 h. The reaction was allowed to cool to room temperature for 24 h. The solid product formed, was collected by filtration and crystallized from ethanol as yellow crystal. Yield: 2.34 g (73%); mp 123˚C - 125˚C; FT-IR vmax (cm−1): 3090 - 3998 (br, OH), 1670(C=O); 1H NMR (DMSO-d6) δ ppm: 7.90 (d, J = 8 Hz, 1H, H-4’), 7.78 (d, J = 8 Hz, 1H, H-7’), 7.47 (t, J = 8 Hz, 1H, H-6’), 7.31 (t, J = 8 Hz, 1H, H-5’), 6.55 (s, 1H, OH, D2O exchangeable), 4.58 (dd, J = 20, 2H, SCH2), 2.98 (d, J = 16, 1H, H-4 pyrazole), 2.82 (d, J = 16, 1H, H-4’pyrazole), 2.01 (s, 3H, CH3), 1.76 (s, 3 H, CH3); 13C NMR ( DMSO-d6) δ ppm: 166.5 (C=O), 163.7 (C-2’), 155.8 (C-3), 152.7 (C-3a’), 134.7 (C-7a’), 126.4 (C-6’), 124.4 (C-5’), 121.8 (C-4’), 121.1 (C-7’), 90.7 (C-5 pyrazole), 52.2 (C-4pyrazole), 37.9 (SCH2),), 25.9 (CH3), 15.9 (CH3). EI-MS m/z [relative intensity]: 321 [M+, 15%], 264 [18%], 230 [3%], 208 [100%], 180 [62%]. Anal. Calcdfor C14H15N3O2S2 (321.42): C, 52.32; H, 4.70; N, 13.07%. Found: C, 52.21; H, 4.55; N, 13.08%.
3.2.3. 2-[(Benzo[d]thiazol-2’-ylthio)methy]-5,7-dimethyl-[1,2,4]triazole-[1,5-a] pyridine-8-carbonitrile (9)
A mixture of 5 (3.21 g, 10.0 mmol) and malononitrile (0.66 g, 10 mmol) in ethanol (20 mL) containing a few drops of piperidine was refluxed for 4 h. The reaction was allowed to cool to room temperature. The solid product formed, was collected by filtration and crystallized from ethanol as brown crystals. Yield: 2.56 g (73%); mp 258˚C - 260˚C; FT-IR vmax (cm−1): 2227 (CN); 1H-NMR (DMSO-d6) δ ppm: 7.94 (d, J = 8 Hz, 1H, H-4’), 7.84 (d, J = 8 Hz, 1H, H-7’), 7.47 (t, J = 8 Hz, 1H, H-6’), 7.36 (t, J = 8 Hz, 1H, H-5’), 7.10 (s,1H, 6-H pyridine), 4.18 (s,2H, SCH2), 2.29 (s,, 3 H, CH3), 2.21 (s, 3 H, CH3); 13C NMR (DMSO-d6) δ ppm: 163.4, 160.7, 153.1, 151.6, 149.5, 143.5, 135.5, 126.8, 125.1, 122.2, 121.8, 116.3, 114.4, 107.8 (arom. Carbons & CN), 30.7 (SCH2), 20.0 (CH3), 17.9 (CH3). EI-MS m/z (relative intensity): 351 [M+, 100%], 318 [23%], 274 [2%], 236 [4%]. Anal. Calcd. for C17H13N5S2 (351.45): C, 58.10; H, 3.73; N, 19.93; Found: C, 58.18; H, 3.83; N, 19.73.
