 Open Journal of Stomatology, 2013, 3, 402-410 OJST http://dx.doi.org/10.4236/ojst.2013.38068 Published Online November 2013 (http://www.scirp.org/journal/ojst/) Cleidocranial dysplasia with a rare mutation: Study of a family with review of literature Ahmet Ercan Sekerci2*, Burhan Balta1, Oğuzhan Bahadir1, Yildiray Sisman2, Munis Dundar1, Turgut Tursem Tokmak3, Stefan Mundlos4 1Department of Medical Genetics, Faculty of Medicine, Erciyes University, Kayseri, Turkey 2Department of Oral Diagnosis and Radiology, Faculty of Dentistry, Erciyes University, Kayseri, Turkey 3Department of Radiodiagnostics, Faculty of Medicine, Erciyes University, Kayseri, Turkey 4Institute of Medical Genetics, Charité-Universitaetsmedizin, Berlin, Germany Email: *aercansekerci@hotmail.com Received 13 September 2013; revised 15 October 2013; accepted 23 October 2013 Copyright © 2013 Ahmet Ercan Sekerci et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Introduction: The present study was aimed at ad- vancing the understanding of the pathogenesis of cleidocranial dysplasia (CCD) by presenting a case study based on history, physical examination, typical radiological features, and molecular analysis and a review of the literature. Methods: This study began with a 23-year-old boy (proband) who was referred to the department of oral and maxillofacial radiol- ogy with chief complaint of the upper-left first molar tooth and routine dental examination. While evalu- ating the panoramic radiograph, the patient had ap- proximately 57 teeth in his both of the jaws. Clinical, radiographical and molecular features of the pro- band, two siblings and their parents were examined and then, DNA analysis was performed. Results: Overall, we present 3 CCD patients with a mutation in the VWRPY motif. The deletion of c. 1754_1757 delTTTG (NM_001024630.2) is determined and it leads to a frame shift mutation and stop codon, p. V585Gfs56X. Conclusions: The present study em- phasized the importance of further clinical and mo- lecular investigation when even a single case of CCD is identified within a family. This is the first study performed in Turkey about a family with a mutation in the VWRPY motif. Genotype-phenotype associa- tion studies in individuals with CCD are necessary to provide important insights into molecular mechanisms associated with this disease. Keywords: Cleidocranial Dysplasia; Mutation; RUNX2; Gene; Impacted Supernumeraries 1. INTRODUCTION Cleidocranial dysplasia (CCD), also known as Marie and Sainton disease [1] or cleidocranial dysostosis, is a rare congenital defect of autosomal dominance inheritance that is characterized by persistently open sutures or de- layed closure of sutures, hypoplasia or aplasia of the clavicles, cone-shaped thorax, short stature, supernumer- ary teeth, delayed eruption of permanent dentition and other skeletal anomalies [2]. Dental anomalies and some degrees of clavicular hy- poplasia seem to be consistent features of the disorder [3]. A variety of dental problems may occur in CCD. This condition is of clinical significance to dentistry due to the involvement of the facial bones, a prolonged retention of deciduous teeth and several impacted permanent succes- sors and supernumerary elements, sometimes accompa- nied by follicular cysts and eruptive pseudocysts [4]. CCD manifests itself as a condition in which teeth fail to erupt, which is thought to be due to the absence of cellu- lar cementum and an increase in the amount of acellular cementum of the roots of the affected teeth [5]. For these reasons, dental management is a significant aspect of the health care of affected persons [6]. This disorder is transmitted as an autosomal dominant trait or it’s caused by a spontaneous genetic mutation and is present at a frequency of one in one million individu- als [7]. It affects both males and females equally [8]. It has been demonstrated that mutation of the runt-related transcription factor 2 gene (RUNX2), located at chromo- some 6p21, is associated with CCD [9]. This gene en- codes a protein necessary for the correct functioning of osteoblastic differentiation and bone formation. RUNX2 has been shown to bind to the DNA sequence element called RUNX-binding site (PyGPyGGT) and to regulate *Corresponding author. OPEN ACCESS 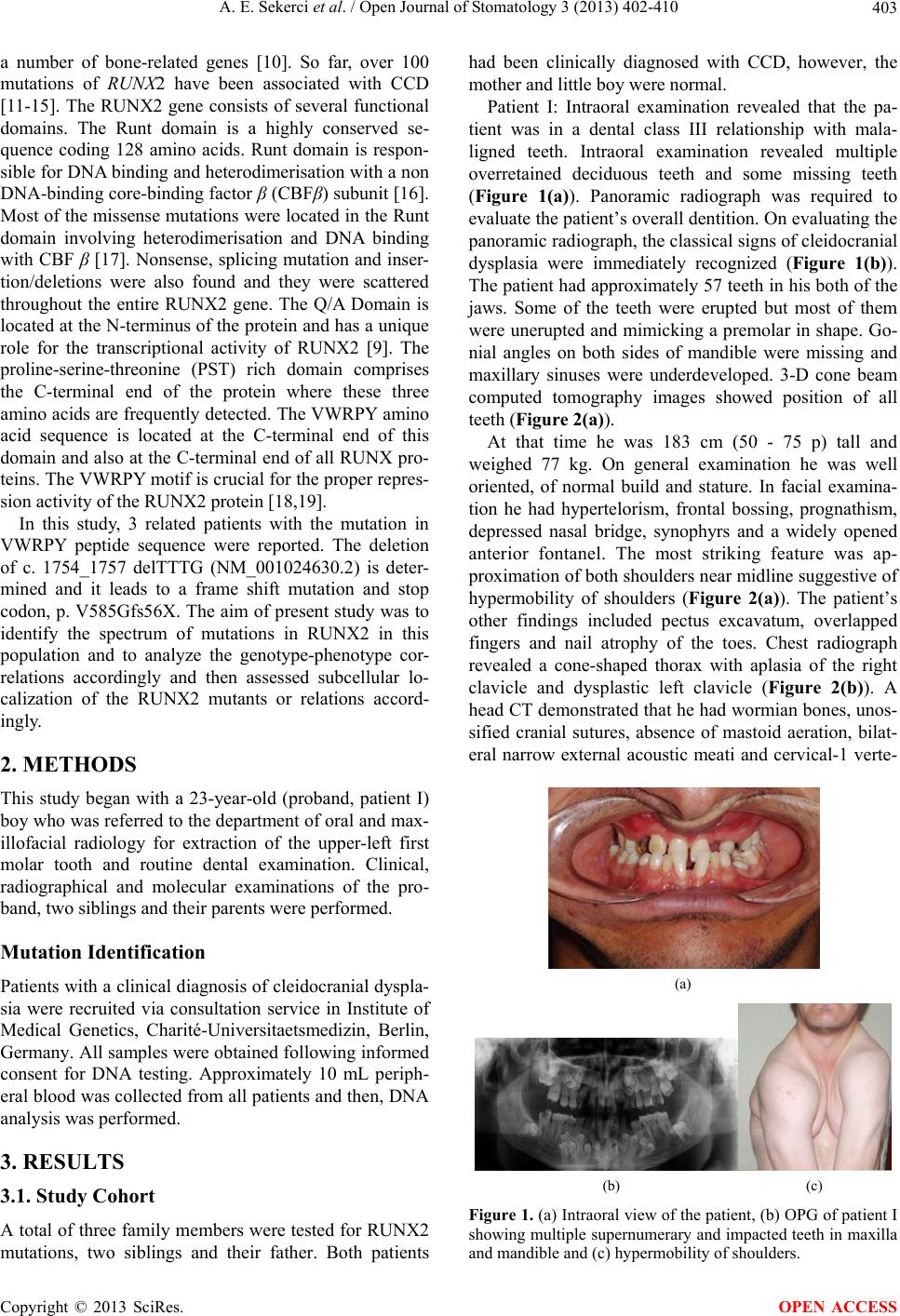 A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 403 a number of bone-related genes [10]. So far, over 100 mutations of RUNX2 have been associated with CCD [11-15]. The RUNX2 gene consists of several functional domains. The Runt domain is a highly conserved se- quence coding 128 amino acids. Runt domain is respon- sible for DNA binding and heterodimerisation with a non DNA-binding core-binding factor β (CBFβ) subunit [16]. Most of the missense mutations were located in the Runt domain involving heterodimerisation and DNA binding with CBF β [17]. Nonsense, splicing mutation and inser- tion/deletions were also found and they were scattered throughout the