Journal of Analytical Sciences, Methods and Instrumentation
Vol.3 No.4(2013), Article ID:38326,10 pages DOI:10.4236/jasmi.2013.34026
Development and Application of a Validated HPLC Method for the Determination of Clindamycin Palmitate Hydrochloride in Marketed Drug Products: An Optimization of the Current USP Methodology for Assay
![]()
1Office of Generic Drugs, Food and Drug Administration, Rockville, USA; 2Division of Product Quality Research, Office of Pharmaceutical Science, Food and Drug Administration, Silver Spring, USA.
Email: Patrick.Faustino@fda.hhs.gov
Copyright © 2013 Geoffrey K. Wu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 7th, 2013; revised October 8th, 2013; accepted October 19th, 2013
Keywords: Clindamycin Palmitate Hydrochloride (CPH); HPLC; Method Validation; Pediatric Dosage Form
ABSTRACT
A simple efficient isocratic reversed-phase HPLC method was developed and validated for the determination of clindamycin palmitate hydrochloride (CPH) and its commercially available oral solution products. Separation was achieved on a Phenomenex Zorbax (Luna) cyano column (150 × 4.6 mm, 5 μm) with a Phenomenex cyano guard cartridge (4 × 3.0 mm) on Agilent 1050 series HPLC system. CPH and its resolution standard lincomycin were eluted isocratically at a flow rate of 1 mL/min with a simplified mobile phase (potassium phosphate buffer (5 mM, pH 3.0)—acetonitrile—tetrahydrofuran (20:75:5, v/v/v)) and detected at 210 nm. The column was maintained at 25˚C. The method was validated according to USP category I requirements. Robustness and forced degradation studies were also conducted. CPH marketed drug products were obtained from a drug distributor and assayed for potency using the validated method. Validation acceptance criteria were met in all cases. The analytical range for CPH was 15 - 500 μg/mL and the linearity was r2 > 0.999 over three days. The method was determined to be specific and robust. Both accuracy (92.0% - 103.8%) and precision (0.67% - 1.52%) were established across the analytical range for low, intermediate and high QC concentrations. Method applicability was demonstrated by analyzing two marketed products of CPH, in which results showed potency >98%. The method was determined to be an enhancement over the current USP methodology for assay as a result of increased efficiency, reduced organic solvents and the elimination of matrix modifiers. This method was successfully applied for the quality assessment of: 1) currently marketed drug products and 2) will in future assess the product quality of novel dosage forms of CPH for pediatric use.
1. Introduction
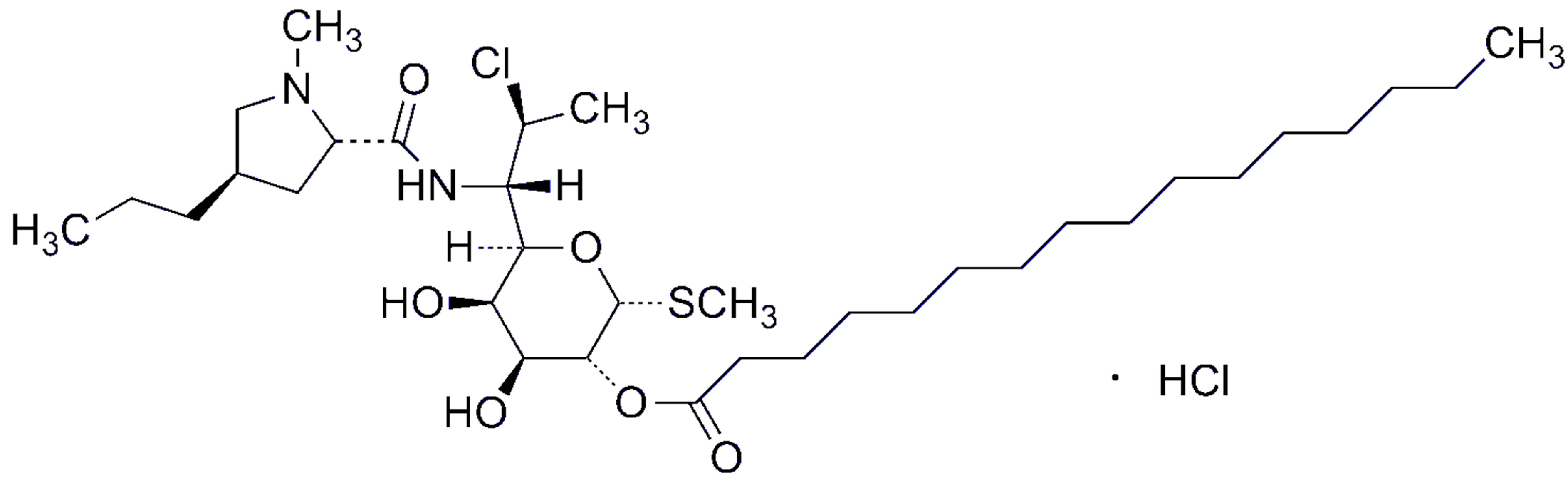
Clindamycin palmitate hydrochloride (CPH, methyl 7- chloro-6,7,8-trideoxy-6-(1-methyl-trans-4-propyl-L-2-pyrrolidinecarboxamido)-1-thio-l-threo-α-d-galactooctopyranoside monohydrochloride, Figure 1(a)" target="_self">Figure 1(a)), is an ester of clindamycin and palmitic acid. Clindamycin (Figure 1(b)" target="_self">Figure 1(b)) is a lincosamide antibiotics first synthesized in 1967. It is active against gram-positive aerobes and highly active against both gram-positive and negative anaerobes [1]. CPH is a white to off-white crystalline solid with a melting point from 168˚C - 178˚C, CPH has a molecular formula of C34H63ClN2O6S·HCl and a molecular weight of 699.85. CPH is a white to off-white amorphous solid with a pKa of 7.6.