3.2.4. (E) 2-[Benzo[d]thiazol-2’-ylthio]-N’-(3-oxobutan-2-ylidine)acetohydrazide (10)
A mixture 3 (2.39 g, 10.0 mmol) and 2,3-butanedione (0.87 g, 10.0 mmol) in ethanol (20 mL) containing a few drops of piperidinewas refluxed for 1 h. The reaction mixture was allowed to cool to room temperature. The product formed was collected by filtration and recrystallized from ethanol as yellow crystals. Yield: 2.64 g (86%); mp 198˚C - 199˚C. FT-IR vmax (cm−1): 3188 - 3448 (br, NH), 1680 (C=O ketone), 1612 (C=O amide); 1H NMR (DMSO-d6) δ ppm: 11.40 (s, 1H, NH, D2O exchangeable), 8.02 (d, J = 8 Hz, 1H, H-4’) 7.81 (d, J = 8 Hz, 1H, H-7’), 7.46 (t, J = 8 Hz, 1H, H-6’), 7.36 (t, J = 8Hz, 1H, H-5’), 4.75 (s, 2H, SCH2), 2.41(s, 3H, COCH3), 1.95 (s, 3H, CH3); 13C NMR (DMSO-d6) δ ppm: 196.9 (C=O ketone), 169.8 (C=O amide), 166.0 (C-2’), 152.4 (C-3a’), 147.4 (C-2), 134.7 (C-7a’), 126.3 (C-6’), 124.6 (C-5’), 121.9 (C-4’), 121.1 (C-7’), 35.3 (SCH2), 24.3 (COCH3), 9.8 (CH3). EI-MS m/z [relative intensity]: 307 [M+, 10%], 264 [100%], 231 [13%], 208 [11%]. Anal. Calcd. for C13H13N3O2S2 (307.39): C, 50.79; H, 4.26; N, 13.67%. Found: C, 50.53; H, 4.39; N, 13.45%.
3.2.5. 2-(Benzo[d]thiazol-2’-ylthio)-N’-(1,3-dioxoisoindolin-2-yl)acetamide (13)
A mixture of 3 (2.39 g 10.0 mmol) and phthalic anhydride (1.48 g, 10 mmol ) in acetic acid (10 mL) was heated under refluxed for 2 - 3 h. The solid product, so formed, was collected by filtration and recrystallized from 1,4-dioxane as white crystal solid. Yield: 2.69 mg (73%); mp 176˚C - 178˚C; FT-IR vmax (cm−1): 3215 (br, NH), 1793 & 1743 (2C=O),1662 (C=O amide); 1H NMR (DMSO-d6) δ ppm : 11.22 (s, 1H, NH, D2O exchangeable), 8.01 (d, J =8 Hz, 1H, H-4’) ,7.94 (d, J = 7.4 Hz, 2H, H-4 & H-7), 7.93 (d, J = 8 Hz, 1H, H-7’), 7.91 (t, J = 7.4 Hz, 2H, H-5 & H-6), 7.48 (t, J = 8 Hz, 1H, H-6’), 7.37 (t, J = 8 Hz, 1H, H-5’), 4.49 (s, 2H, SCH2); 13C NMR (DMSO-d6) δ ppm: 166.5 (2C=O amide ) 165.3 (C=O amide ), 164.9 (C-2’), 152.5 (C-3a’), 135.2 (C-5 & C6), 134.9 (C-7a’), 129.4 (C-4a & C-7a), 126.4 (C-6’), 124.5 (C-5’), 123.7 (C-4 & C-7), 121.8 (C-4’), 121.3 (C-7’), 34.3 (SCH2). EI-MS m/z [relative intensity]: 369 [M+, 295 [11%], 250 [9%], 208 [100%]. Anal. Calcd. for C17H11N3O3S2 (369.42): C, 55.27; H, 3.00; N, 11.37%. Found: C, 54.90; H, 3.00; N, 11.22%.
3.2.6. 2-(Benzo[d]thiazol-2’-ylthio)-N’-(3-oxocylohex-1-enyl)acetohydrazide hydrate (15)
A mixture of 3 (2.39 g 10.0 mmol) and 1,3-cyclohexanedione (1.12 g, 10.0 mmol) in ethanol (20 mL) was refluxed for 2 - 3 h. The reaction mixture was allowed to cool to room temperature. The product formed was collected by filtration and recrystallized from ethanol as yellow crystals. Yield: 2.63 g (79%); mp 185˚C - 187˚C; FT-IR vmax (cm−1):3433 (OH), 3198 (NH), 1695(C=O), 1657 (C=O); NMR (DMSO-d6) δ ppm: 10.29 (s, 1H, NH, D2O exchangeable), 8.96 (s, 1H, NH, D2O exchangeable), 8.02 (d, J = 8 Hz, 1H, H-4’), 7.96 (d, J = 8 Hz, 1H, H-7’), 7.47 (t, J = 8 Hz, 1H, H-6’), 7.37 (t, J = 8 Hz, 1H, H-5’), 5.15 (s, 1H, H-2) 4.23 (s, 2H, SCH2), 2. 32 (t, J = 8, 2 H, H-4), 2.11 (t, J = 8, 2 H, H-6) 1.8 (t , J = 8, 2H, H-5); 13C NMR (DMSO-d6) δ ppm: 196.2 (C=O ketone), 170.5 (C=O amide), 166.0 (C-2), 164.6 (C-1), 152 (C-3a’), 135.1 (C-7a’), 126.0 (C-6’), 124.0 (C-5’), 122.1 (C-4’), 121 (C-7’), 99.7 (C-2), 37.1 (SCH2), 26.0 (C-4), 22.1 (C-6), 19.2 (C-5). EI-MS m/z [relative intensity]: 333 [M+-H2O, 10%], 315 [7%], 223 [3%], 208 [47%], 180 [56%]. Anal.Calcd.for C15H17N3O3S2 (351.34): C, 51.26; H, 4.88; N, 11.92%. Found: C, 51.08; H, 4.74; N, 11.92%.
