 American Journal of Analyt ical Chemistry, 2011, 2, 18-26 doi:10.4236/ajac.2011.21003 Published Online February 2011 (http://www.SciRP.org/journal/ajac) Copyright © 2011 SciRes. AJAC Rapid HPLC Method for Monitoring Relevant Residues of Pharmaceuticals Products in Environmental Samples Juana Rodríguez Flores, Ana Maria Contento Salcedo, Lorena Muñoz Fernández Department of Analytical Ch emistry and Foods Technology, University of Castilla-La Mancha, Ciudad Real, Spain E-mail: juana.rflores@uclm.es Received September 3, 2010; revised December 20, 2010; accepted January 10, 2011 Abstract This work presents a multi-residue analytical method based on solid phase extraction (SPE) followed by high-performance liquid chromatographic (HPLC) with diode array (DAD) detection for the simultaneous determination of a group of pharmaceutical products that include ten antidepressants and three anticanceri- genic in environmental samples (water and soil). Baseline separation of the studied compounds was obtained on an ultrabase C18 (4.6 mm i.d. × 150 mm, 5 μm particle) column using acetonitrile:phosphate buffer pH 2.5 (35:65, v/v) as mobile phase with a flow rate of 1.5 mL/min. Different aspects including linearity, accuracy, precision and detection and quantification limits were examined in order to validate the proposed method. Detection limits between 1 and 50 ng/mL were obtained for all the target compounds. This method was ap- plied to the analysis of environmental samples as waters and soils of different precedence. Prior, the HPLC determination the samples were purified and enriched using SPE or liquid-liquid extraction (LLE) of the tar- get compounds. Keywords: High-Performance Liquid Chromatographic (HPLC), Breast Cancer, Antidepressant, Environmental Samples 1. Introduction The occurrence of residues of pharmaceuticals in the aquatic environment has attracted considerable interest in recent years [1]. The disposal of unused medication via the toilet seems to be of minor importance but many of the pharmaceuticals applied in human medical care are not completely eliminated in the human body. Often they are excreted only slightly transformed or even unchanged mostly conjugated to polar molecules. These conjugates can easily be cleaved during sewage treatment and the original pharmaceutically active compounds (PhACs) will then be released into the aquatic environment mostly by effluents from municipal sewage treatment plants (STPs). Several investigations have shown some evi- dence that substances of pharmaceutical origin are often not eliminated during wastewater treatment and also not biodegraded in the environment [2]. Under recharge conditions, residues of pharmaceutically active com- pounds may also leach into groundwater aquifers. Thus, they have already been reported to occur in ground and drinking water samples from water works using bank filtration or artificial groundwater recharge downstream from municipal STPs. The presence of PhACs from human medical care in groundwater may, however, also be caused by other sources such as landfill leachates [3-5] or manufacturing residues [6]. Nowadays, and especially in the industrial- ized countries, strong regulations and advanced manu- facturing practices shall prevent such spills. In the past, regulations were not as strong and in several cases the release of production residues was either tolerated or even accepted. But the occurrence of pharmaceutical residues in the environment may also be caused by agri- culture applying large amounts of PhACs as veterinary drugs and feed additives in livestock breeding. Figure 1 shows possible sources and pathways for the occurrence of PhAc residues in the environment. The elimination efficiency of conventional wastewater treatment does not provide a significant reduction of the concentration of pharmaceuticals and their metabolites before they are to re-enter the environment as literature data prove. With the knowledge that wastewater treat- ment