During the development of pediatric preparations, a series of clindamycin esters were synthesized to minimize its bitter taste. As a successful 2-palmitate ester derivative, CPH is microbiologically inactive; however, it can be readily cleaved to clindamycin by enzymes located in small-intestine and has been shown to demonstrate similar therapeutic strength [2]. It has been used to treat a wide spectrum of bacterial infections, for instance, infections of respiratory tract, skin and soft-tissue infections, osteomyelitis, gynecological infections, as well as
 (a)
(a)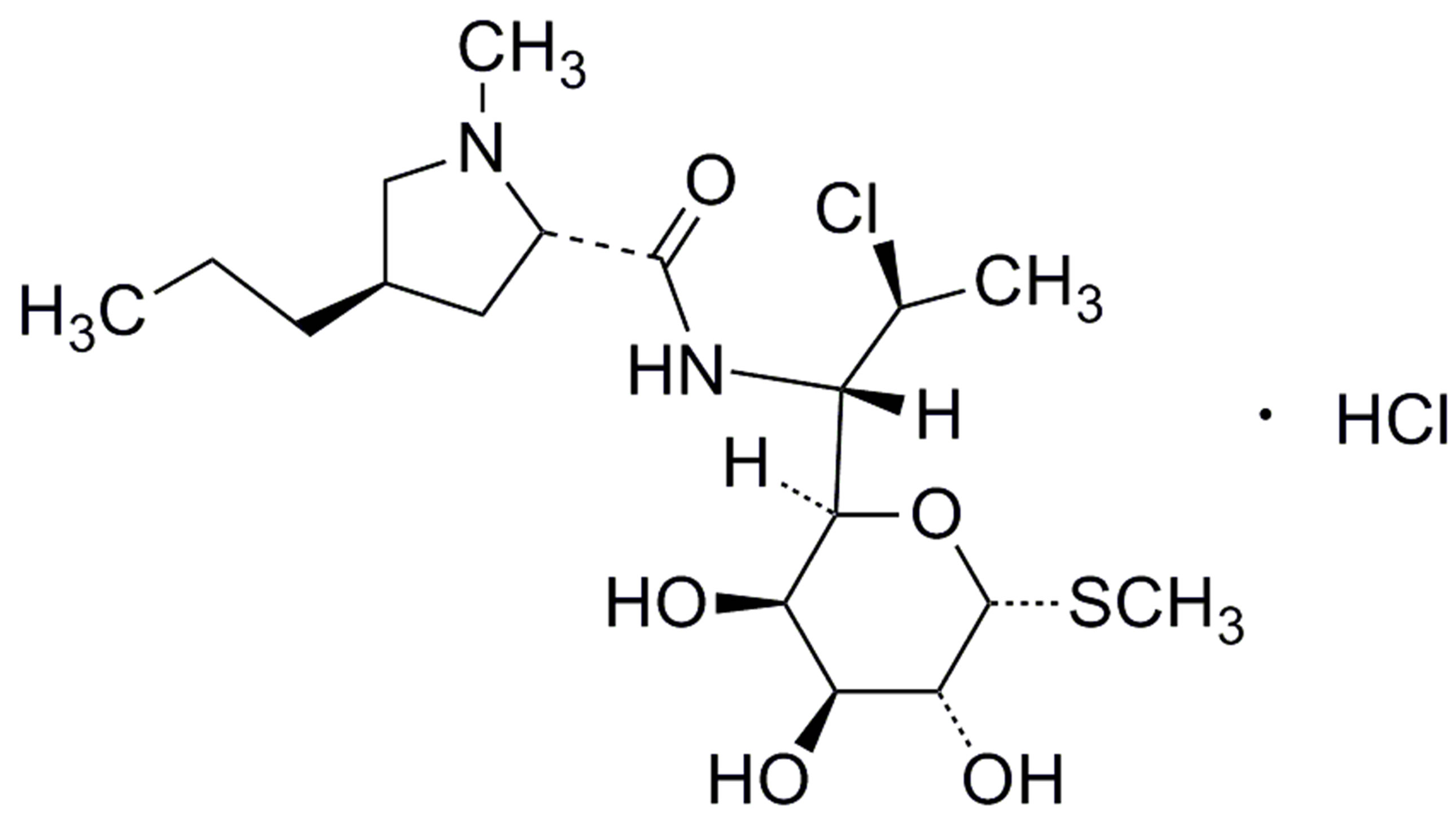 (b)
(b)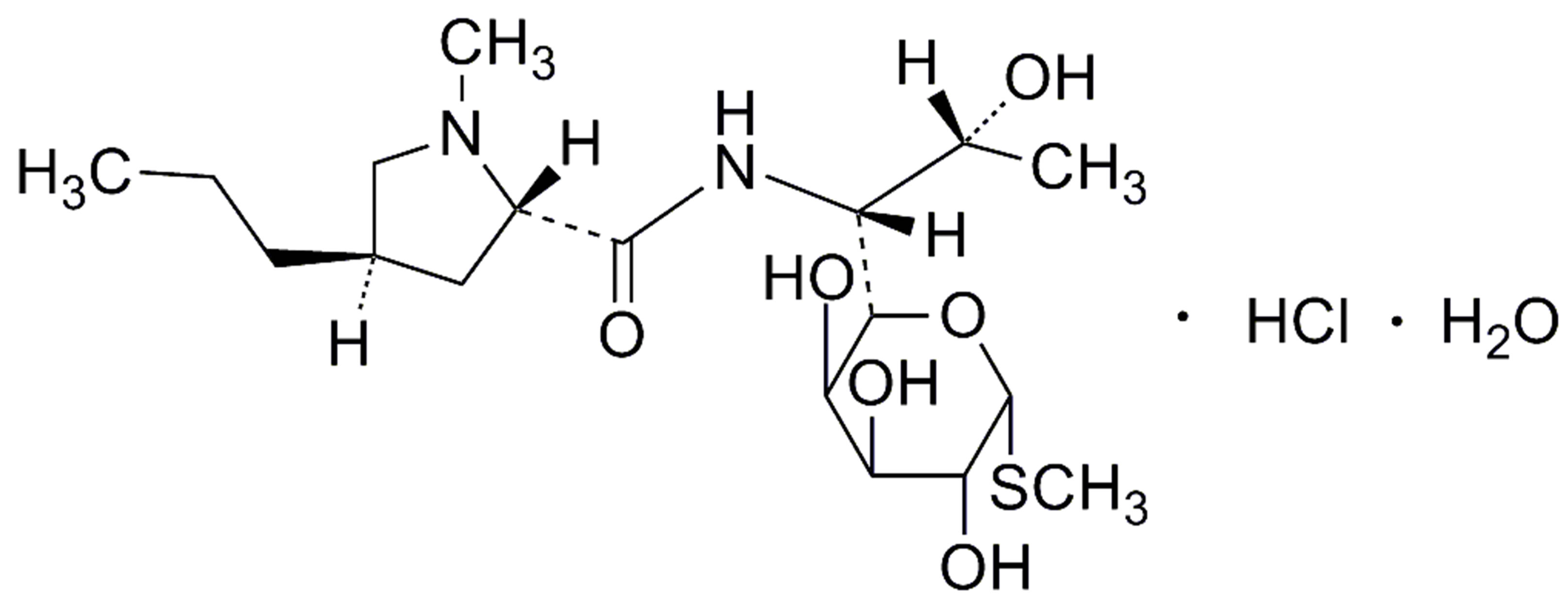 (c)
(c)
Figure 1. Chemical structures of clindamycin palmitate hydrochloride (a), clindamycin hydrochloride (b) and lincomycin hydrochloride (c). (a) Clindamycin palmitate hydrochloride (CPH). (b) Clindamycin hydrochloride. (c) Lincomycin hydrochloride monohydrate.
bacterial endocarditis.
In contrast to the other two clinically available clindamycin salts (i.e., clindamycin hydrochloride and clindamycin phosphate), there are only two HPLC analytical methods reported for CPH analysis and neither of them is ideal to implement or simple in their execution. Both methods required complex mobile phase compositions, with high organic content while both yielding longer retention times. The first method is a compendial method from the United State Pharmacopoeia using a C18 reversed-phase chromatographic column with refractive index (RI) detector, and with a mobile phase containing ~92% of methanol with a total run time of 30 min [3]. This is an adaptation from a method first published in 1978 by Brown [4]. The method is still being widely used by other researchers with minor modifications [5]. However, the use of RI detector can significantly compromise method robustness since the detector is sensitive to various environmental factors, such as small changes to ambient temperature, and operational factors such as flow rate, column pressure and non-compatibility with gradient elution and ultimately difficult to implement. The second method reported by Raju and colleagues [6] was an isocratic HPLC method using UV detector (210 nm) also using methanol as a major (90%, v/v%) component in the mobile phase. However, the mobile phase did not have buffering capacity and triethylamine (TEA) was used as ion pairing agent complicating the mobile phase preparation. TEA acts as a base causing the mobile phase pH to increasee, which negatively impacts the robustness of non-buffer mobile phase systems. Additionally TEA can not be easily washed out of reverse phase columns typically requiring 100 - 200 column volumes, thereby limiting column use and ultimately column lifetime.
Developing a practical HPLC method for CPH is challenging due to its amphiphlic nature and highly pH-dependent solubility in aqueous medium [7]. In its free base form, when pH is close to or above its pKa (7.6), CPH is extremely insoluble. For instance at pH 7.4 it has an aqueous solubility of <0.0002 mg/mL. Once the pH drops to around 4, its solubility increases to 0.238 mg/ mL; if micelle formation is considered, its solubility can increase to 50 mg/mL at pH 3.6. Such a solubility variation was observed in our laboratory studies. This solubility phenomenon is significantly different from the widely used hydrochloride salt where solubility is reported to be greater than 500 mg/mL in water [8]. Additionally the ionic strength and the presence of a high concentration of inorganic salts also have a significant impact on reducing CPH aqueous solubility. However, CPH is freely soluble in methanol and acetonitrile [5,9].