3.2.7. (E)-2-[Benzo[d]thiazol-2’-ylthio]-N’-(2-thiophene-2-yl-methylene)acetohydrazide (17)
A mixture of 3 (2.39 g, 10.0 mmol), and thiophene-2-carbaldehyde (1.12 g, 10.0 mmol) in methanol (20 mL) was refluxed for 1 h. The reaction mixture was allowed to cool to room temperature. The product formed was collected by filtration and crystallized from ethanol as yellow crystals. Yield: 2.53 g (76%); mp 134˚C - 136˚C; FT-IR vmax (cm−1): 3295 (NH); 1658 (CO); 1H-NMR (DMSO-d6) δ ppm: 11.74 (s, 1H, D2O exchangeable, NH), 8.25 (s, 1 H, HC=N), 7.10 - 8.00 (m, 7H, Ar-H), 4.63 (s, 2H, SCH2); 13C NMR (DMSO-d6) δ ppm: 167.9 (C=O), 165.3 (C-2’), 152.5 (-3a’), 142.4, 139.1, 134.8, 131.3, 130.7, 128.7, 127.8, 126.3, 124.4, 121.8 (arom. & HC=N), 35.8 (SCH2). EI-MS m/z [relative intensity]: 333 [M+, 12%], 259 [4%], 223 [3%], 208 [100%], 180 [71%]. Anal. Calcd. for C14H11N3OS3 (333.38): C, 50.43; H, 3.33; N, 12.60%. Found: C, 50.33; H, 3.27; N, 12.61%.
3.2.8. 2-(Benzo[d]thiazol-2’-ylthio)-N’-(acetyl)acetohydrazide (19)
A mixture of 3 (2.39 g 10.0 mmol) and triethyl orthoformate (1.6 g, 10.0 mmol) in acetic anhydride (20 mL) was refluxed for 2 h. The reaction was allowed to cool to room temperature, then poured in ice cold water. The solid product formed was collected by filtration and crystallized from ethanol as white crystals. Yield: 1.9 g (71%); mp 202˚C - 204˚C; FT-IR vmax (cm−1): 3205 (2NH), 1685, 1641 (2C=O amide);1H-NMR (DMSO-d6) δ ppm: 10.28 (s, 1H, NH, D2O exchangeable), 9.99 (s, 1H, NH, D2O exchangeable), 8.02 (d, J = 8 Hz, 1H, H-4’), 7.85 (d, J = 8 Hz, 1H, H-7’), 7.48 (t, J = 8 Hz, 1H, H-6’), 7.38 (t, J = 8Hz, 1H, H-5’), 4.23 (s, 2H, SCH2), 1.92 (s, 3H, CH3); 13C-NMR (DMSO-d6): δC 167.9 (C=O), 165.9 (C=O), 165.4 (C-2’), 152.5 (C-3a’), 134.8 (C-7a’), 126.4 (C-6’), 124.6 (C-5’), 121.9 (C-4’), 121.2 (C-7’), 34.7 (SCH2), 20.5 (CH3). EI-MS m/z [relative intensity]: 281 [M+, 12%], 236 [4%], 208 100%], 180 [37%]. Anal. Calcd. for C11H11N3O2S2 (281.35): C, 46.96; H, 3.94; N, 14.93%. Found: C, 46.81; H, 3.95; N, 14.92%.