entire RUNX2 gene. The Q/A Domain is located at the N-terminus of the protein and has a unique role for the transcriptional activity of RUNX2 [9]. The proline-serine-threonine (PST) rich domain comprises the C-terminal end of the protein where these three amino acids are frequently detected. The VWRPY amino acid sequence is located at the C-terminal end of this domain and also at the C-terminal end of all RUNX pro- teins. The VWRPY motif is crucial for the proper repres- sion activity of the RUNX2 protein [18,19]. In this study, 3 related patients with the mutation in VWRPY peptide sequence were reported. The deletion of c. 1754_1757 delTTTG (NM_001024630.2) is deter- mined and it leads to a frame shift mutation and stop codon, p. V585Gfs56X. The aim of present study was to identify the spectrum of mutations in RUNX2 in this population and to analyze the genotype-phenotype cor- relations accordingly and then assessed subcellular lo- calization of the RUNX2 mutants or relations accord- ingly. 2. METHODS This study began with a 23-year-old (proband, patient I) boy who was referred to the department of oral and max- illofacial radiology for extraction of the upper-left first molar tooth and routine dental examination. Clinical, radiographical and molecular examinations of the pro- band, two siblings and their parents were performed. Mutation Identification Patients with a clinical diagnosis of cleidocranial dyspla- sia were recruited via consultation service in Institute of Medical Genetics, Charité-Universitaetsmedizin, Berlin, Germany. All samples were obtained following informed consent for DNA testing. Approximately 10 mL periph- eral blood was collected from all patients and then, DNA analysis was performed. 3. RESULTS 3.1. Study Cohort A total of three family members were tested for RUNX2 mutations, two siblings and their father. Both patients had been clinically diagnosed with CCD, however, the mother and little boy were normal. Patient I: Intraoral examination revealed that the pa- tient was in a dental class III relationship with mala- ligned teeth. Intraoral examination revealed multiple overretained deciduous teeth and some missing teeth (Figure 1(a)). Panoramic radiograph was required to evaluate the patient’s overall dentition. On evaluating the panoramic radiograph, the classical signs of cleidocranial dysplasia were immediately recognized (Figure 1(b)). The patient had approximately 57 teeth in his both of the jaws. Some of the teeth were erupted but most of them were unerupted and mimicking a premolar in shape. Go- nial angles on both sides of mandible were missing and maxillary sinuses were underdeveloped. 3-D cone beam computed tomography images showed position of all teeth (Figure 2(a)). At that time he was 183 cm (50 - 75 p) tall and weighed 77 kg. On general examination he was well oriented, of normal build and stature. In facial examina- tion he had hypertelorism, frontal bossing, prognathism, depressed nasal bridge, synophyrs and a widely opened anterior fontanel. The most striking feature was ap- proximation of both shoulders near midline suggestive of hypermobility of shoulders (Figure 2(a)). The patient’s other findings included pectus excavatum, overlapped fingers and nail atrophy of the toes. Chest radiograph revealed a cone-shaped thorax with aplasia of the right clavicle and dysplastic left clavicle (Figure 2(b)). A head CT demonstrated that he had wormian bones, unos- sified cranial sutures, absence of mastoid aeration, bilat- eral narrow external acoustic meati and cervical-1 verte- (a) (b) (c) Figure 1. (a) Intraoral view of the patient, (b) OPG of patient I showing multiple supernumerary and impacted teeth in maxilla and mandible and (c) hypermobility of shoulders. Copyright © 2013 SciRes. OPEN ACCESS 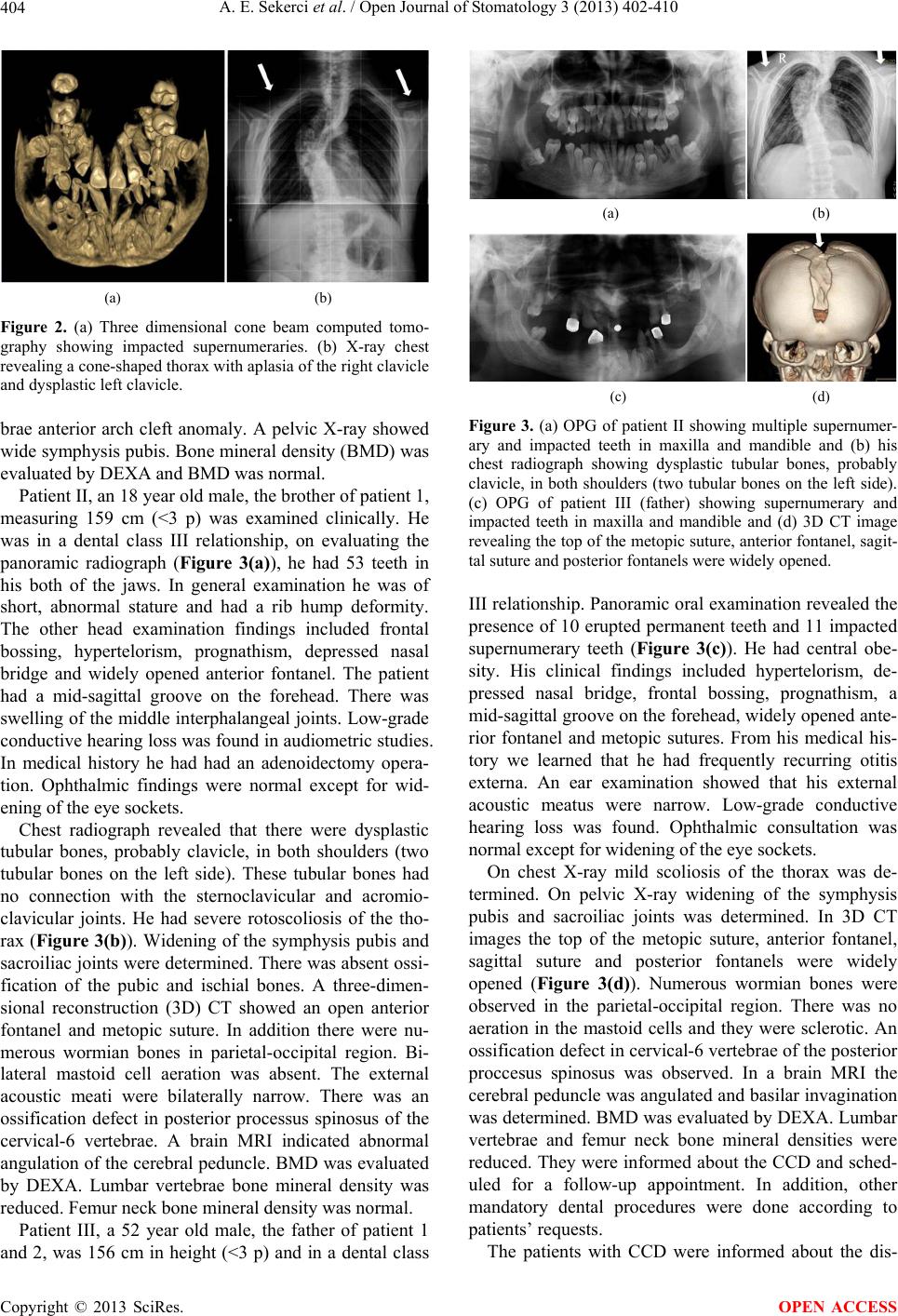 A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 404 (a) (b) Figure 2. (a) Three dimensional cone beam computed tomo- graphy showing impacted supernumeraries. (b) X-ray chest revealing a cone-shaped thorax with aplasia of the right clavicle and dysplastic left clavicle. brae anterior arch cleft anomaly. A pelvic X-ray showed wide symphysis pubis. Bone mineral density (BMD) was evaluated by DEXA and BMD was normal. Patient II, an 18 year old male, the brother of patient 1, measuring 159 cm (<3 p) was examined clinically. He was in a dental class III relationship, on evaluating the panoramic radiograph (Figure 3(a)), he had 53 teeth in his both of the jaws. In general examination he was of short, abnormal stature and had a rib hump deformity. The other head examination findings included frontal bossing, hypertelorism, prognathism, depressed nasal bridge and widely opened anterior fontanel. The patient had a mid-sagittal groove on the forehead. There was swelling of the middle interphalangeal joints. Low-grade conductive hearing loss was found in audiometric studies. In medical history he had had an adenoidectomy opera- tion. Ophthalmic