In order to optimize the USP method and provide a more robust means for assessing product quality (e.g., content uniformity and dissolution) control, a novel HPLC-UV method with well controlled mobile phase pH, low ionic strength, robust performance, and ease of system setup was developed and validated here. To demonstrate the system suitability, lincomycin, the synthesis starting material and major impurity, was used in our study as a resolution and impurity standard [5,9]. Other possible impurities in CPH drug substance include clindamycin hydrochloride which was assayed in our system. Sample detection was performed by a UV detector at 210 nm wavelength; phosphate buffer (pH 3.0) was employed to precisely control the acidity of the mobile phase; and no ion pairing agent was required for chromatographic selectivity or resolution. The method can selectively detect CPH and its related impurities with acceptable resolution. This simple and robust method will be a substantial improvement to the current USP method to evaluate product quality and drug stability.
2. Experimental
2.1. Materials
Clindamycin palmitate hydrochloride and lincomycin (resolution standard, Figure 1(c)) certified reference standards were purchased from the United States Pharmacopeia (Rockville, MD). Clindamycin palmitate hydrochloride drug substance was purchased from RIA International LLC (East Hanover, NJ). Clindamycin HCl was purchased from TCI (Tokyo, Japan). Clindamycin palmitate hydrochloride drug products were purchased from Bradley Drug-Washington Wholesale Drug Exchange (Bethesda, MD). Nylon syringe filters were purchased from Sun SRI (Rockwood, TN). HPLC grade monobasic potassium phosphate and ACS grade phosphoric acid were purchased from Fisher Scientific (Fairlawn, NJ). HPLC grade acetonitrile was purchased from Burdick and Jackson (Muskegon, MI). HPLC ready 18 MOhm water was obtained, in-house, from a Milli-Q Gradient A-10 water purification system, Millipore Corp. (Bedford, MA).
2.2. Instrumentation and Chromatographic Conditions
Hewlett Packard 1050 series (Agilent Technologies, Wilmington, DE) HPLC system was used for the analysis. It is equipped with a quaternary pump, online degasser, column heater, autosampler, and diode array detector (DAD). ChemStation software (Agilent Technologies, Wilmington, DE) was used to perform data collection and analysis. Separation was achieved on a Phenomenex Luna cyano column (150 × 4.6 mm, 5 µm) in series with a Phenomenex cyano SecurityGuard column (4.0 × 3.0 mm, Phenomenex, Torrance, CA). The elution was isocratic with a mobile phase of 5 mM KH2PO4 (pH 3.0)—acetonitrile—tetrahydrofuran (20:75:5, v/v/v). The column temperature was maintained at 25˚C. The injection volume was 30 µL with UV detection at 210 nm.
2.3. Preparation of Standard Solutions
CPH stock solution I of 10 mg/mL was prepared in phosphate buffer (5 mM, pH 3.0) using the USP reference standard. Calibration standard solutions at seven levels were prepared by serially diluting the stock solution I with the HPLC mobile phase to concentrations of 15, 25, 50, 100, 250, 375, and 500 μg/mL over the analytical range of 15 - 500 μg/mL.
CPH stock solution II of 10 mg/mL was prepared in phosphate buffer (5 mM, pH 3.0) using USP reference standard. Quality control (QC) standard solutions were prepared daily by diluting the stock solution II with the HPLC mobile phase to the final QC concentrations of 15, 100 and 500 μg/mL.
Lincomycin stock solution I of 10 mg/mL was prepared in phosphate buffer (5 mM, pH 3.0) using the USP reference standard. This solution was later used to prepare a mixture of CPH and lincomycin for daily system suitability tests. Clindamycin hydrochloride stock solution of 10 mg/mL was prepared in the same way.
2.4. Sample Preparation from the Marketed Products
Two CPH products (brand-name and generic), both in a powder form for oral solution for pediatric patients, were designated as Aand B-powder respectively and assayed for potency. For each product, powder equivalent to about 5 mg of CPH was accurately weighed on an analytical balance and transferred to a 10 mL volumetric flask. About 8 mL of HPLC mobile phase was added to the flask and the content was sonicated for 5 min. Additional mobile phase was added to adjust the volume to 10 mL. The resulting solution was filtered via a 0.20 μm nylon filter into standard analytical glass vials. The filtrate, containing ~500 μg/mL CPH, was further diluted in mobile phase to 250 and 125 μg/mL. All three concentrations (125, 250 and 500 μg/mL) were injected into the HPLC for potency measurements.
2.5. Method Validation
The method was validated according to the United States Pharmacopeia requirements for Category I for assay [10]. The following validation characteristics were evaluated: linearity, range, accuracy, precision, specificity, limit of quantitation and robustness.
System suitability standard solution which contained 100 μg/mL CPH and 10 µg/mL lincomycin as a resolution standard was prepared by diluting and mixing CPH and lincomycin stock solutions with mobile phase. System suitability was determined from six replicate injections of the system suitability standard before sample analysis. The acceptance criteria were: relative standard deviation (RSD) for peak area less than 2.0%, greater than 2000 plates per column (13,333 plates per meter), a peak width at half height of less than 0.25 minute, a resolution for CPH and lincomycin greater than 1.5, a USP tailing factor less than 2.0. Equation for calculating resolution (R) is list below:
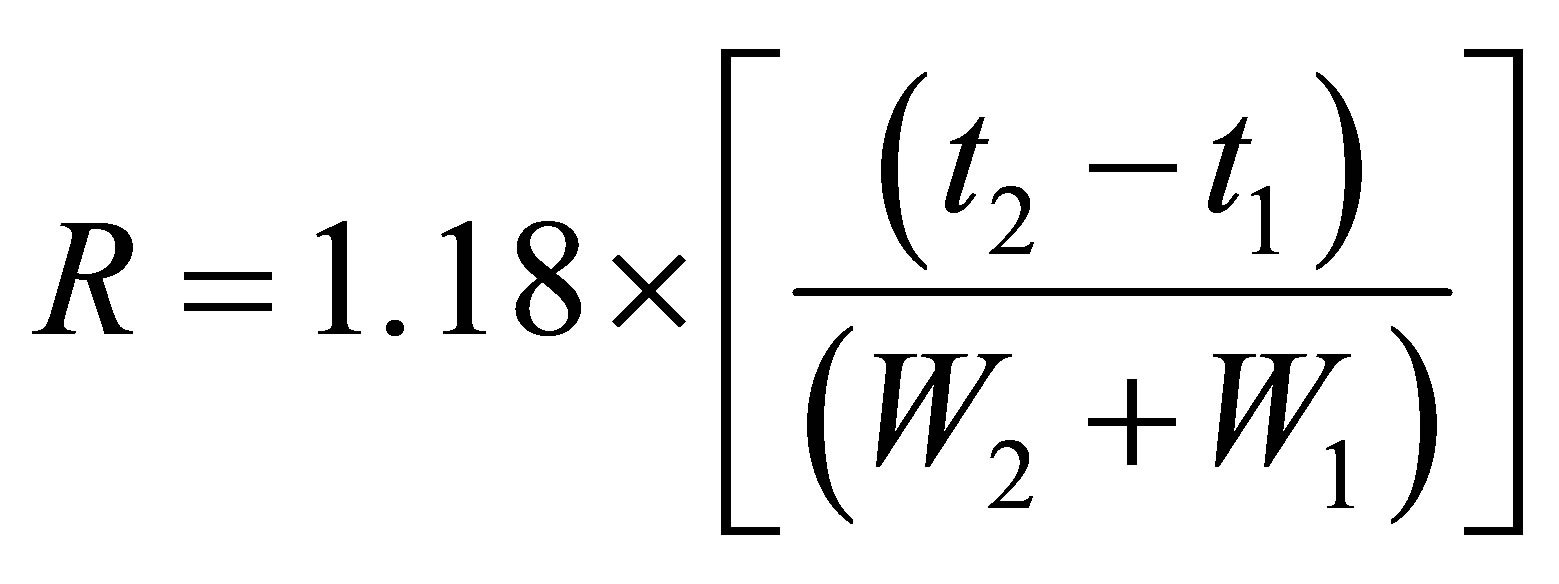
where t2 and t1 were respective retention times for CPH and lincomycin, and W2 and W1 were the peak widths at half height. Acquired results were used to evaluate critical operational parameters of the chromatographic system to ensure adequate resolution and reproducibility immediately prior to sample analysis.