3.2.9. 2-(Benzo[d]thiazol-2’-ylthio-)-N’-(2-chloroacetyl)acetohydrazide (20)
A mixture 3 (2.39 g, 10.0 mmol) and chloroacetyl chloride (1.12 g, 10.0 mmol) in DMF (20 mL) was heated for 2 h. The mixture was allowed to cool to room temperature. The solid product formed was collected by Filtration and crystallized from ethanol as yellow crystals. Yield: 2.28 g (82%); mp 162˚C - 164˚C; FT-IR vmax (cm−1): 3174 (2NH), 1659 (2C=O amide); 1H-NMR (DMSO-d6) δ ppm: 10.56 (s, 1H, NH, D2O exchangeable) 10.52 (s, 1H,NH, D2O exchangeable), 8.03 - 7.35 (m, 4H, ArH), 4.52 (s, 2H, CH2), 4.26 (s, 2H, CH2); 13C-NMR (DMSO- d6) δC ppm: 165.7 (C=O), 165.3 (C=O), 162.3 (C-2’), 152.5 (C-3a’), 135.8 (C-7a’), 126.4 (C-6’), 124.6 (C-5’), 121.8 (C-4’), 121.2 (C-7’), 40.8 (CH2Cl), 34.6 (SCH2). EI-MS m/z [relative intensity]: 316 [M+, 13%], 279 [2%], 241 [2%], 208 [100%]. Anal. Calcd. for C11H10ClN3O2S2 (315.80): C, 41.84; H, 3.19; N, 13.31%. Found: C, 41.72; H, 3.23; N, 13.43%.
3.2.10. N’[-2-(Benzo[d]thiazol-2’-ylthio)acetyl]cinnamohydrazide (21)
A mixture of 3 (2.39 g 10.0 mmol) and cinnamoyl chloride (1.66 g, 10.0 mmol) in 1,4 dioxane (20 mL) containing a few drops of triethylamine was refluxed for 2 h .The solid product formed, was collected by filtration and crystallized from ethanol as white crystals. Yield: 2.80 g (76%); mp 164˚C - 166˚C; FT-IR vmax (cm−1): 3207 (2NH), 1672 (C=O), 1641 (CO-amide); 1H-NMR (DMSO-d6) δH: 10.60 (s, 1H,NH, D2O exchangeable), 10.39 (s, 1H, NH, D2O exchangeable), 8.03 (d, J = 8 Hz, 1H, H-4’), 7.87 (d, J = 8 Hz, 1H, H-7’), 7.88 - 7.37 (m, 8H, Ar-H & vinylic-H), 6.70 (d, 1H, J = 16, vinylic-H), 4.28 (s, 2H, SCH2); 13C-NMR (DMSO-d6) δC ppm: 165.8 & 165.0 (2C=O), 163.3, 152.5, 140.3, 134.8, 134.5, 129.8, 129.0, 127.7, 126.4, 124.6, 121.8, 121.2, 119.2 (arom. carbons) 34.7 (CH2). EI-MS m/z [relative intensity]: 369 [M+, 7%], 208 [51%] 180 [9%], 148 [3%], 131 [100%]. Anal. Calcd. for C18H15N3O2S2 (369.46): C, 58.52; H, 4.09; N, 11.37%. Found: C, 58.32; H, 4.09; N, 11.39%.
3.2.11. 5-[(Benzo[d]thiazol-2’-ylthio)methyl]-4-phenyl-2H-1,2,4-triazole-3(4H)-thione (22)
A mixture of 3 (2.39 g 10.0 mmol) and phenyl isothiocyanate (1.99 g, 10.0 mmol) in ethanol (20 mL) and sodium hydroxide solution (2 g in 20 mL water) was refluxed for 2 h. The reaction mixture was allowed to cool to room temperature then acidify with 10% HCl. The product formed was collected by filtration and crystallized from ethanol as yellow crystals. Yield: 2.99 g (84%); mp 140˚C - 142˚C; FT-IR vmax (cm−1): 3061(NH); 1H-NMR (DMSO-d6) δH ppm: 7.17 - 7.95 (m, 10H, ArH & NH), 4.54 (s, 2H, SCH2). EI-MS [relative intensity]: 356 [M+, 71%], 323 [8%] 265 [3%], 224 [7%].190 [12%]. Anal. Calcd. for C16H12N4S3 (356.46): C 53.91; H, 3.39; N, 15.72%. Found: C, 53.97; H, 3.46; N, 15.92%.