plants discharge pharmaceuticals into surface wa-  J. R. FLORES ET AL. Copyright © 2011 SciRes. AJAC 19 Figure 1. Scheme showing possible sources and pathways for the occurrence of pharmaceutical residues in the aquatic environment. ters like rivers and lakes [7,8] as well as into the sea [9,10], strategies for an advanced elimination of these pollutants became overdue. However effluents from wastewater treatment plants (WWTPs) can be considered one of the most important sources of pharmaceuticals in the environment. In the recent years, the presence of pharmaceuticals in WWTPs effluents have been reported [11-16]. Concen- tration of pharmaceuticals in the environment, their temporary evolution and their possible synergic and an- tagonist effects depend not only in the amount dis- charged from WWTPs but also on the geographical area and climate conditions. Because of that the study of the concentration evolution of these pharmaceuticals, related to climate conditions in the studied area, is necessary to evaluate the risk of negative environment effects. Accurate and sensitive methods are necessary for the determination of pharmaceuticals in environmental sam- ples, in order to, evaluate the amount of these compounds that are being discharged into the aquatic environment. HPLC-MS [12,13,17-22] and GC-MS [14,15,23] have been used to determine pharmaceutical in water samples. However, when analyzing highly contaminated samples, such as wastewater, a suppression of the electrospray ionization is likely to occur. Besides, these instruments are still very expensive and consequently not widely dis- tributed. On the contrary, almost, every laboratory has a common HPLC system, thus our alternative proposed in to apply HPLC with diode array detector to determine pharmaceutical residues in environmental samples. Antidepressants and anticancerigenic are class of phar- maceuticals that is extensively used in industrialized countries and they can be too environmental pollutants and cause adverse effects on ecological systems. So, in this paper, a method for routine determination of ten antidepressants (fluvoxamine, fluoxetine, citalo- pram, trazodone, paroxetine, sertraline, clomipramine, imipramine, doxepine, venlafaxine) and three antican- cerigenic used for breast cancer (letrozole, anastrozole, exemestane) in environmental samples is proposed. The method involves sample pre-treatment by solid phase extraction (SPE) for water samples and analytical determination by HPLC with diode array (DAD) detec- tor. The aim of this work was to provide an accurate, sen- sitive and inexpensive alternative to the use of HPLC- MS in the routine determination of these pharmaceuticals. The proposed method has been conveniently validated in waters of several precedence and soils. 2. Experimental 2.1. Chemicals and Reagents All chemicals and solvents used were of analytical re- agent grade. All reagents were from Panreac (Barcelona,  20 J. R. FLORES ET AL. Spain). Clomipramine (CLO), citalopram (CIT) and fluvox- amine (FLV) were supplied by Tocris. Imipramine (IMI) and letrozole (LE) were kindly supplied by Novartis Laboratories. Fluoxetine (FLX), paroxetine (PAR), tra- zodone (TRA) and anastrazole (ANA) were purchased from Sigma-Aldrich, Glaxosmithkline, Farma-Leporl and Astrazeneca laboratories respectively. Exemestane (EXE) and sertraline (SER) were supplied by Pfizer and doxepine (DOX) was purchased from Farmasierra S.A. Standard stock solutions were prepared by dissolving the appropriate amount of the pure substance in 100 mL to give a final concentration of 100 mg/L. LE and EXE were dissolved with ethanol-water 50/50 (v/v); TRA, CIT, FLV, IMI, VEN, ANA and FLX were dissolved with Milli-Q water and DOX, CLO, PAR and SER with methanol. All the solutions were stored under refrigera- tion at 4˚C. Working standard solutions were prepared daily by dilution of the stock standard solution with Milli-Q wa- ter. Mobile phases and buffer solution were prepared from analytical-grade-reagent Na2HPO4, NaH2PO4 and H3PO4 from Panreac (Barcelona, Spain) and HPLC-grade ace- tonitrile from Panreac. The buffers and acetonitrile solu- tions were filtered through 0.45 μm filters (HNWP mem- brane filters). This type of membrane filter was pur- chased from Millipore. 