Standard calibration curves were prepared with seven calibrators over a concentration range of 15 - 500 μg/mL for CPH. Linear least square regression analysis was then employed to correlate peak areas and drug concentrations. The resultant standard curves were evaluated for intraand inter-day reproducibility.
Accuracy and precision of the method were determined for the drug substance by analyzing QC standard samples at three concentrations levels of CPH (15, 100, and 500 μg/mL) over the analytical range. The method precision was established by injecting five standard QC samples at each concentration level for the intra-day precision and on three days for the intermediate precision. Precision was expressed by the relative standard deviation (RSD, %) of the analyte peak areas. Accuracy was expressed as percent recovery (%) by comparing the amount determined from the quality control standards against the respective nominal values. Accuracy of the method was also tested on two drug products at three concentrations with 3 replicates at each level, using the ICH Q2R1 approach of standard addition [11]. In this approach the following changes were made to the procedures described in section 2.4: only 50% of the nominal amount of drug product was placed in the flask and then the drug product was spiked with CPH stock solution III up to the target concentrations. The target concentrations were 125, 250 and 500 μg/mL of CPH in total; in addition, samples containing only 50% of the nominal amount of drug product without spiking were prepared and analyzed at three corresponding levels. Percent recovery (%) of CPH was calculated by comparing the known spiked amount of CPH to the amount CPH detected after subtracting the un-spiked (50% product) samples.
Method robustness was evaluated by analyzing the system suitability standard and evaluating system suitability parameters after varying the HPLC pump flow rate (± 2.5%), auto-sampler injector volume (± 10%), column compartment temperature (± 2˚C), mobile phase pH (± 0.1) and organic composition (20% regarding tetrahydrofuran content), respectively.
Method specificity was evaluated by analyzing a mixture of CPH drug substance and lincomycin (a major synthetic precursor of CPH), and two drug products versus sample blank. In addition, CPH standards were subjected to forced degradation under different conditions: elevated temperature (55˚C for 24 hours), light (365 nm for 24 hours at room temperature), 10% peroxide (for 24 hours at room temperature), acidic (0.1 M HCl for 4 hours at room temperature), and basic (0.1 M NaOH for 4 hours, room temperature). Resulting HPLC chromatograms were compared to determine if CPH was coeluting with lincomycin or other degradation products or excipient. Full spectrum (200 - 560 nm) chromatograms were acquired and Agilent ChemStation peak purity algorithms were also used to evaluate specificity.
3. Results and Discussion
3.1. Optimization of HPLC Method
Similar to other derivatives of lincomycin, CPH has a relatively weak UV absorption with an absorbance peak maxima around 206 nm. Different from the other lincomycin derivatives, CPH is an amphiphilic molecule due to the hydrophobicity of palmitate (14 carbon chain) and the basic nature of clindamycin. Its amphililicity leads to an anomalous solubility feature: it can only be reasonably dissolved in aqueous medium at a pH below 3.7 (53.2 mg/mL) because of micelle formation; above this pH, it is practically insoluble (e.g., 0.001 mg/mL at pH 5.8) [7]. During solubility studies there was difficulty dissolving CPH in aqueous medium and micelle formation was observed. These observations were contradictory to most of the publications and even USP material safety data sheet (MSDS), which claimed that CPH is freely soluble [12]. Both matters complicate developing a simple and robust HPLC method for CPH. This may explain why there are only two major groups of analytical methods available for CPH in the scientific literature.
The first group of methods is represented by the compendial method recorded in USP36/NF31 (CPH monograph); other similar methods were developed following minor modifications to this USP method. Acetate organic salts were used to constitute the mobile phases in these methods and this rendered RI detector as the only choice due to the high UV cutoff (~230 nm) of these salts. However, the well-known disadvantages of RI detector are its sensitivity to environmental influence, and long equilibration periods which may negatively impact routine operations. Recently another HPLC method was reported using UV detector at 210 nm for CPH analysis, in which the mobile phase did not possess any buffering capacity [6]. A common feature for all the aforementioned methods is that a high percentage (>90%) of methanol was used in mobile phase. Unfortunately like acetate salts, methanol has a UV cutoff around 210 nm, which is the chosen detection wavelength in the last method. Therefore, an HPLC method void of these problems is necessary to best assess the product quality of CPH. To this end, our goal was to develop a simple and robust HPLC method, ideally possessing buffering capacity and using UV detection at the absorbance maxima.
Initially a C18 column, used in all the published methods, was chosen and a series of mobile phase screening was conducted (Table 1). The mobile phase (MP1), composed of H2O/methanol (10:90) supplemented with 0.5% triethylamine (TEA), reported by Raju et al. was first evaluated [6]. CPH could be eluted with acceptable symmetry, but the CPH retention was prohibitively long (~33 min) on a Phenomenex Luna C18 (2) column (250
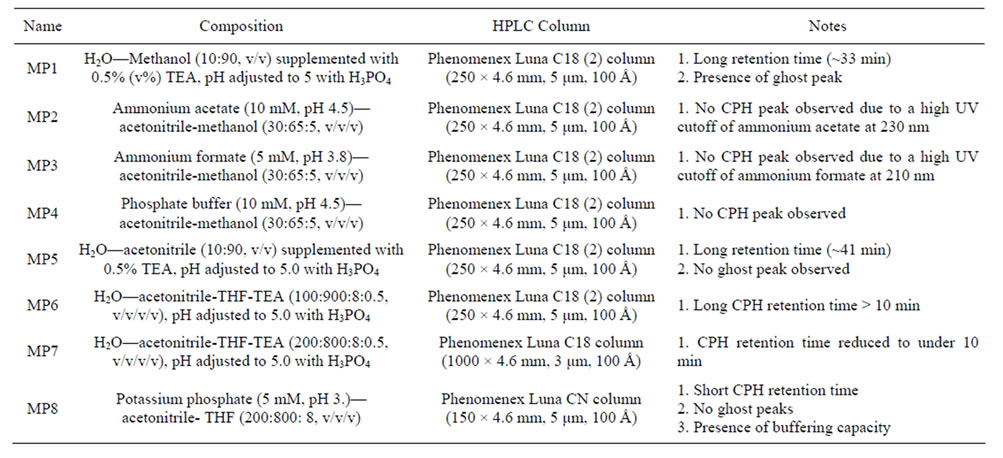
Table 1. List of major mobile phases and HPLC columns screened.
× 4.6 mm, 5 um, 100 Å) with a flow rate of 1 mL/min at ambient temperature. More importantly, ghost peaks had been observed and the source of these peaks was highly possible the mobile phase components, where the sample elution may have been incomplete. Following the reported preparation, pH (5.0) of the mobile phase was simply adjusted by titrating the mixture using phosphoric acid, although this procedure is not recommended since a pH meter generally does not measure properly in the presence of a large amount of organic solvent. This is also important, suggesting that ionization from TEA can significantly increase the pH of the MP and the pKa of CHP is 7.6.
In order to introduce buffering capacity and eliminate the ghost peaks, aqueous buffers were used to replace DI water and acetonitrile was employed to substitute methanol. The additional mobile phases that were screened include: ammonium acetate (10 mM, pH 4.5)—acetonitrile—methanol (30:65:5, v/v/v, MP2) and ammonium formate (5 mM, pH 3.8)—acetonitrile—methanol (30:65: 5, v/v/v, MP3); no major peak of CPH could be observed since these two buffers have high UV cutoffs at 230 and 210 nm, respectively. Phosphate buffer is generally recognized as the most ideal buffer with a low UV cutoff < 200 nm. Therefore a mobile phase (MP4), composed of phosphate buffer (pH 4.5, 10 mM)—acetonitrile—methanol (30:65:5, v/v/v) was examined; however, no CPH peak was observed either. These mobile phases were not supplemented with triethylamine (TEA), which could be critical to the successful elution of CPH on C18 columns.