findings were normal except for wid- ening of the eye sockets. Chest radiograph revealed that there were dysplastic tubular bones, probably clavicle, in both shoulders (two tubular bones on the left side). These tubular bones had no connection with the sternoclavicular and acromio- clavicular joints. He had severe rotoscoliosis of the tho- rax (Figure 3(b)). Widening of the symphysis pubis and sacroiliac joints were determined. There was absent ossi- fication of the pubic and ischial bones. A three-dimen- sional reconstruction (3D) CT showed an open anterior fontanel and metopic suture. In addition there were nu- merous wormian bones in parietal-occipital region. Bi- lateral mastoid cell aeration was absent. The external acoustic meati were bilaterally narrow. There was an ossification defect in posterior processus spinosus of the cervical-6 vertebrae. A brain MRI indicated abnormal angulation of the cerebral peduncle. BMD was evaluated by DEXA. Lumbar vertebrae bone mineral density was reduced. Femur neck bone mineral density was normal. Patient III, a 52 year old male, the father of patient 1 and 2, was 156 cm in height (<3 p) and in a dental class (a) (b) (c) (d) Figure 3. (a) OPG of patient II showing multiple supernumer- ary and impacted teeth in maxilla and mandible and (b) his chest radiograph showing dysplastic tubular bones, probably clavicle, in both shoulders (two tubular bones on the left side). (c) OPG of patient III (father) showing supernumerary and impacted teeth in maxilla and mandible and (d) 3D CT image revealing the top of the metopic suture, anterior fontanel, sagit- tal suture and posterior fontanels were widely opened. III relationship. Panoramic oral examination revealed the presence of 10 erupted permanent teeth and 11 impacted supernumerary teeth (Figure 3(c)). He had central obe- sity. His clinical findings included hypertelorism, de- pressed nasal bridge, frontal bossing, prognathism, a mid-sagittal groove on the forehead, widely opened ante- rior fontanel and metopic sutures. From his medical his- tory we learned that he had frequently recurring otitis externa. An ear examination showed that his external acoustic meatus were narrow. Low-grade conductive hearing loss was found. Ophthalmic consultation was normal except for widening of the eye sockets. On chest X-ray mild scoliosis of the thorax was de- termined. On pelvic X-ray widening of the symphysis pubis and sacroiliac joints was determined. In 3D CT images the top of the metopic suture, anterior fontanel, sagittal suture and posterior fontanels were widely opened (Figure 3(d)). Numerous wormian bones were observed in the parietal-occipital region. There was no aeration in the mastoid cells and they were sclerotic. An ossification defect in cervical-6 vertebrae of the posterior proccesus spinosus was observed. In a brain MRI the cerebral peduncle was angulated and basilar invagination was determined. BMD was evaluated by DEXA. Lumbar vertebrae and femur neck bone mineral densities were reduced. They were informed about the CCD and sched- uled for a follow-up appointment. In addition, other mandatory dental procedures were done according to patients’ requests. The patients with CCD were informed about the dis- Copyright © 2013 SciRes. OPEN ACCESS  A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 Copyright © 2013 SciRes. 405 OPEN ACCESS Table 1. Used primers. ease. Mandatory dental procedures were done according to patients’ requests. They didn’t accept treatment pro- cedures involve a combination of maxillofacial surgery, orthodontics, and prosthodontics. The proband’s little brother and mother clinically and radiographically were normal. 3.2. Genetic Analysis The cDNA sequence of CBFA1 was determined by a combination of nested RT-PCR with lymphoblast RNA and genomic sequencing of PAC clones. For the analysis of lymphoblast RNA, PCR primers were designed to amplify the 1.4 kb cDNA in 2 overlapping fragments, from the start of the Runt domain in exon 1 to the stop codon in exon 7. The primers for the first/second round PCR were AML3F1/AML3F2 and CBFA-7/CBFA-6, respectively. In the second round, the 5’ or 3’ primer was replaced by AML3F5 or AML3R4, respectively. The characterization of the 5’ end of CBFA1, as well as all the intron-exon boundaries, were determined by direct sequencing (using an ABI sequencer) of the PAC clones with appropriate exon primers. These primers were de- rived from published sequences by the sequencing of RT–PCR products, or from conserved segments in the mouse. The primers used are shown in Table 1. 