2.2. Chromatographic Conditions A Thermo FinniganTM Surveyor® Plus HPLC system with diode-array detector was utilized. The system was monitored by means of a computer equipped with Chrom Quest 5.0 software, which was used for all measurements and data treatment. Compounds were separated on a 4.6 mm i.d. × 150 mm, 5 μm particle, ultrabase C18 reversed phase column (Análisis Vínicos, Ciudad Real, Spain) with acetonitrile and 70 mM phosphate buffer, pH 2.5 (35:65, v/v), as mobile phase. Isocratic elution was per- formed at a flow rate of 1.5 mL/min. The volume injected was 20 μL. Use of diode-array detection enabled extrac- tion of chromatograms at different wavelengths. The op- timization process was made monitoring the analytes at 230 nm and the validation procedure at 215 and 230 nm. All the analyses were made by duplicate and peak areas were used for the quantification. 2.3. Treatment of the Samples The environmental samples objects of study were water samples from different origin (tap, sea and wastewater) and ground samples. Water samples were collected using glass bottles pre- rinsed with ultra-pure water. Extraction phase-solid (SPE) was used. The SPE car- tridges (Sep-pack Plus tC18, waters) were conditioned using 5 mL of methanol and 5 mL of 10 mM phosphate buffer solution (pH 7.0). The water samples were trans- ferred to the SPE cartridges through a Teflon tube using a vacuum manifold system (Supelco VisiprepTM Sep- pack system, Madrid, Spain) coupled to a vacuum pump (Millipore XF 54 23050). The wastewater samples, prior to extraction were fil- tered through 0.45 μm of size pore filters. Concerning tap and sea water samples the pH was adjusted to 6.5 with HCl and only tap water samples was necessary to fit the ionic strength to 50 mM by addition of NaCl. After the conditioning step, water samples (50 mL) were percolated through the cartridges at a flow rate of 10 mL/min. Only for wastewater samples, the loaded cartridges were washed with 8 mL of 10 mM phosphate buffer (pH 7.0) and 2 mL of methanol:water (30:70, v/v). Finally, the elution was performed with 2 mL of metha- nol. The soil samples belong to the province of Ciudad Real (Spain) and were collected using bottles of polyeth- ylene. First, it was come to the breakage of aggregates with a wood mortar. Later, the soil samples were intro- duced in a furnace to dry them and afterwards were ex- tended until the humidity balances with the one of the atmosphere, removing from time to time. Finally these samples were sifted with a mortar until reduce the size of soil particles. Next, 0.5 g of the soil samples was placed in a conical bottom glass tube and 5 mL of methanol was added. Af- ter, 10 min of vertical agitation the samples were centri- fugated (5000 rpm, 10 min) and the supernatant was transferred into another conical glass tube, this process was repeated three times. Finally the extract was evapo- rated under nitrogen stream and reconstituted with 5 mL of methanol. 3. Results and Discussion 3.1. Optimization of Chromatographic Conditions To optimize the chromatographic separation of the thir- teen analytes studied, several of preliminary experiments was performed testing different mobile phases consisting of methanol, acetonitrile or mixture of both as organic phase and different buffer solution at various concentra- tion. The optimal separation of 13 compounds studied was achieved using an isocratic elution with 70 mM phos- Copyright © 2011 SciRes. AJAC 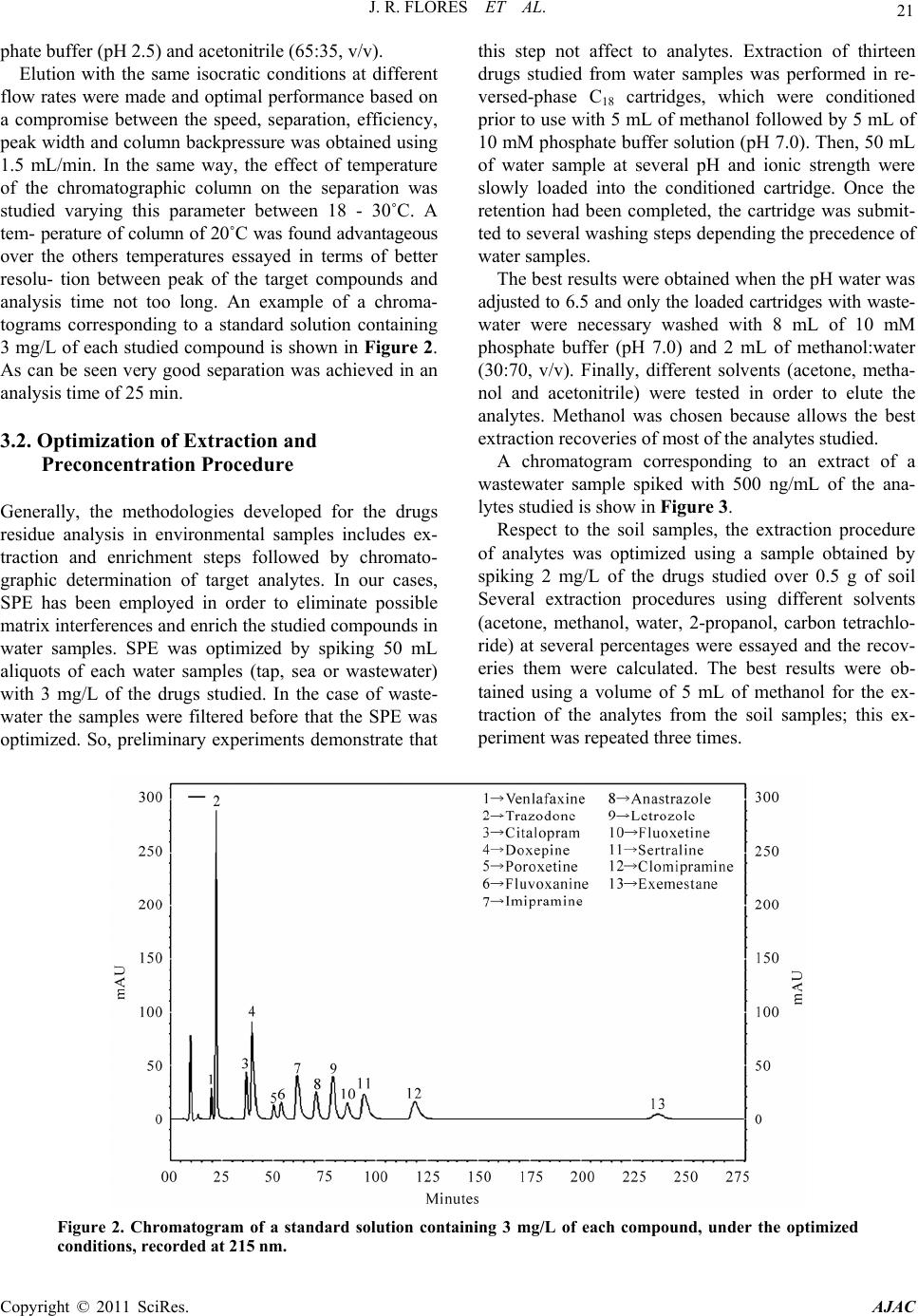 J. R. FLORES ET AL. Copyright © 2011 SciRes. AJAC 21 phate buffer (pH 2.5) and acetonitrile (65:35, v/v). Elution with the same isocratic conditions at different flow