So TEA was reintroduced and the following mobile phase combinations were tested. The first one (MP5) consisted of H2O—acetonitrile (10:90, v/v) supplemented with 0.5% TEA with pH 5.0 adjusted by phosphoric acid. Comparing against the published mobile phase, the only difference was that acetonitrile was used to replace methanol. Results showed that a major peak of CPH could be observed around 41 min when isocratically eluted at a flow rate of 1 mL/min at 30˚C; more importantly, no ghost peaks were observed since acetonitrile has low UV cutoff (~190 nm). Using MP5 while increasing column temperature to 35˚C; surprisingly this only shortened the retention time from 41 to 40 min. In order to further reduce the retention time, tetryhydrofuran (THF), an organic modifier, was added to the current mobile phase. The influence of THF content was examined and the volume percentages screened of were 1%, 2%, 5%, 8% and 10%. And the CPH retention time was reduced to 37, 35, 26, 17 and 19 min, respectively. Therefore, 8% of THF supplement was found appropriate; however, TEA seemed indispensible for efficient elution.
Thus a more promising mobile phase (H2O—acetonitrile-THF-TEA, 100:900:8:0.5, v/v/v/v, MP6) was identified; but the percentage of acetonitrile was too high. So the ratio was adjusted to 200:800 (H2O—acetonitrile, v/v, MP7); and this new mobile phase combination resulted in a more efficient CPH retention time around 22 min. Using this new mobile phase as a working reference, we migrated to a shorter Phenomenex Luna C18 column (100 × 4.6 mm, 3 μm, 100 Å) aiming to reducing CPH retention time to under 10 min. With this HPLC condition, CPH was eluted out at 8.6 min. Further adjustment of H2O—acetonitrile to 100:900 and 300:700 was conducted; the CPH retention time shifted to 6.7 and 17 min, respectively. These results were satisfactory; however, the mobile phase composition was still complex without a buffering capacity.
To better address these issues, the optimization of the stationary phase and HPLC column selection were performed. As observed, CPH bound to C18 stationary phase tightly so that TEA was necessary to form an ion pair to achieve efficient elution. A major factor contributing to this strong interaction is the fourteen carbon chain of palmitate. In order to reduce the affinity of CPH to the column, a cyano stationary phase with lower hydrophobicity was selected since it is more suitable for efficient elution of basic amines. To ensure the consistency, a Phenomena Luna CN column (150 × 4.6 mm, 5 μm, 100 Å) was selected. Stationary phase change allowed for significant modifications to the mobile phase composition: DI water was replaced by buffer with a ratio to acetonitrile as 200:800 (v/v) and TEA was eliminated; while 8% of THF was kept as an organic modifier. Given the low UV-cutoff at 200 nm, potassium phosphate buffer (5 mM, pH 3.0) was employed and results showed that this mobile phase could provide better peak symmetry and less baseline noise. Under the current chromatographic condition, CPH had a retention time of 5.5 min when isocratically eluted with a flow rate of 1 mL/min at 25˚C.
Therefore, to decrease the chromatographic elution time we chose to analyze CPH at an acidic pH (pH = 3.0) due to the hydrophobicity of palmitate and the basic nature of clindamycin. At this pH, CPH is positively charged and relatively easier to be eluted off a reverse phase column. Additionally, this pH is >4 units away from the pKa of CPH ensuring the constant composition of the ionic species. The selection of a cyano stationary phase further facilitated the efficient elution of CPH with an ideal elution time and a lower organic content.
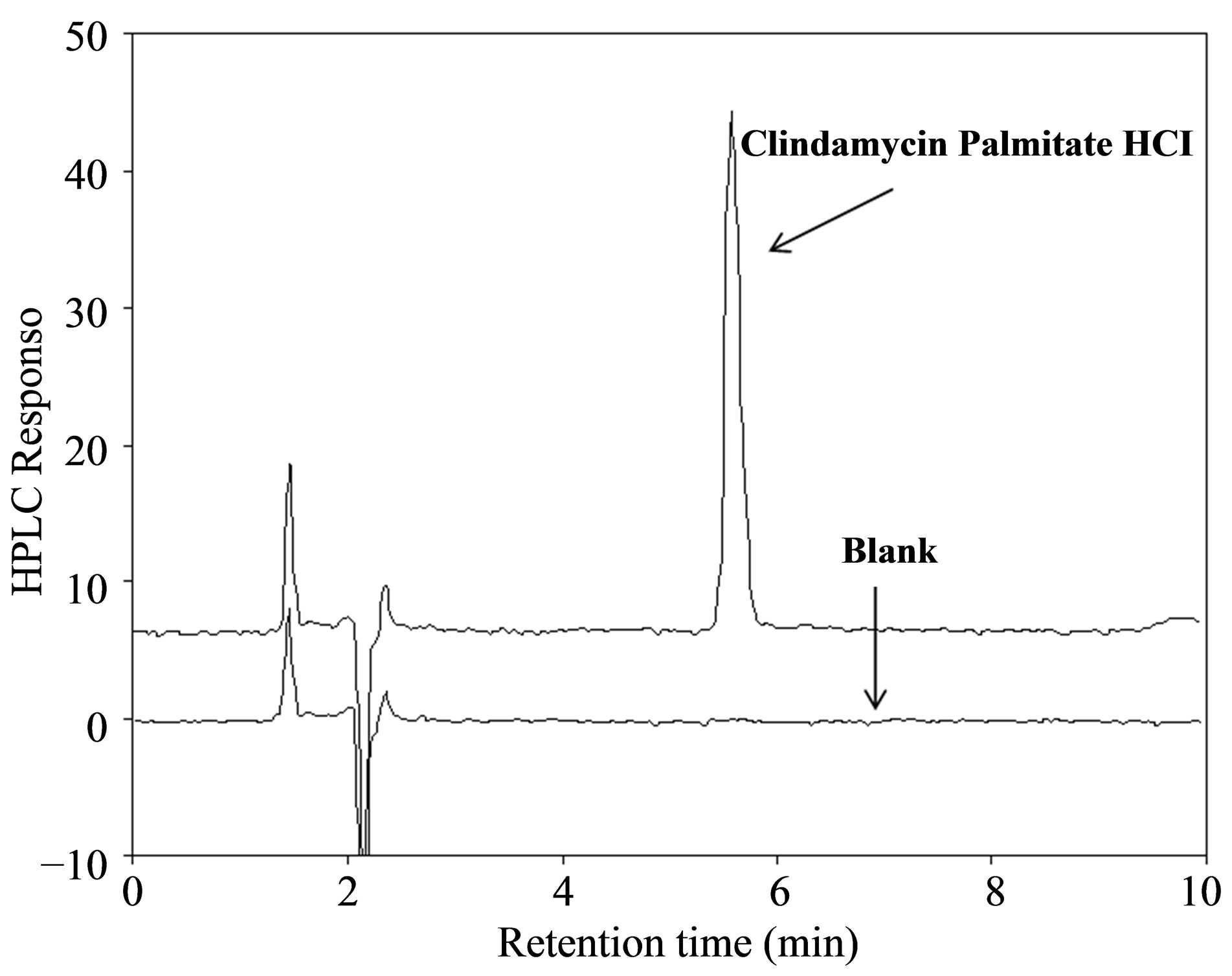
In summary, a novel HPLC analysis method for CPH is established on a Phenomenex Luna CN column (150 × 4.6 mm, 5 μm, 100 Å) performed at 25˚C. After minor adjustment, the mobile phase was composed of phosphate buffer (5 mM, pH 3.0)—acetonitrile—tetrahydrofuran (20:75:5, v/v/v). CPH was eluted isocratically at a flow rate of 1 mL/min with a run time of 10 min; its retention time was around 5.6 min, detected at 210 nm (Figure 2(a)). This method was a significant improvement over the previous methods. First, a simple mobile phase was formulated without using ion pairing agent (e.g., triethylamine). Second, it possessed a buffering capacity to ensure the ionized state of CPH and retention time reproducibility. Third, the commonly available UV detector, instead of RI detector, was adopted to ensure the robustness of the analysis. Fourth, acetonitrile with a low UV cutoff (~190 nm) was employed to replace methanol (UV cutoff @ 240 nm) so that the interference from the mobile phase was minimized. Lastly, this