4. DISCUSSION Dental findings in patients with CCD are characterized by abnormal dentition, including multiple supernumerary teeth, retention of primary teeth, eruption failure of per- manent teeth, malocclusion, irregular forms of dentition, wide spacing in the lower incisor area, supernumerary tooth germs, parallel-sided ascending rami, cysts in their gums that usually form around extra teeth [20] and in- crease in odontogenesis leading to excessive number of supernumerary teeth [21]. Suggested factors of over re- tained deciduous teeth are lack of eruption potential and lack of cellular cementum on roots of permanent teeth, delayed mineralization of teeth, physical barrier-abnor- mal density of bone overlying the succedaneous teeth, and failure of bony crypt to resorb. Suggested etiology for supernumerary teeth is incomplete or severely de- layed resorption of the dental lamina, which is then reac- tivated at the time of crown completion in the normal permanent teeth [22]. Delayed or arrested eruption has also been attributed to lack of cellular cementum. Less commonly alterations of hard dental tissues, eruption cysts, a high propensity for caries and narrow high palate have been found [23]. The most consistent dental find- ings in individuals with CCD are the presence of the sec- ond permanent molar with the primary dentition (80%), wide spacing in the lower incisor area, supernumerary tooth germs (70%), and parallel-sided ascending rami [20]. Individuals with CCD are more likely to have an underbite and to have cysts in their gums that usually form around extra teeth [24]. In present sudy, the siblings have multiple impacted teeth in maxilla and mandible. Underdevelopment of the maxilla and relative mandibu- lar prognathism are common [25]. There is a predisposi- tion to develop numerous supernumerary teeth, particu- larly in mandibular premolar and maxillary anterior re- gions [26]. Other than delayed, the permanent molars generally erupt without incident [27] as siblings in the present sdudy. The differential diagnosis of CCD includes osteogene- sis imperfecta (frequent fractures), pycnodysostosis (skeletal density), congenital hypothyroidism (disturbed thyroid metabolism). Gardner syndrome, Hallerman- Streiff syndrome (narrow face, hypotrichosis, mi- crophthalmia), and the orofaciodigital syndrome type I [6], Crane-Heise syndrome, mandibuloacral dysplasia, Sense Antisense Exon 0 AML3F8/5-GAGAATGCTAACTCGCCTCCAG AML3R8/5-GAGACCACCCGAGTCAGTGAGTCG CBFA-9/5-CCACGACAACCGCACCATG MCBFA-1/5-ATGCGTATTCCTGTAGATCCG Exon 1 MCBFA-3/5 -AGCGACGTGAGCCCGGTG CBFA-8/5-CATGGTGCGGTTGTCGTGG Exon 2 AML3F2/5 -CGGAGAGGTACCAGATGGGAC AML3R1/5 -GTCCCATCTGGTACCTCTCCG; Exon 3 AML3F2/5 -CGGAGAGGTACCAGATGGGAC AML3R1/5 -GTCCCATCTGGTACCTCTCCG Exon 4 AML3F4/5 -ACAAATCCTCCCCAAGTAGCT AML3R2/5 -TGATAGGTAGCTACTTGGGGA Exon 5 AML3F5/5 -TACCACCCCGCTGTCTTCCAC AML3R4/5 -GAAATGCGCCTAGGCACATCG Exon 6 AML3F7/5 -ATGATGACACTGCCACCTCTG AML3R7/5 -TGCCTGGCTCTTCTTACTGAG AML3R5/5 –GGTTGGAGAAGCGGCTCTCAG CBFA-6/5 –CACTGGGCCACTGCTGAG Exon 7 CBFA-7/5 -GATACGTGTGGGATGTGGC 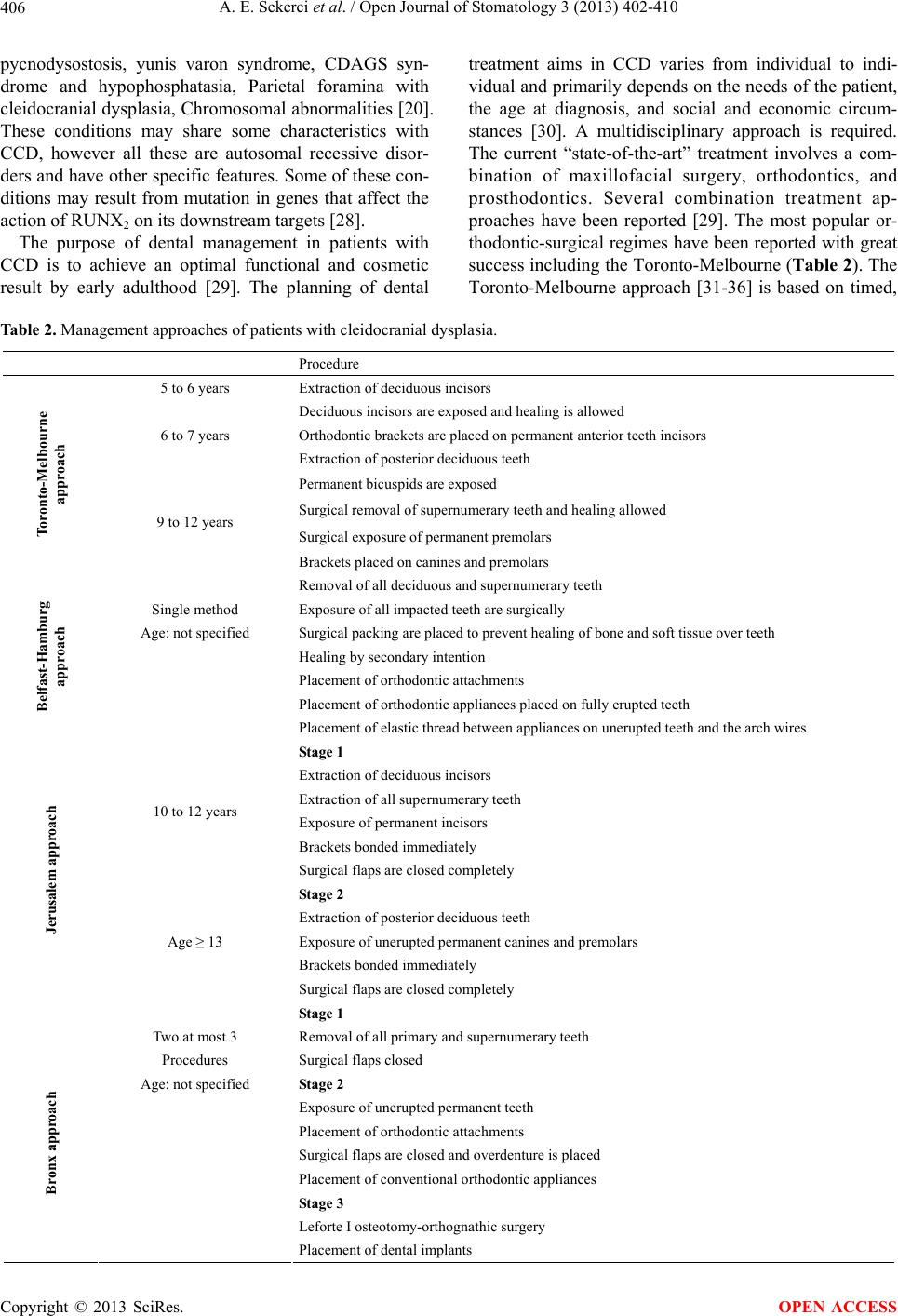 A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 406 pycnodysostosis, yunis varon syndrome, CDAGS syn- drome and hypophosphatasia, Parietal foramina with cleidocranial dysplasia, Chromosomal abnormalities [20]. These conditions may share some characteristics with CCD, however all these are autosomal recessive disor- ders and have other specific features. Some of these con- ditions may result from mutation in genes that affect the action of RUNX2 on its downstream targets [28]. The purpose of dental management in patients with CCD is to achieve an optimal functional and cosmetic result by early adulthood [29]. The planning of dental treatment aims in CCD varies from individual to indi- vidual and primarily depends on the needs of the patient, the age at diagnosis, and social and economic circum- stances [30]. A multidisciplinary approach is required. The current “state-of-the-art” treatment involves a com- bination of maxillofacial surgery, orthodontics, and prosthodontics. Several combination treatment ap- proaches have been reported [29]. The most popular or- thodontic-surgical regimes have been reported with great success including the Toronto-Melbourne (Table 2). The Toronto-Melbourne approach [31-36] is based on timed, Table 2. Management approaches of patients with cleidocranial dysplasia. Procedure 5 to 6 years Extraction of deciduous incisors Deciduous incisors are exposed and healing is allowed Orthodontic brackets arc placed on permanent anterior teeth incisors 6 to 7 years Extraction of posterior deciduous teeth Permanent bicuspids are exposed Surgical removal of supernumerary teeth and healing allowed Surgical exposure of permanent premolars Toronto-Melbourne approach 9 to 12 years Brackets placed on canines and premolars Removal of all deciduous and supernumerary teeth Single method Exposure of all impacted teeth are surgically Age: not specified Surgical packing are placed to prevent healing of bone and soft tissue over teeth Healing by secondary intention Placement of orthodontic attachments Placement of orthodontic appliances placed on fully erupted teeth Belfast-Hamburg approach Placement of elastic thread between appliances on unerupted teeth and the arch wires Stage 1 Extraction of deciduous incisors Extraction of all supernumerary teeth