rates were made and optimal performance based on a compromise between the speed, separation, efficiency, peak width and column backpressure was obtained using 1.5 mL/min. In the same way, the effect of temperature of the chromatographic column on the separation was studied varying this parameter between 18 - 30˚C. A tem- perature of column of 20˚C was found advantageous over the others temperatures essayed in terms of better resolu- tion between peak of the target compounds and analysis time not too long. An example of a chroma- tograms corresponding to a standard solution containing 3 mg/L of each studied compound is shown in Figure 2. As can be seen very good separation was achieved in an analysis time of 25 min. 3.2. Optimization of Extraction and Preconcentration Procedure Generally, the methodologies developed for the drugs residue analysis in environmental samples includes ex- traction and enrichment steps followed by chromato- graphic determination of target analytes. In our cases, SPE has been employed in order to eliminate possible matrix interferences and enrich the studied compounds in water samples. SPE was optimized by spiking 50 mL aliquots of each water samples (tap, sea or wastewater) with 3 mg/L of the drugs studied. In the case of waste- water the samples were filtered before that the SPE was optimized. So, preliminary experiments demonstrate that this step not affect to analytes. Extraction of thirteen drugs studied from water samples was performed in re- versed-phase C18 cartridges, which were conditioned prior to use with 5 mL of methanol followed by 5 mL of 10 mM phosphate buffer solution (pH 7.0). Then, 50 mL of water sample at several pH and ionic strength were slowly loaded into the conditioned cartridge. Once the retention had been completed, the cartridge was submit- ted to several washing steps depending the precedence of water samples. The best results were obtained when the pH water was adjusted to 6.5 and only the loaded cartridges with waste- water were necessary washed with 8 mL of 10 mM phosphate buffer (pH 7.0) and 2 mL of methanol:water (30:70, v/v). Finally, different solvents (acetone, metha- nol and acetonitrile) were tested in order to elute the analytes. Methanol was chosen because allows the best extraction recoveries of most of the analytes studied. A chromatogram corresponding to an extract of a wastewater sample spiked with 500 ng/mL of the ana- lytes studied is show in Figure 3. Respect to the soil samples, the extraction procedure of analytes was optimized using a sample obtained by spiking 2 mg/L of the drugs studied over 0.5 g of soil Several extraction procedures using different solvents (acetone, methanol, water, 2-propanol, carbon tetrachlo- ride) at several percentages were essayed and the recov- eries them were calculated. The best results were ob- tained using a volume of 5 mL of methanol for the ex- traction of the analytes from the soil samples; this ex- periment was repeated three times. Figure 2. Chromatogram of a standard solution containing 3 mg/L of each compound, under the optimized conditions, recorded at 215 nm.  22 J. R. FLORES ET AL. Figure 3. Chromatogram corresponding: A) blank of wastewater B) extracts from wastewater analyzed spiked with 0.5 mg/L for all analytes. All the extraction procedures are exhaustively descri- bed in the Experimental section. 3.3. Validation of the Method The proposed method was adequately validated in all the environmental samples object of study. Validation was performed by measuring peak areas at the wavelength of maximum absorbance of each of the analytes, to maximize sensitivity. The wavelength used was 230 nm for trazodone, citalopram, doxepine, par- oxetine, fluvoxamine, imipramine, fluoxetine, sertraline and clomipramine, and 215 nm for venlafaxine, anastro- zole, letrozole and exemestane. In order to evaluate the precision of the proposed method within-laboratory repeatability and reproducibil- ity were estimated. To ensure