 (a)
(a)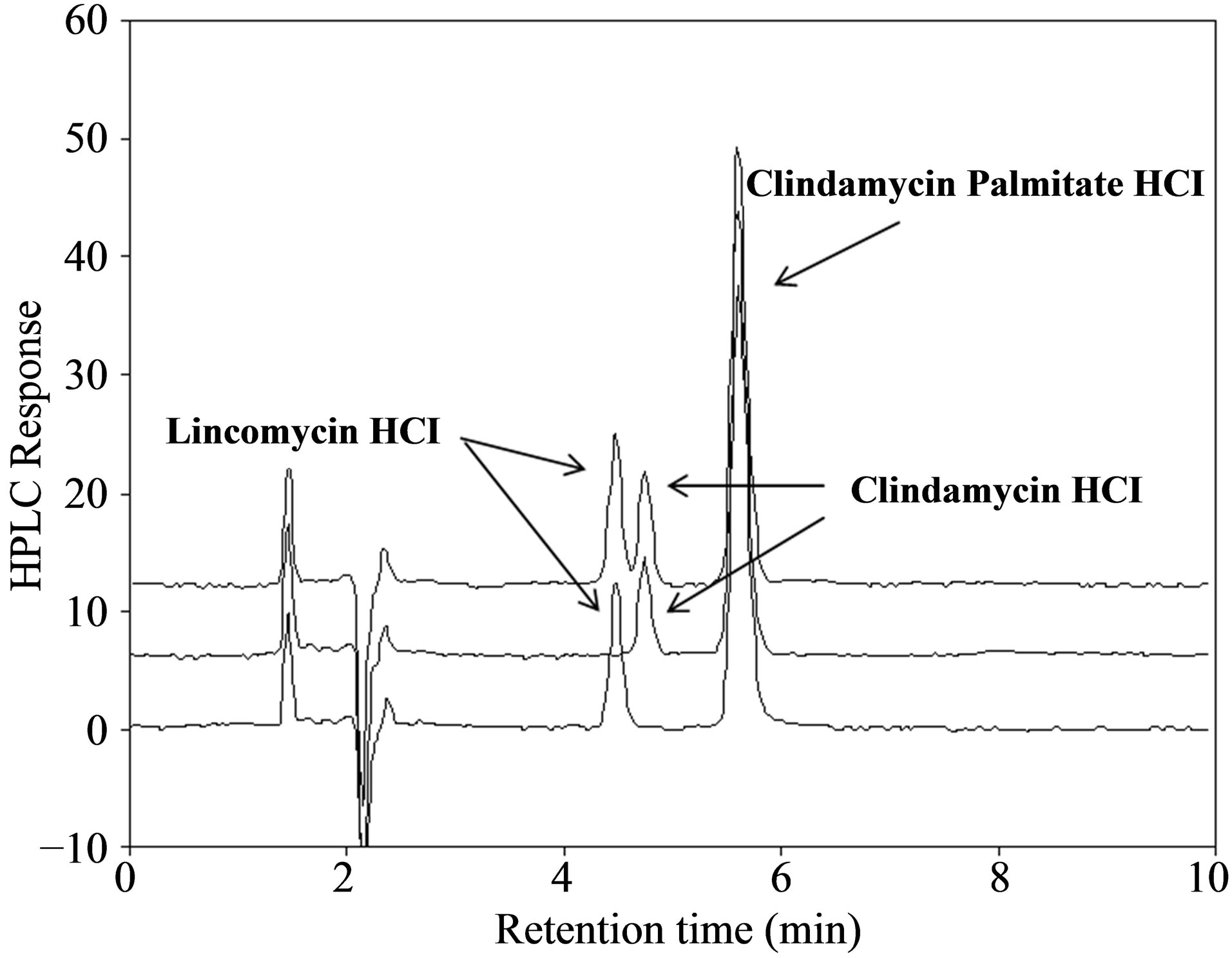 (b)
(b)
Figure 2. HPLC chromatograms of blank mobile phase and clindamycin palmitate hydrochloride USP reference standard (a), and system suitability samples (b). Retention times for lincomycin, clindamycin HCl and clindamycin palmitate hydrochloride were 4.47, 4.74 and 5.60 min, respectively.
method had enhanced selectivity over CPH and its related synthetic impurity and is a potential analytical tool to evaluate product stability.
3.2. Method Validation
The following method validation characteristics were addressed for CPH: accuracy, precision, specificity, linearity, range, and robustness. The method validation for CPH meets the requirements for USP Category I [10].
3.3. System Suitability
The system suitability test is designed to ensure the validity of the analytical procedure as well as the conformation of the resolution between different peaks of interest. All critical parameters tested met the acceptance criterion on all days, as listed in Table 2. Adequate resolution of >1.5 between CPH and lincomycin demonstrated the method specificity in the presence of its synthetic precursor, as shown in Figure 2(b). Clindamycin HCl, another possible impurity, was also well separated from CPH.
3.4. Linearity and Range
Linearity of the method was confirmed by preparing standard curves for the analytical range of 15 - 500 μg/ mL for CPH for three days. The results, summarized in Table 3, demonstrated an excellent correlation between analyte peak area and concentration over the analytical range with r2 ≥ 0.999 using a least squares linear regression function.
3.5. Accuracy and Precision
Accuracy and precision was established across the analytical range for CPH. The intraand inter-day accuracy and precision were calculated from the QC samples for CPH. Results for the intra-day accuracy and precision of CPH were summarized in Table 4. In order to meet the experimental requirements, the acceptance criterion for low concentrations was set to be ± 10%, and ± 5% for medium and high concentrations. In the future use of this method, this design would allow us to capture the very early dissolution time points for pediatric dosage forms. The above analytical range will be necessary to achieve our analytical objectives and able to successfully and accurately analyze these dosage forms to best evaluate their release characteristics. Moreover, we were aware that even small increase of the lower limit of quantitation would significantly impact the accuracy in a positive manner.
The accuracy results for CPH in the two drug products were summarized in Table 5. Results for the accuracy of CPH tested in drug products Aand Bpowder at three concentrations levels (125, 250 and 500 μg/mL) by the technique of standard addition ranged from 100.62% to 104.82%. The recovery was within 100 ± 5% for all samples.
3.6. Robustness
To ensure the HPLC method insensitive to minor changes in the experimental conditions, it is important to demonstrate robustness of the method. As pointed out by Dejaegher and colleagues [13], the following responses were evaluated: CPH retention time, peak area, capacity factor, USP tailing, resolution between CPH and lincomycin, and theoretical plate numbers. Results were summarized in Table 6 and none of the alterations caused a significant change in these responses and all responses met the acceptance criteria for system suitability. Therefore the method is robust and reliable to quantitate CPH.
3.7. Specificity and Forced Degradation
Full spectrum data acquisition was performed for system suitability standard (Figure 2) and two products as well to examine for co-eluting peaks (Figure 3). After peak

Table 2. System suitability test (n = 6).
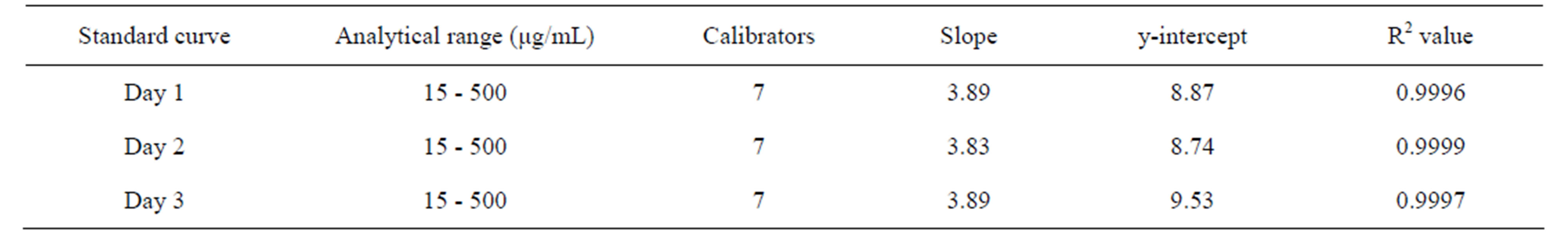
Table 3. Parameters and linearity of clindamycin palmitate hydrochloride calibration curves.