Exposure of permanent incisors Brackets bonded immediately 10 to 12 years Surgical flaps are closed completely Stage 2 Extraction of posterior deciduous teeth Exposure of unerupted permanent canines and premolars Brackets bonded immediately Jerusalem approach Age ≥ 13 Surgical flaps are closed completely Stage 1 Two at most 3 Removal of all primary and supernumerary teeth Procedures Surgical flaps closed Age: not specified Stage 2 Exposure of unerupted permanent teeth Placement of orthodontic attachments Surgical flaps are closed and overdenture is placed Placement of conventional orthodontic appliances Stage 3 Leforte I osteotomy-orthognathic surgery Bronx approach Placement of dental implants Copyright © 2013 SciRes. OPEN ACCESS  A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 407 serial extraction of deciduous teeth and depends on the extent to which the roots of the permanent teeth have developed. The Belfast-Hamburg approach is a single surgical procedure that limits the number of surgeries to a single episode. All deciduous and supernumerary teeth are extracted and all unerupted permanent teeth are ex- posed simultaneously under general anesthesia. A third approach, the Jerusalem approach is based on at least 2 surgical interventions, the timing of which is dependent on the root development of the permanent dentition. The Bronx approach uses 2, and at most 3, surgical interven- tions. Kalliala [27] suggested that removal of primary or su- pernumerary teeth does not usually promote eruption of unerupted permanent teeth. However, Golan et al. [20] proposed that supernumerary teeth in patients with CCD should be diagnosed and removed as early as possible because the supernumerary teeth will always impede normal eruption of permanent teeth. Following alignment of all permanent teeth, any underlying skeletal discrep- ancy (most commonly a Class III skeletal malocclusion) can be corrected through orthognathic surgery after the completion of growth [Lefort 1 maxillary osteotomy]. The complete overlay denture has numerous advantages for the pediatric patient. Enhanced mastication and es- thetics are the more obvious benefits and speech also may be improved. The alveolar bone is maintained by the retention of teeth compared to its loss when teeth are extracted [37]. Sato [38] suggests the use of a three-dimensional method of locating the position of impacted supernumer- ary teeth insisting upon the importance of the removal of the supernumerary teeth and the planning of an ortho- dontic treatment that will allow for occlusion of the re- tained teeth. In our case, cone-beam computed tomogra- phy scans of Patient I was performed. In present case, the protocol involves timely extraction of deciduous teeth, staged surgical removal of supernumerary teeth, expo- sure of selected unerupted permanent teeth and ortho- dontic forced eruption. Then Lefort I maxillary osteot- omy corrected through orthognathic surgery, but both he and other patients with CCD did not accepted unfortu- nately for any treatment planning. Mutations in RUNX2 have a high penetrance and ex- treme variability, ranging from isolated dental anomalies to fully manifesting disease with poorly ossified cranium and absence of clavicles [39]. In experimental studies, mice carrying one mutated RUNX2 allele show all the hallmarks of CCD, like hypoplasia of the clavicles and patent fontanels. When both copies of the RUNX2 gene are inactivated, bone development cannot proceed be- cause of a block in differentiation of the mesenchymal precursor cells toward osteoblasts [40]. This shows that RUNX2 gene is one of the main regulatory genes for bone formation. Although RUNX2 is a key regulatory gene for bone formation, low BMD is not one of the main findings in CCD. In fact we can say that it is rarely reported in CCD. In present report, patients II and III have decreased BMD levels but patient I was normal with regard to BMD. A remarkable point is that the two patients with scoliosis also have low BMD levels. Thus it is possible that there could be a correlation between BMD and scoliosis; but of course this must be supported with further research in more patients. Many researchers have tried on this subject to find a genotype-phenotype correlation in CCD [17]. However there is no reliable data on this subject. Yoshida et al. suggested that there is a correlation, inverse ratio, be- tween height and the number of supernumerary teeth [17]. However this correlation could not be supported in subsequent studies. In our study, patient I had 29 im- pacted teeth and was 183 cm in height (75 - 90 p). Pa- tient II was 159 cm (<3 p) tall and had 23 impacted teeth. Patient III was 156 cm (<3 p) tall and had 11 impacted teeth. Patient I was the most severely affected sibling in respect to supernumerary teeth but his height was in the normal range (75 - 90 p). The other two patients had fewer supernumerary teeth than patient I but their heights were under 3P. Our report