correct quantification of studied analytes, in the environmental samples a spiked extract at 0.12 mg/L of each sample was injected nine times in the same day and nine times in different days. Comparison of the two sets of data was carried out by applying the Snedecor F-test on relative standard devia- tion (RSD) values obtained for migration times and peak areas. In terms of repeatability, it is remarkable that all the relative standard deviations were lower than 3.95% for peak areas and 0.89% for retention times. In terms of reproducibility, the comparison of the averages by means of the Snedecor F-test did not provide any significant difference between both series for a signification level of 0.05 (n = 18). Limits of detection (LOD) and limits of quantification (LOQ) were calculated using the maximal sensitivity allowed by the system and calculating the standard de- viation (SD) of this response. LOD and LOQ were esti- mated by multiplying the SD of blanks by a factor of 3 and 10, respectively. Under these conditions LODs and LOQs obtained were subsequently validated separately by the analysis of six standards prepared at their respect- tive concentrations of all the compounds. The linearity in the response was studied using ma- trix-matched calibration solutions prepared by spiking environmental samples extracts at six concentration lev- els for every compound, ranging from 35 to 1500 ng/mL in the samples. The linear regression equations were calculated using the least-squares method and coeffi- cients of correlation values higher than 0.99 were ob- tained. In Table 1 analytical parameters obtained with the proposed method for the analysis of studied com- pounds in wastewater are shown. Similar results were obtained for the other environmental samples studied. In order to test the accuracy of the proposed method the environmental samples object of study were spiked with studied compounds at several concentrations levels. These samples were analysed using the extraction and chromatographic procedure optimized in this work. Sig- nals obtained from spiked samples were compared with the peak areas obtained by injecting standard solutions directly. Recoveries obtained from soil and wastewater samples spiked at several concentration levels are shown in Table 2. As can be seen, good recoveries ranged be- tween 85 and 100% were obtained for wastewater. Simi- lar results were achieved when tap and sea water were analysed. However, with the extraction procedure opti- mized in this work, poor extraction recoveries of citalo- pram were obtained in soil samples. Whereas recoveries Copyright © 2011 SciRes. AJAC 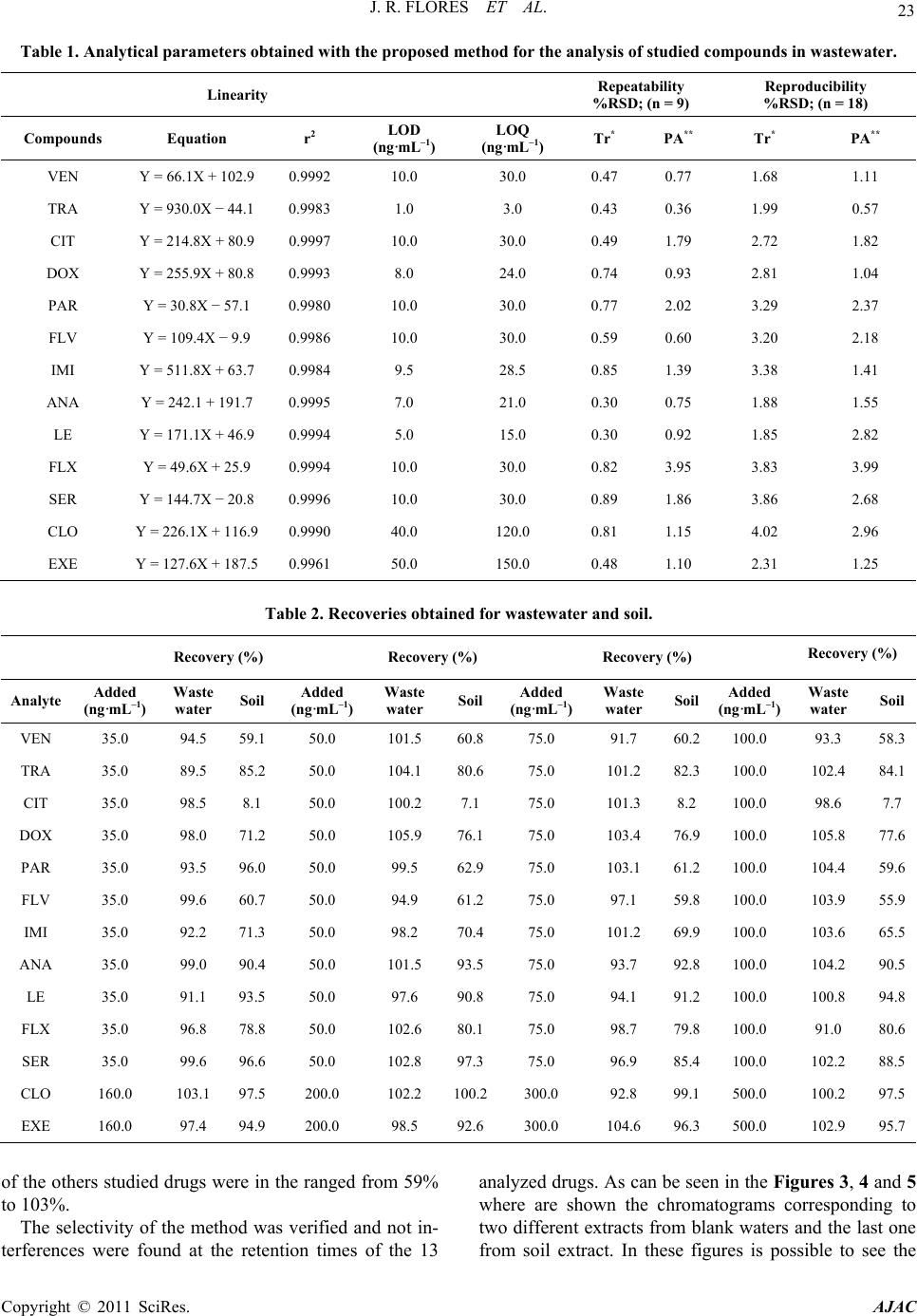 J. R. FLORES ET AL. 23 Table 1. Analytical parameters obtained with the propose d method for the analysis of studied compounds in wastewater. Linearity Repeatability %RSD; (n = 9) Reproducibility %RSD; (n = 18) Compounds Equation r2 LOD (ng·mL–1) LOQ (ng·mL–1) Tr* PA** Tr* PA** VEN Y = 66.1X + 102.9 0.9992 10.0 30.0 0.47 0.77 1.68 1.11 TRA Y = 930.0X − 44.1 0.9983 1.0 3.0 0.43 0.36 1.99 0.57 CIT Y = 214.8X + 80.9 0.9997 10.0 30.0 0.49 1.79 2.72 1.82 DOX Y = 255.9X + 80.8 0.9993 8.0 24.0 0.74 0.93 2.81 1.04 PAR Y = 30.8X − 57.1 0.9980 10.0 30.0 0.77 2.02 3.29 2.37 FLV Y = 109.4X − 9.9 0.9986 10.0 30.0 0.59 0.60 3.20 2.18 IMI Y = 511.8X + 63.7 0.9984 9.5 28.5 0.85 1.39 3.38 1.41 ANA Y = 242.1 + 191.7 0.9995 7.0 21.0 0.30 0.75 1.88 1.55 LE Y = 171.1X + 46.9 0.9994 5.0 15.0 0.30 0.92 1.85 2.82 FLX Y = 49.6X + 25.9 0.9994 10.0 30.0 0.82 3.95 3.83 3.99 SER Y = 144.7X − 20.8 0.9996 10.0 30.0 0.89 1.86 3.86 2.68 CLO Y = 226.1X + 116.9 0.9990 40.0 120.0 0.81 1.15 4.02 2.96 EXE Y = 127.6X + 187.5 0.9961 50.0 150.0 0.48 1.10 2.31 1.25 Table 2. Recoveries obtained for wastewater and soil. Recovery (%) Recovery (%) Recovery (%) Recovery (%) Analyte Added (ng·mL–1) Waste water Soil Added (ng·mL–1) Waste water Soil Added (ng·mL–1)Waste water Soil Added (ng·mL–1) Waste water Soil VEN 35.0 94.5 59.1 50.0 