3.2.12. (E)-Dimethyl 2-[2-(-2(benzo[d]thiazol-2’-ylthio)acetyl)hydrazono] succinate (23)
Mixture of 3 (2.39 g, 10.0 mmol) and dimethylacetylene dicarboxylate (1.12 g, 10.0 mmol) in 1,4 dioxane (20 mL) containing a few drops of triethylamine was refluxed for 3 h. The mixture was allowed to cool to room temperature. The solid product formed, was collected by filtration and crystallized from methanol as pale yellow crystals. Yield: 2.85 g (75%); mp 147˚C - 148˚C; FT-IR vmax (cm−1): 3182 (NH), 1749,1710 (2C=O ester), 1683 (C=O amide); 1H NMR (DMSO-d6) δppm: 11.74 (s, 1H, NH, D2O exchangeable), 7.98 (d, J = 8 Hz, 1H, H-4’), 7.84 (d, J = 8 Hz, 1H, H-7’), 7.45 (t, J = 8 Hz, 1H, H-6’), 7.35 (t, J = 8 Hz, 1H, H-5’), 4.71 (s, 2H, SCH2), 3.83 (s, 2H, CH2), 3.82 (s, 3H, OCH3), 3.67 (s, 3H, OCH3); 13C-NMR (DMSO-d6) δC/ppm: 170.0 (amide C=O), 166.4 (ester C=O), 165.2 (ester C=O), 164.5 (C-2’), 152.9 (C-3a’), 136.3 (C=N), 135.2 (C-7a’), 126.8 (C-6’), 125.0 (C-5’), 122.3 (C-4’), 121.6 (C-7’), 53.1 (OCH3), 52.6 (OCH3), 35.8 (SCH2), 32.6 (COCH2). EI- MS m/z [relative intensity]: 381 [M+, 10%], 323 [71%], 272 [2%], 250 [12%], 208 [41%]. Anal. Calcd. for C15H15N3 O5S2 (381.32): C, 47.23; H, 3.96; N, 11.02%. Found: C, 47.13; H, 3.85; N, 11.21%.
3.3. Single Crystal X-Ray Diffraction Studies
Crystal structure of compounds 6 [10] , 10 [12] , 13 [13] , 15 [14] and 23 [16] , as well as their packing pattern were successfully developed by single crystal X-ray diffraction analysis. The molecular structure information of all these molecules, obtained from the X-diffraction method is in perfect agreement with their predicted synthetic protocol and other characterization techniques like 1HNMR and 13C NMR and mass spectroscopy.
Table 6 summarizes the crystal nature, unit cell descriptions and crystallographic refinement parameters of all crystal samples studied in this work.
4. Conclusions
We have corrected the wrong literature structures formed from reactions 2-(benzo[d]thiazol-2’-ylthio) acetohy-
Table 6. Precise crystal data for 6, 10, 13, 15 and 23.
drazide (3) with acetyl acetone and phthalic anhydride. We have also reported that both compounds 3 and 5 were utilized as a key intermediate for the synthesis of a novel heterocyclic compounds. We also reported the synthesis of a novel series of condensation and substituted derivatives of 2-(benzo[d]thiazol-2’-ylthio) acetohydrazide in good yield. The structures of the newly synthesized compounds were confirmed by elemental analysis, 1H-NMR, 13C-NMR spectra, Ms, and X-ray crystallographic investigations.
Acknowledgements
This work financed by the University of Kuwait research grant SC10/13. We are grateful to the Faculty of Science, Chemistry Department, and the SAF facility for the spectral and analytical data (Project GS01/10, GS03/08, GS01/03, GS 01/05). The CCDC file numbers for all crystal data are given in the reference section.
Author Contributions
F.A. is the Principal Investigator, designed the research work, collected the analysis of the spectroscopic data and wrote the manuscript. A.A. is the research associate performed the work in the laboratory. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare, they have no conflict of interests regarding the publication.
Cite this paper
Fatima Al-Omran,Adel Abou El-Khair, (2016) Synthesis, Spectroscopy and X-Ray Characterization, of Novel Derivatives of Substituted 2-(Benzothiazol-2’-ylthio)acetohydrazide. International Journal of Organic Chemistry,06,31-43. doi: 10.4236/ijoc.2016.61004
References
- 1. Mahran, M.A., El-Nassry, S.M.F., Allam, S.R. and Elzawawy, L.A. (2003) Synthesis of Some New Benzothiazole Derivatives as Potential Antimicrobial and Antiparasitic Agents. Die Pharmazie, 58, 527-530.