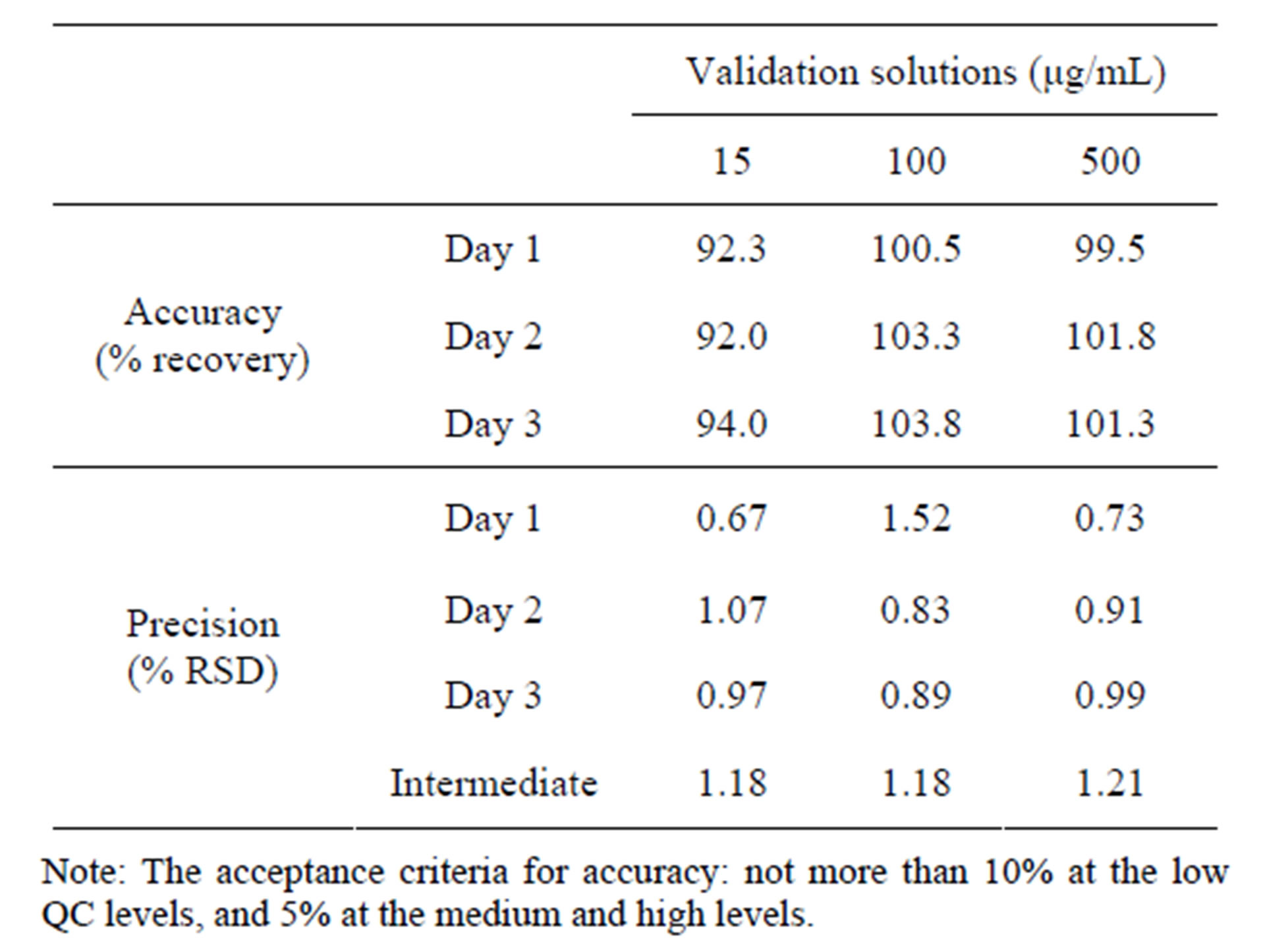
Table 4. Clindamycin palmitate hydrochloride accuracy and precision: drug substance (% recovery, n = 5).

Table 5. Clindamycin palmitate hydrochloride recovery: drug product (% recovery, n = 3) via standard addition test.
purity analysis, results demonstrated that no co-eluting peaks of CPH and lincomycin was observed; no impurities were detected in CPH peak; and excipients from the products were determined not co-eluted with CPH by observation following a spectral peak purity CPH evaluation. Under the current stress-test conditions, CPH was stable under the stress conditions of heat, light and oxidation but labile in presence of acid and base. However, no co-eluting peaks were observed from these stressed samples. Together with all the aforementioned observations, we determined that this method is specific for CPH analysis.
3.8. Analysis of the Marketed Products
In the market there is only one solid dosage form available for CPH, flavored granules for oral solution with a single dose strength of 75 mg/5 mL upon reconstitution. Currently, there are one brand-name and two generic products being marketed. To ensure the representativeness, both brand-name (one product) and generic product (one of the two) were purchased and analyzed by this validated method. Representative chromatograms were shown in Figure 3. Results for CPH potency are summarized in Table 7. All products had a potency of >98%. Although only two drug products were tested due to the limited number of products available, this method demonstrated strong applicability for product potency analysis.
In addition, this method has been proved applicable for potency analysis of novel CPH formulations prepared in house. It also demonstrated an applicability to quantitate CPH release from these novel formulations during dissolution testing (data not published).
4. Conclusion
A simple and efficient and robust reverse-phase HPLC method for clindamycin palmitate hydrochloride was found to be accurate, precise, specific, and linear across
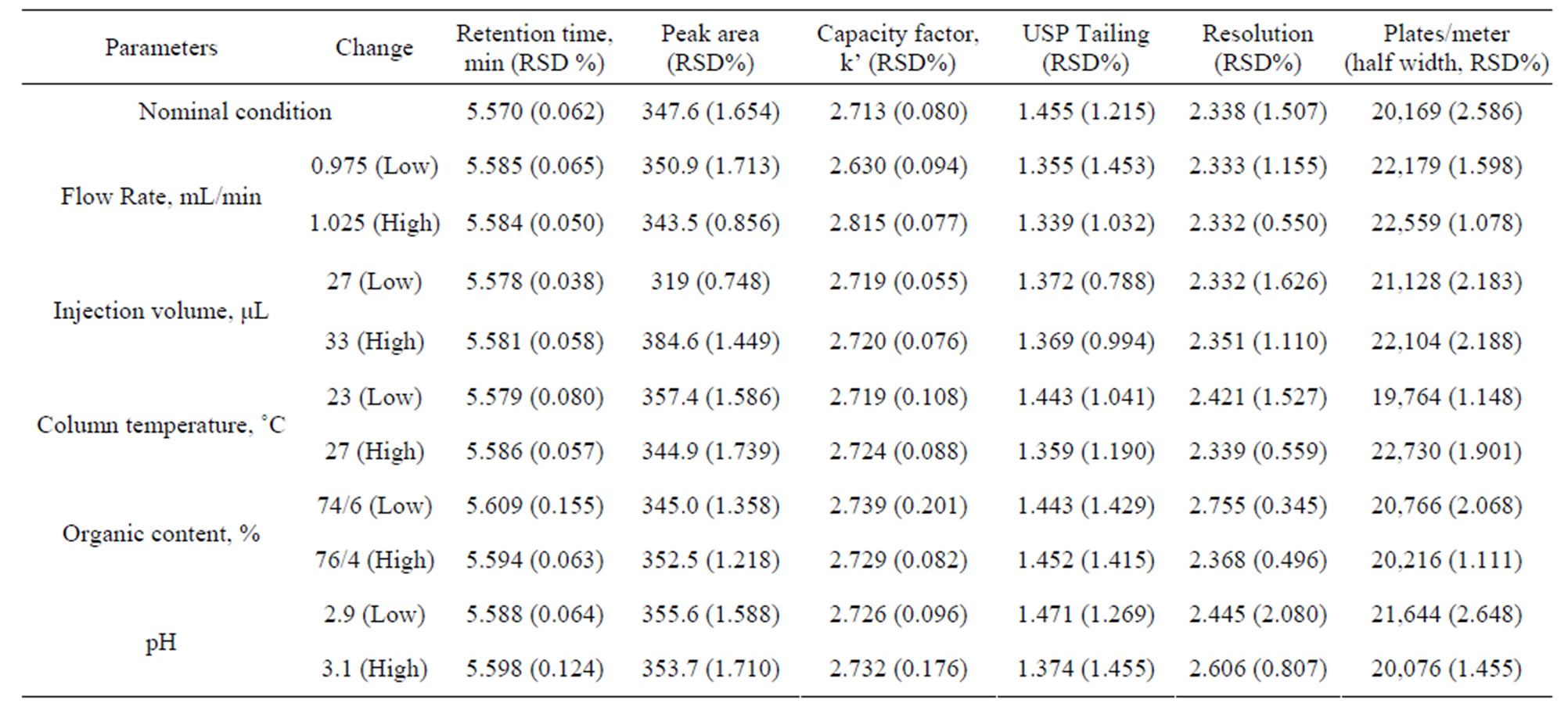
Table 6. Robustness test results for the HPLC method for clindamycin palmitate hydrochloride analysis.