101.5 60.875.0 91.7 60.2 100.0 93.3 58.3 TRA 35.0 89.5 85.2 50.0 104.1 80.675.0 101.2 82.3 100.0 102.4 84.1 CIT 35.0 98.5 8.1 50.0 100.2 7.1 75.0 101.3 8.2 100.0 98.6 7.7 DOX 35.0 98.0 71.2 50.0 105.9 76.175.0 103.4 76.9 100.0 105.8 77.6 PAR 35.0 93.5 96.0 50.0 99.5 62.975.0 103.1 61.2 100.0 104.4 59.6 FLV 35.0 99.6 60.7 50.0 94.9 61.275.0 97.1 59.8 100.0 103.9 55.9 IMI 35.0 92.2 71.3 50.0 98.2 70.475.0 101.2 69.9 100.0 103.6 65.5 ANA 35.0 99.0 90.4 50.0 101.5 93.575.0 93.7 92.8 100.0 104.2 90.5 LE 35.0 91.1 93.5 50.0 97.6 90.875.0 94.1 91.2 100.0 100.8 94.8 FLX 35.0 96.8 78.8 50.0 102.6 80.175.0 98.7 79.8 100.0 91.0 80.6 SER 35.0 99.6 96.6 50.0 102.8 97.375.0 96.9 85.4 100.0 102.2 88.5 CLO 160.0 103.1 97.5 200.0 102.2 100.2300.0 92.8 99.1 500.0 100.2 97.5 EXE 160.0 97.4 94.9 200.0 98.5 92.6300.0 104.6 96.3 500.0 102.9 95.7 of the others studied drugs were in the ranged from 59% to 103%. The selectivity of the method was verified and not in- terferences were found at the retention times of the 13 analyzed drugs. As can be seen in the Figures 3, 4 and 5 where are shown the chromatograms corresponding to two different extracts from blank waters and the last one from soil extract. In these figures is possible to see the Copyright © 2011 SciRes. AJAC  24 J. R. FLORES ET AL. Figure 4. Chromatogram corresponding: A) blank of seawater B) extracts from seawater analyzed spiked with 0.5 mg/L for all analytes. Figure 5. Chromatogram corresponding: A) blank of soil B) extracts from soil analyzed spiked with 2 mg/L for all compounds. differences after an spiked of different amounts of our drugs in waters (0.5 mg/L) and soil (2 mg/L). The selec- tivity was also determined, by measurement of peaks homogeneity using the techniques of normalization and comparison of spectra from different peak sections and absorbance measures at two wavelengths [24]. Both te- chniques proved again to have a high level of purity of the peak corresponding to the studied compounds in all the samples. In conclusion the proposed method showed a good selectivity for the environmental samples chosen for our analysis. 4. Applications To demonstrate the applicability of the described method in this work, several environmental samples as water of different precedence (tap, sea and waste) and soils be- long to different zones from the province of Ciudad Real Copyright © 2011 SciRes. AJAC  J. R. FLORES ET AL. 25 were analyzed. The water samples analysed were sam- ples in different weather stations. All the samples were fortified with ranged between 35 and 1000 ng/mL con- centrations of studied compounds and submitted to the analytical procedure described for each case in this work. The recoveries obtained in all the water samples ana- lyzed were ranged between 90 and 109% for all com- pounds studied. Chromatogram of a sea water sample fortified with 500 ng/mL of drugs studied is shown in Figure 4 as ex- ample. As can be observed in the figure the high selectivity provided by the proposed method allows a reliable iden- tification of the target compounds in wastewater and obtaining similar results in all the water samples ana- lyzed. Respect to soil samples analyzed, good recoveries were obtained (ranged between 59 and 103%) for all the target compounds except for citalopram that was around 8%. A chromatogram corresponding to a spiked soil sam- ple is shown in Figure 5. Good selectivity was observed too, in this type of samples. 