- 2. Wang, W., Zhang, G.-P., Song, B.-A., Wang, H., Jin, L.-H., Hu, D.-Y. and Yang, S. (2007) Synthesis and Anti-Tobacco Mosaic Virus Activity of O,O’-Dialkyl-α-(substituted benzothia-zol-2-yl)amino-(substituted phenylmethyl) Phosphonate. Chinese Journal of Chemistry, 26, 279-284.
http://sioc-journal.cn/Jwk_yjhx/EN/Y2007/V26/I02/279 - 3. Kaur, H., Kaur, S., Singh, I., Saxena, K.K. and Kumar, A. (2010) Synthesis, Characterization and Biological Activity of Various Substituted Benzothiazole Derivatives. Digest Journal of Nanomaterials and Biostructures, 5, 67-76.
- 4. Wardakhan, W.W., El-Sayed, N.N. and Mohareb, R.M. (2013) Synthesis and Anti-Tumor Evaluation of Novel Hydrazide and Hydrazide-Hydrazone Derivatives. Acta Pharmaceutica, 63, 45-57.
http://dx.doi.org/10.2478/acph-2013-0004 - 5. Mohareb, R.M., Fleita, D.H. and Sakka, O.K. (2011) Novel Synthesis of Hydrazide-Hydrazone Derivatives and Their Utilization in the Synthesis of Coumarin, Pyridine, Thiazole and Thiophene Derivatives with Antitumor Activity. Molecules, 16, 16-27.
http://dx.doi.org/10.3390/molecules16010016 - 6. Al-Omran, F., Abou El-Khair A. (2013)Studies and X-rays Determinations with 2-(Acetonylthio)benzothiazole: Synthesis of 2-(Benzothiazol-2-ylthio)-1-phenylethanone and 2-(Acetonylthio)benzothiazole by C-S Bond Cleavage of 2-(Acetonylthio)benzothiazole in KOH. Journal Heterocyclic Chemistry, 51, 62-70.
http://dx.doi.org/10.1002/jhet.1693 - 7. Al- Omran, F., Mohareb, R.M. and Abou El-khair, A. (2011) Synthesis and E/Z Configuration Determination of Novel Derivatives of 3-Aryl-2-(benzothiazol-2'-ylthio) acrylonitrile, 3-(Benzothiazol-2'-ylthio)-4-(furan-2''- yl)-3-buten-2-one and 2-(1-(Furan-2''-yl)-3'-oxobut-1''-en-2-ylthio)-3-phenylquinazolin-4(3H)-one. Molecules, 16, 6129-6147.
http://dx.doi.org/10.3390/molecules16076129 - 8. Mohareb, R.M. and Al-Omran, F. (2012) Reaction of Pregnenolone with Cyanoacetylhydrazine. Novel Synthesis of Hydrazide-Hydazone, Pyrazole, Pyridine, Thiazole, Thiophene Derivatives and Their Cytotoxicity Evaluations. Steroids, 77, 1551-1559.
http://dx.doi.org/10.1016/j.steroids.2012.09.007 - 9. AL-Saadi, M.A.E. and AL-Bayati, R.H.I. (2006) Synthesis and Characterization of Some New Derivatives of 2-Mercapto Benzothiazole and Evaluation Their Biological Activity. National Journal of Chemistry (NJC), 23, 390-404.
- 10. The CCDC File 1012540 Contains the Supplementary Crystallographic Data for Compounds 6 in This Paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk
- 11. Liu, X.-H., Sun, Z.-H., Yang, M.-Y., Tan, C.-X., Weng, J.-Q., Zhang, Y.-G. and Ma, Y. (2014) Microwave Assistant One Pot Synthesis, Crystal Structure, Antifungal Activities and 3D-QSAR of Novel 1,2,4-Triazolo [4,3-a] pyridines. Chemical Biology & Drug Design, 84, 342-347.
http://dx.doi.org/10.1111/cbdd.12323 - 12. The CCDC file 1012539 contains the supplementary crystallographic data for compounds 10 in this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk
- 13. The CCDC file 1012538 contains the supplementary crystallographic data for compound 13 in this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk
- 14. The CCDC file 1012541 contains the supplementary crystallographic data for compound 15 in this report. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk
- 15. Sun, G.-X., Sun, Z.-H., Yang, M.-Y., Liu, X.-H., Ma, Y. and Wei, Y.-Y. (2013) Design, Synthesis, Biological Activities and 3D-QSAR of New N,N'-Diacylhydrazines Containing 2,4-Dichlorophenoxy Moieties. Molecules, 18, 14876-14891.
http://dx.doi.org/10.3390/molecules181214876 - 16. The CCDC file 1032664 contains the supplementary crystallographic data for compound 23 in this report. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk
NOTES

*Corresponding author.