is not in agreement with Yo- shida et al.’s suggestion [17]. The Runt-related transcription factor, RUNX, genes are key regulators for bone [40] and hematopoietic [41,42] formation. RUNX2 is a member of this family and has a crucial role in bone formation [40]. This pro- tein has several domains. These are; Q/A domain, the length of this domain is important for the transcriptional activity of RUNX2 [9,43]; Nuclear Localization Signal (NLS), reponsible for the nuclear transportation of RUNX2 protein [44]; the proline-serine-threonine Rich Domain, essential for the transcriptional activity of RUNX2 protein [45]; the last five amino acids of the whole protein is known as the VWPRY motif. This motif has a crucial role for the repression activity of Runt do- main. The Runt domain is highly conserved peptide mo- tif in all RUNX proteins in humans and other vertebrates. It is the active DNA-binding site of RUNX proteins [16]. The Runt domain has two distinct repression activities, one is dependent on intact VWRPY sequence and the other is unrelated with this conserved sequence [46]. VWRPY-dependent repression activity needs co-rep- ressor proteins and an intact VWRPY motif to reveal its effect properly. The Groucho protein is the prototype of the co-repressor family which was first defined in Dro- sophila. Transducin-like enhancers of split (TLE) pro- teins are human homologs of Groucho. As co-repressors, Gro/TLE family of proteins does not bind to DNA di- rectly. Instead, it interacts with the Runt domain and the VWRPY motif of the Runt related transcription factors Copyright © 2013 SciRes. OPEN ACCESS  A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 408 like RUNX1 and RUNX2 to repress gene transcription. For a consistent interaction between the Runt and Gro/TLE protein, the C-terminal 49 amino acids of the Runt domain and the conserved VWRPY motif are nec- essary [18,46]. In addition, two translocations in Acute Myeloid Leukemia, t (3; 21) and t (8; 21), cause rear- rangement in the RUNX1 gene which is homolog of RUNX2. As a result of these rearrangements, the fusion protein maintains an intact Runt domain but loses the VWRPY motif. Thus, it is predicted that fusion proteins can be deficient with regard to TLE-mediated transcrip- tional repression [47,48]. In experimental studies, the WRPY tetra-peptide sequence was deleted from the C-terminus and this showed that a mutant protein cannot interact with Groucho whereas intact proteins can [46]. The mutation in this report is quite similar to this ex- perimental study. The valine (V) amino acid in VWRPY turns into glycine by a frame shift mutation. Until 2005, there was no reported patient of mutation in VWPRY. Napierala et al. [49] first identified the 1754_1757del TTTG mutation (NM_0010 24630.2) in one patient. This deletion results in a frame shift and stop codon p. V585Gfs56X. Herein we report another three patients affected by this mutation. 5. CONCLUSION In conclusion, we can say that an intact VWRPY motif is crucial for the transcriptional repression activity of RUNX2 proteins and, as a result of mutation in the VWRPY peptide sequence, the transcriptional repression activity of the RUNX2 protein, which is essential for osteogenesis, disappeared via the breaking interaction between RUNX2 and TLE proteins. In the management of CCD patients, a molecular genetic analysis would be helpful. The clinician has a key role in identifying CCD patients and arranging a mutation search by a genetic counseling. REFERENCES [1] Marie, P. and Sainton P. (1898) Sur la dysostose cleido- cranienne hereditaire. Revista de Neurología, 6, 835-838. [2] Mundlos, S. (1999) Cleidocranial dysplasia: Clinical and molecular genetics. Journal of Medical Genetics, 36, 177-182. [3] Quack, I., Vonderstrass, B., Stock, M., Aylsworth. A.S., Becker, A., Brueton. L., et al. (1999) Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. The American Journal of Human Genetics, 65, 1268-1278. http://dx.doi.org/10.1086/302622 [4] Figueroa, A.A. and Friede, H. (2000) Craniofacial growth in unoperated craniofacial malformations. The Cleft Pal- ate—Craniofacial Journal, 37, 431-432. http://dx.doi.org/10.1597/1545-1569(2000)037<0431:CG IUCM>2.0.CO;2 [5] Counts, A.L., Rohrer, M.D., Prasad, H. and Bolen, P., (2001) An assessment of root cementum in cleidocranial dysplasia. The Angle Orthodontist, 71, 293-298. [6] Roberts, T., Stephen, L. and Beighton, P. (2013) Cleido- cranial dysplasia: A review of the dental, historical, and practical implications with an overview of the South Af- rican experience. Oral Surgery, Oral Medicine, Oral Pa- thology and Oral Radiology, 115, 46-55. http://dx.doi.org/10.1016/j.oooo.2012.07.435 [7] Dalessandri, D., Laffranchi, L., Tonni, I., Zotti, F., Pi- ancino, M.G., Paganelli, C., et al. (2011) Advantages of cone beam computed tomography (CBCT) in the ortho- dontic treatment planning of cleidocranial dysplasia pa- tients: A case report. Head & Face Medicine, 27, 