5. Conclusions In the present work, a simple and fast multi-residue method based on SPE step followed by an HPLC-DAD determination has been developed for the simultaneous extraction and analysis of ten antidepressants (fluvox- amine, fluoxetine, citalopram, trazodone, venlafaxine, paroxetine, doxepine, sertaline, imipramine and clomi- pramine) and three aromatase inhibitors (letrozole, an- astrazole and exemestane) in environmental samples. It has been shown that this method represents an easy and fast analytical approach, viable for routine analysis, us- ing instrumental very simple, available in almost every laboratory. The proposed method was exhaustively validated in terms of linearity, accuracy, specificity and precision in environmental samples. Quantitative recoveries were ob- tained for all the target compounds ranging from 59 and 103% except citalopram determination which is ex- tracted from soil samples with recoveries around of 8%. Linearity with R2 > 0.994 and precision of with the RSD (%) between 0.30% and 3.99% was very satisfactory. The SPE step optimized allows not only the elimina- tion of hydrophobic interferences but also an important sample preconcentration which results in LOD(s) in wa- ter samples between 1 and 50 ng/mL. HPLC-DAD is an inexpensive analytical technique compared to HPLC-MS and, because of this is a useful and affordable alternative to HPLC-MS for routine analysis of pharmaceuticals. It can be a useful tool to known the amount of these com- pounds discharged from wastewater treatment plants (WWTPs) to the aquatic environment and to evaluate the effect of WWTPs in the elimination of pharmaceutical compounds. In conclusion, methods are being developed to en- hance the capabilities for measuring emerging chemical contaminants and their associated degradation products in the environment. Therefore, prioritization of com- pounds investigated as the analyzed drugs requires care- ful evaluation of the potential for their environmental occurrence and persistence, potential health effects, and the appropriate level at which they should be measured. 6. References [1] T. Heberer, “Occurrence, Fate, and Removal of Pharma- ceutical Residues in the Aquatic Environment: A Review of Recent Research Data,” Toxicology Letters, Vol. 131, 2002, pp. 5-17. doi:10.1016/S0378-4274(02)00041-3 [2] T. A. Ternes, “Occurence of Drugs in German Sewage Treatment Plant and Rivers,” Water Research, Vol. 32, 1998, pp. 3245-3260. [3] W. P. Eckel, B. Ross and R. K. Isensee, “Pentobarbital Found in Ground Water,” Ground Water, Vol. 31, 1993, pp. 801-804. doi:10.1111/j.1745-6584.1993.tb00853.x [4] J. V. Holm, K. Rügge, P. L. Bjerg and T. H. Christensen, “Occurrence and Distribution of Pharmaceutical Organic Compounds in the Groundwater Downgradient of a Land- fill (Grindsted, Denmark),” Environmental Science & Technology, Vol. 29, 1995, pp. 1415-1420. doi:10.1021/es00005a039 [5] M. Ahel and I. Jelicic, “Phenazone Analgesics in Soil and Groundwater below a Municipal Solid Waste Landfill,” In: C. G. Daughton and T. Jones-Lepp, Eds., Pharmaceu- tical and Personal Care Products in the Environment: Scientific and Regulatory Issues. Symposium Series 791, American Chemical Society, Washington DC, 2001, pp. 100-115. [6] K. Reddersen, T. Heberer and U. Dünnbier, “Identifica- tion and Significance of Phenazone Drugs and Their Me- tabolites in Ground and Drinking Water,” Chemosphere, Vol. 49, 2002, pp. 539-544. doi:10.1016/S0045-6535(02)00387-9 [7] K. Fent, A. A. Weston and D. Caminada, “Ecotoxicology of Human Pharmaceuticals,” Aquatic Toxicology, Vol. 76, 2006, pp. 122-159. doi:10.1016/j.aquatox.2005.09.009 [8] F. Sacher, F. T. Lange, H. J. Brauch and I. Blankenhorn, “Pharmaceuticlas in Groundwaters Analytical Methods and Results of a Monitoring Program in Baden-Würt- temberg Germany,” Journal of Chromatography A, Vol. 938, 2001, pp. 199-210. doi:10.1016/S0021-9673(01)01266-3 [9] G. L. Brun, M. Bernier, R. Losier, K. Doe, P. Jackman and H. B. Lee, “Pharmaceutically Active Compounds in Atlantic Canadian Sewage Treatment Plant Effluents and Copyright © 2011 SciRes. AJAC  J. R. FLORES ET AL. Copyright © 2011 SciRes. AJAC 26 Receiving Waters, and Potential for Environmental Ef- fects as Measured by Acute and Chronic Aquatic Toxic- ity,” Environmental Toxicology and Chemistry, Vol. 25, 2006, pp. 2163-2176. doi:10.1897/05-426R.1 [10] C. D. Metcalfe, X. S. Miao, B. G. Koening and J. Struger, “Distribution of Acidic and Neutral Drugs in Surface Waters Near Sewage Treatment Plants in