 (a)
(a) (b)
(b)
Figure 3. HPLC chromatograms of clindamycin palmitate hydrochloride products: A-powder (a) and B-powder (b). CPH had a retention time of 5.60 min.
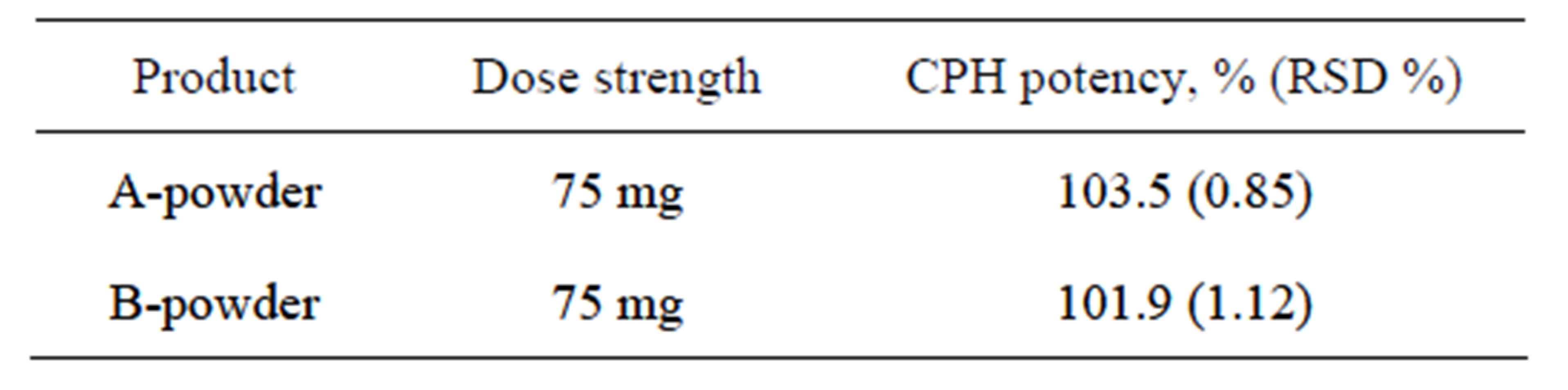
Table 7. HPLC results of clindamycin palmitate hydrochloride products (n = 3).
the analytical range. The significant advantages of this method, over existing methods including the current USP methodology, were the simplicity of mobile phase preparation, buffering capacity, reduced consumption of organic solvents and avoidance of ion pairing agents. The method was robust and demonstrated to be selective for the determination of CPH in the presence of its primary synthetic impurity in drug substance and products. The current method represents a significant improvement over the current USP methodology for CPH (USP CPH monograph). The method was successfully applied for the assessment of product quality of various marketed CPH dosage forms. It is also promising to be further developed into a selective stability indicating method if impurities standards are readily available [14].
5. Acknowledgements
This study was supported in part by the NIH pediatric formulation initiative and intramural FDA research funds. The authors would like to acknowledge Gretchen Whitesell’s editorial assistance.
REFERENCES
- A. A. Forist, R. M. DeHaan and C. M. Metzler, “Clindamycin Bioavailability from Clindamycin-2-Palmitate and Clindamycin-2-Hexadecylcarbonate in Man,” Journal of Pharmacokinetics and Biopharmaceutics, Vol. 1, No. 2, 1973, pp. 89-98. http://dx.doi.org/10.1007/BF01059623
- M. Cimbollek, N. Nies, A. Liebendorfer, et al., “The Potential of the Prodrug Clindamycin Palmitate as an Implantable Slow Release form of the Antibotic Clindamycin for Heart Valves,” Journal of Controlled Release, Vol. 33, No. 1, 1995, pp. 47-53. http://dx.doi.org/10.1016/0168-3659(94)00061-X
- United States Pharmacopeia, “Clindamycin Palmitate Hydrochloride,” United States Pharmacopeia (36) and National Formulary (31), 2013, pp. 3028-3029.
- L. W. Brown, “High-Pressure Liquid Chromatographic Assays for Clindamycin, Clindamycin Phosphate, and Clindamycin Palmitate,” Journal of Pharmaceutical Sciences, Vol. 67, No. 9, 1978, pp. 1254-1257. http://dx.doi.org/10.1002/jps.2600670917
- C. Bharathi, P. Jayaram, J. S. Raj, et al., “Identification, Isolation and Characterization of Impurities of Clindamycin Palmitate Hydrochloride,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 48, No. 4, 2008, pp. 1211- 1218. http://dx.doi.org/10.1016/j.jpba.2008.08.011
- C. B. V. N. Raju, G. Panda, G. N. Rao, et al., “HPLC-UV Assay Method for Clindamycin Palmitate Hydrochloride as Drug Substance and Oral Solution,” Analytical Letters, Vol. 41, No. 11, 2008, pp. 2033-2043. http://dx.doi.org/10.1080/00032710802209334
- E. L. Rowe, “Anomalous Solution Behavior of 2-Palmitate Esters of Lincomycin and Clindamycin,” Journal of Pharmaceutical Sciences, Vol. 68, No. 10, 1979, pp. 1292-1296. http://dx.doi.org/10.1002/jps.2600681025
- L. W. Brown and W.F. Beyer, “Clindamycin Hydrochloride,” In: K. Florey and G. Rafik Bishara, Ed., Analytical Profiles of Drug Substances, Acedemic Press, Inc., London, 1981, pp. 75-91.
- S. M. Wang, S. S. Bu, H. M. Liu, et al., “Separation and Characterization of Clindamycin Phosphate and Related Impurities in Injection by Liquid Chromatography/Electrospray Ionization Mass Spectrometry,” Rapid Communications in Mass Spectrometry, Vol. 23, No. 6, 2009, pp. 899-906. http://dx.doi.org/10.1002/rcm.3935
- United States Pharmacopeia, “General Information <1225> Validation of Compendial Procedures,” United States Pharmacopeia (36) and National Formulary (31), 2013, pp 983-988.
- “Guidance for Industry Q2B Validation of Analytical Procedures: Methodology,” International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), 1996.
- United States Pharmacopeia, “Materials Safety Data Sheet: Clindamycin Palmitate Hydrochloride,” 2007. http://www.usp.org/pdf/EN/referenceStandards/msds/1137005.pdf
- B. Dejaegher and Y.V. Heyden, “Ruggedness and Robustness Testing,” Journal of Chromatography A, Vol. 1158, No. 1-2, 2007, pp. 138-157. http://dx.doi.org/10.1016/j.chroma.2007.02.086
- M. Bakshi and S. Singh, “Development of Validated Stability-Indicating Assay Methods—Critical Review,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 28, No. 6, 2002, pp. 1011-1040. http://dx.doi.org/10.1016/S0731-7085(02)00047-X