6. [8] Mohan, R.P., Suma, G.N., Vashishth, S. and Goel, S. (2010) Cleidocranial dysplasia: Clinico-radiological il- lustration of a rare case. Journal of Oral Science, 52, 161- 166. http://dx.doi.org/10.2334/josnusd.52.161 [9] Mundlos, S., Otto, F., Mundlos, C., Mulliken, J.B., Ayls- worth, A.S., Albright, S., et al. (1997) Mutations involve- ing the transcription factor CBFA1 cause cleidocranial dysplasia. Cell, 89, 773-779. http://dx.doi.org/10.1016/S0092-8674(00)80260-3 [10] Li, Y.L. and Xiao, Z.S. (2007) Advances in Runx2 regu- lation and its isoforms. Medical Hypotheses, 68, 169-175. http://dx.doi.org/10.1016/j.mehy.2006.06.006 [11] Shen, Z., Zou, C.C., Yang, R.W. and Zhao, Z.Y. (2009) Cleidocranial dysplasia: Report of 3 cases and literature review. Clinical Pediatrics, 48, 194-198. http://dx.doi.org/10.1177/0009922808323107 [12] Li, Y., Pan, W., Xu, W., He, N., Chen, X., Liu, H., et al. (2009) RUNX2 mutations in Chinese patients with clei- docranial dysplasia. Mutagenesis, 24, 425-431. http://dx.doi.org/10.1093/mutage/gep025 [13] Zhang, C., Zheng, S., Wang, Y., Zhao, Y., Zhu, J. and Ge, L. (2010) Mutational analysis of RUNX2 gene in Chinese patients with cleidocranial dysplasia. Mutagenesis, 25, 594. http://dx.doi.org/10.1093/mutage/geq044 [14] Wang, G.X., Sun, R.P. and Song, F.L. (2010) A novel RUNX2 mutation (T420I) in Chinese patients with clei- docranial dysplasia. Genetics and Molecular Research, 9, 41-47. http://dx.doi.org/10.4238/vol9-1gmr685 [15] Fang, C.Y., Xue, J.J., Tan, L., Jiang, C.H., Gao, Q.P., Liang, D.S. and Wu, L.Q. (2011) A novel single-base de- letion mutation of the RUNX2 gene in a Chinese family with cleidocranial dysplasia. Genetics and Molecular Research, 10, 3539-3544. http://dx.doi.org/10.4238/2011.December.14.5 [16] Ogawa, E., Maruyama, M., Kagoshima, H., Inuzuka, M., Lu, J., Satake, M., et al. (1993) PEBP2/PEA2 represents a new family of transcription factor homologous to the products of the Drosophila runt and the human AML1 gene. Proceedings of the National Academy of Sciences, 90, 6859-6863. http://dx.doi.org/10.1073/pnas.90.14.6859 [17] Yoshida, T., Kanegane, H., Osato, M., Yanagida, M., Miyawak, T., Ito, Y., et al. (2002) Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype corre- lations. The American Journal of Human Genetics, 71, Copyright © 2013 SciRes. OPEN ACCESS  A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 409 724-738. http://dx.doi.org/10.1086/342717 [18] Chen, G. and Courey, A.J. (2000) Groucho/TLE family proteins and transcriptional repression. Gene, 249, 1-16. http://dx.doi.org/10.1016/S0378-1119(00)00161-X [19] Levanon, D., Goldstein, R.E., Bernstein, Y., Tang, H., Goldenberg, D., Stifani, S., et al. (1998) Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proceedings of the National Academy of Sciences, 95, 11590-11595. http://dx.doi.org/10.1073/pnas.95.20.11590 [20] Golan, I., Baumert, U., Hrala, B.P. and Mussig, D. (2003) Dentomaxillofacial variability of cleidocranial dysplasia: clinicoradiological presentation and systematic review. Dentomaxillofacial Radiology, 32, 347-354. http://dx.doi.org/10.1259/dmfr/63490079 [21] Quinn, P.D., Lewis, J. and Levin, L. (1992) Surgical management of a patient with cleidocranial dysplasia: A case report. Special Care Dentistry, 12, 131-133. http://dx.doi.org/10.1111/j.1754-4505.1992.tb00429.x [22] Alves, N. and De Caso, R. (2008) Cleidocranial dyspla- sia—A case report. International Journal of Morphology, 26, 1065-1068. http://dx.doi.org/10.4067/S0717-95022008000400043 [23] Fukuta, Y., Totsuka, M., Fukuta, Y., Takeda, Y., Yoshida, Y., Niitsu, J., et al. (2001) Histological and analytical studies of a tooth in a patient with Cleidocranial dysplasia. Journal of Oral Science, 43, 85-89. http://dx.doi.org/10.2334/josnusd.43.85 [24] McNamara, C.M., O’Riordan, B.C., Blake, M. and Sandy, J.R. (1999) Cleidocranial dysplasia: Radiological ap- pearances on dental panoramic radiography. Dentomax- illofacial Radiology, 28, 89-97. http://dx.doi.org/10.1038/sj.dmfr.4600417 [25] Winter. G.R. (1943) Dental conditions in cleidocranial dysostosis. American Journal of Orthodontics and Oral Surgery, 29, 61-89. http://dx.doi.org/10.1016/S0096-6347(43)90045-6 [26] Jensen, B.L. and Kreiborg, S. (1990) Development of the dentition in cleidocranial dysplasia. Journal of Oral Pa- thology & Medicine, 19, 89-93. http://dx.doi.org/10.1111/j.1600-0714.1990.tb00803.x [27] Kalliala, E. and Taskinen, P.J. (1962) Cleidocranial dy- sostosis: Report of six typical cases and one atypical case. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology, 14, 808. http://dx.doi.org/10.1016/0030-4220(62)90331-6 [28] Goto, T., Aramaki, M., Yoshihashi, H., Nishimura, G., Hasegawa, Y., Takahashi. T., et al. (2004) Large fontan- els are a shared features of haploinsufficiency of RUNX2 and its coactivator CBFB. Congenital Anomalies, 44, 225-229. http://dx.doi.org/10.1111/j.1741-4520.2004.00043.x [29] Becker, A., Lustman, J. and Shteyer, A. (1997) Cleido- cranial dysplasia: Part 1-General principles of the ortho- dontic and surgical treatment modality. American Journal of Orthodontics and Dentofacial Orthopedics, 111, 28-33. http://dx.doi.org/10.1016/S0889-5406(97)70298-1 [30] D’Alessandro, G., Tagariello, T. and Piana, G. (2010) Craniofacial changes and treatment of the stomatognathic system in subjects with cleidocranial dysplasia. European Journal of Paediatric Dentistry, 11, 39-43. [31] Smylski, P.T., Woodside, D.G. and Harnett, B.E. (1974) Surgical and orthodontic treatment of cleidocranial dy- sostosis. International Journal of Oral Surgery, 3, 380- 385. http://dx.doi.org/10.1016/S0300-9785(74)80002-5 [32] Hall, R.K. and Hyland, A.L. (1978) Combined surgical and orthodontic management of the oral abnormalities in children with cleidocranial dysplasia. International Jour- nal of Oral Surgery, 7, 267-273. http://dx.doi.org/10.1016/S0300-9785(78)80093-3 [33] Richardson, A. and Swinson, T. (1987) Combined ortho- dontic and surgical approach to cleidocranial dysostosis. Transactions European Orthodontic Society, 63, 23. [34] Behlfelt, K. (1987) Cleido-cranial dysplasia: Diagnosis and treatment concept. Transactions European Ortho- dontic Society, 63, 25. [35] Petropoulos, V.C., Balshi, T.J., Balshi, S.F., et al. (2004) Treatment of a patient with cleidocranial dysplasia using osseointegrated implants: A patient report. The Interna- tional Journal of Oral & Maxillofacial Implants, 19, 282- 287. [36] Kelly, E. and Nakamoto, R.Y. (1974) Cleidocranial dy- sostosis—A prosthodontic problem. Journal of Prosthetic Dentistry, 31, 518-526. http://dx.doi.org/10.1016/0022-3913(74)90173-5 [37] Daskalogiannakis, J., Piedade, L., Lindholm, T.C., Sán- dor, G.K. and Carmichael, R.P. (2006) Cleidocranial Dysplasia: 2 Generations of management. Journal of the Canadian Dental Association, 72, 337-342. [38] Sato, K., Sugawara, J., Mitani, H. and Kawamura, H. (1998) Use of selectively colored stereolithography for diagnosis of impacted supernumerary teeth for a patient with cleidocranial dysplasia. The International Journal of Adult Orthodontics & Orthognathic Surgery, 13, 163-167. [39] Golan, I., Preising, M., Wagener, H., Baumert, U., Nied- erdellmann, H., Lorenz, B. and Mussig, D. (2000) A novel missense mutation of the CBFA1 gene in a family with cleidocranial dysplasia (CCD) and variable expres- sivity. Journal of Craniofacial Genetics and Develop- mental Biology, 20, 113-120. [40] Komori, T., Yagi, H., Nomura, S., Yamaguchi, A., Sasaki, K., Deguchi, K., Shimizu, Y., et al. (1997) Targeted dis- ruption of Cbfa1 results in a complete lack of bone for- mation owing to maturational arrest of osteoblasts. Cell, 89, 755-764. http://dx.doi.org/10.1016/S0092-8674(00)80258-5 [41] Okuda, T., van Deursen, J., Hiebert, S.W., Grosveld, G. and Downing, J.R. (1996) AML1, the target of multiple chromosomal translocations in human leukemia, is essen- tial for normal fetal liver hematopoiesis. Cell, 84, 321- 330. http://dx.doi.org/10.1016/S0092-8674(00)80986-1 [42] Speck, N.A., Stacy, T., Wang, Q., North, T., Gu, T.L., Miller, J., Binder, M., et al. (1999) Core-binding factor: A central player in hematopoiesis and leukemia. Cancer Research, 59, 1789-1793. [43] Thirunavukkarasu, K., Mahajan, M., McLarren, K.W., Copyright © 2013 SciRes. OPEN ACCESS  A. E. Sekerci et al. / Open Journal of Stomatology 3 (2013) 402-410 Copyright © 2013 SciRes. 410 OPEN ACCESS Stifani, S. and Karsenty, G. (1998) Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Molecular and Cellular Biology, 18, 4197-4208. [44] Kanno, T., Kanno, Y., Chen, L.F., Ogawa, E., Kim, W.Y. and Ito, Y. (1998) Intrinsic transcriptional activation-inhi- bition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Molecular and Cellular Bi- ology, 18, 2444-2454. [45] Zhang, Y.W., Yasui, N., Ito, K., Huang, G., Fuji, M., Hanai, J., et al. (2000) A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proceedings of the National Academy of Sciences, 7, 10549-10554. http://dx.doi.org/10.1073/pnas.180309597 [46] Aronson, B.D., Fisher, A.L., Blechman, K., Caudy, M. and Gergen, J.P. (1997) Groucho-dependent and inde- pendent repression activities of Runt domain proteins. Molecular and Cellular Biology, 17, 5581-5587. [47] Miyoshi, H., Shimizu, K., Kozu, T., Maseki, N., Kaneko, Y. and Ohki, M. (1991) t(8;21) breakpoints on chromo- some 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proceedings of the National Academy of Sciences, 88, 10431-10434. http://dx.doi.org/10.1073/pnas.88.23.10431 [48] Nucifora, G., Birn, D.J, Erickson. P., Gao, J., LeBeau, M.M., Drabkin, H.A., et al. (1993) Detection of DNA re- arrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood, 81, 883-888. [49] Napierala, D., Garcia-Rojas, X., Sam, K., Wakui, K., Chen, C., Mendoza-Londono, R., et al. (2005) Mutations and promoter SNPs in RUNX2, a transcriptional regula- tor of bone formation. Molecular Genetics and Metabo- lism, 86, 257-268. http://dx.doi.org/10.1016/j.ymgme.2005.07.012.
|