the Lower Great Lakes, Canada,” Environmental Toxicology and Chemistry, Vol. 22, 2003, pp. 2881-2889. doi:10.1897/02-627 [11] T. A. Ternes, “Occurrence of Drugs in German Sewage Treatment Plants and Rivers,” Water Resources, Vol. 32, 1998, pp. 3245-3260. doi:10.1016/S0043-1354(98)00099-2 [12] T. A. Ternes, M. Bonerz and T. Schidt, “Determination of Neutral Pharmaceuticals in Wastewater and Rivers by Liquid Chromatography-Electrospray Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 938, 2001, pp. 175-185. doi:10.1016/S0021-9673(01)01205-5 [13] M. Farré, I. Ferrer, A. Ginebreda, M. Figueras, L. Oliv- ella, L. Tirapu, M. Vilanova and D. Barceló, “Determina- tion of Drugs in Surface Water and Wastewater Samples by Liquid Chromatography-Mass Spectrometry: Methods and Preliminary Results Including Toxicity Studies with Vibrio Fischeri,” Journal of Chromatography A, Vol. 938, 2001, pp. 187-197. doi:10.1016/S0021-9673(01)01154-2 [14] I. Rodríguez, J. B. Quintana, J. Carpinteiro, A. M. Carro, R. A. Lorenzo and R. Cela, “Determination of Acidic Drugs in Sewage Water by Gás Chromatography-Mass Spectrometry as Tert-Butyldimethylsilyl Derivatives,” Journal of Chromatography A, Vol. 985, 2003 pp. 265-274. doi:10.1016/S0021-9673(02)01528-5 [15] I. Rodríguez, J. Carpinteiro, J. B. Quintana, A. M. Carro, R. A. Lorenzo and R. Cela, “Solid-Phase Microextraction with On-Fiber Derivation for the Analysis of Anti-Inflamatory Drugs in Water Samples,” Journal of Chromatography A, Vol. 1024, 2004, pp. 1-8. doi:10.1016/j.chroma.2003.10.049 [16] C. González-Barreiro, M. Lores, M. C. Cassis and R. Cela, “Simultaneous Determination of Neutral and Acidic Pharmaceuticals in Wastewater by High-Performance Liquid Chromatography-Post-Column Photochemically Induced Fluorimetry,” Journal of Chromatography A, Vol. 993, 2003, pp. 29-37. doi:10.1016/S0021-9673(03)00392-3 [17] W. Gebhardt and H. F. Schröder, “Liquid Chromatogra- phy-(Tandem) Mass Spectrometry for the Follow-Up of the Elimination of Persistent Pharmaceuticals During Waste-Water Treatment Applying Biological Wastewater Treatment and Advanced Oxidation,” Journal of Chro- matography A, Vol. 1160, 2007, pp. 34-43. doi:10.1016/j.chroma.2007.05.075 [18] C. Nebot, S. W. Gibb and K. G. Boyd, “Quantification of Human Pharmaceuticals in Water Samples by High Per- formance Liquid Chromatography-Tandem Mass Spec- trometry,” Analytica Chimica Acta, Vol. 598, 2007, pp. 87-94. doi:10.1016/j.aca.2007.07.029 [19] S. L. MacLeod, P. Sudhir and C. S. Wong, “Stereoisomer Analysis of Wastewater-Derived Β-Blockers, Selective Serotonin Re-Uptake Inhibitors, and Salbutamol by High-Performance Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1170, 2007, pp. 23-33. doi:10.1016/j.chroma.2007.09.010 [20] J. Radjenovic, M. Petrovic and D. Barceló, “Advanced Mass Spectrometric Method Applied to the Study off Ate and Renoval of Pharmaceuticals in Wastewater Treat- ment,” Trends in Analytical Chemistry, Vol. 26, 2007, pp. 1132-1144. doi:10.1016/j.trac.2007.10.002 [21] T. Vasskog, T. Anderssen, S. Pedersen-Bjergaard, R. Kallenborn and E. Jensen, “Occurrence of Selective Se- rotonin Reuptake Inhibitors in Sewage and Receiving Waters at Spitsbergen and in Norway,” Journal of Chro- matography A, Vol. 1185, 2008, pp. 194-205. doi:10.1016/j.chroma.2008.01.063 [22] K. H. Langford and K. V. Thomas, “Determination of Pharmaceutical Compounds in Hospital Effluents and Their Contribution to Wastewater Treatment Works,” Environment International, Vol. 35, 2009, pp. 766-770. doi:10.1016/j.envint.2009.02.007 [23] M. J. Gómez, A. Agüera, M. Mezcua, J. Hurtado, F. Mo- cholí and A. R. Fernández-Alba, “Simultaneous Analysis of Neutral and Acidic Pharmaceuticals as Well as Related Compounds by Gas Chromatography-Tandem Mass Spectrometry in Wastewater,” Talanta, Vol. 73, 2007, pp. 314-320. doi:10.1016/j.talanta.2007.03.053 [24] L. Huber, “Applications of Diode-Array Experience,” Hewlett Packard, Waldbronn, 1989, Publication No. 12-5953-2330.
|