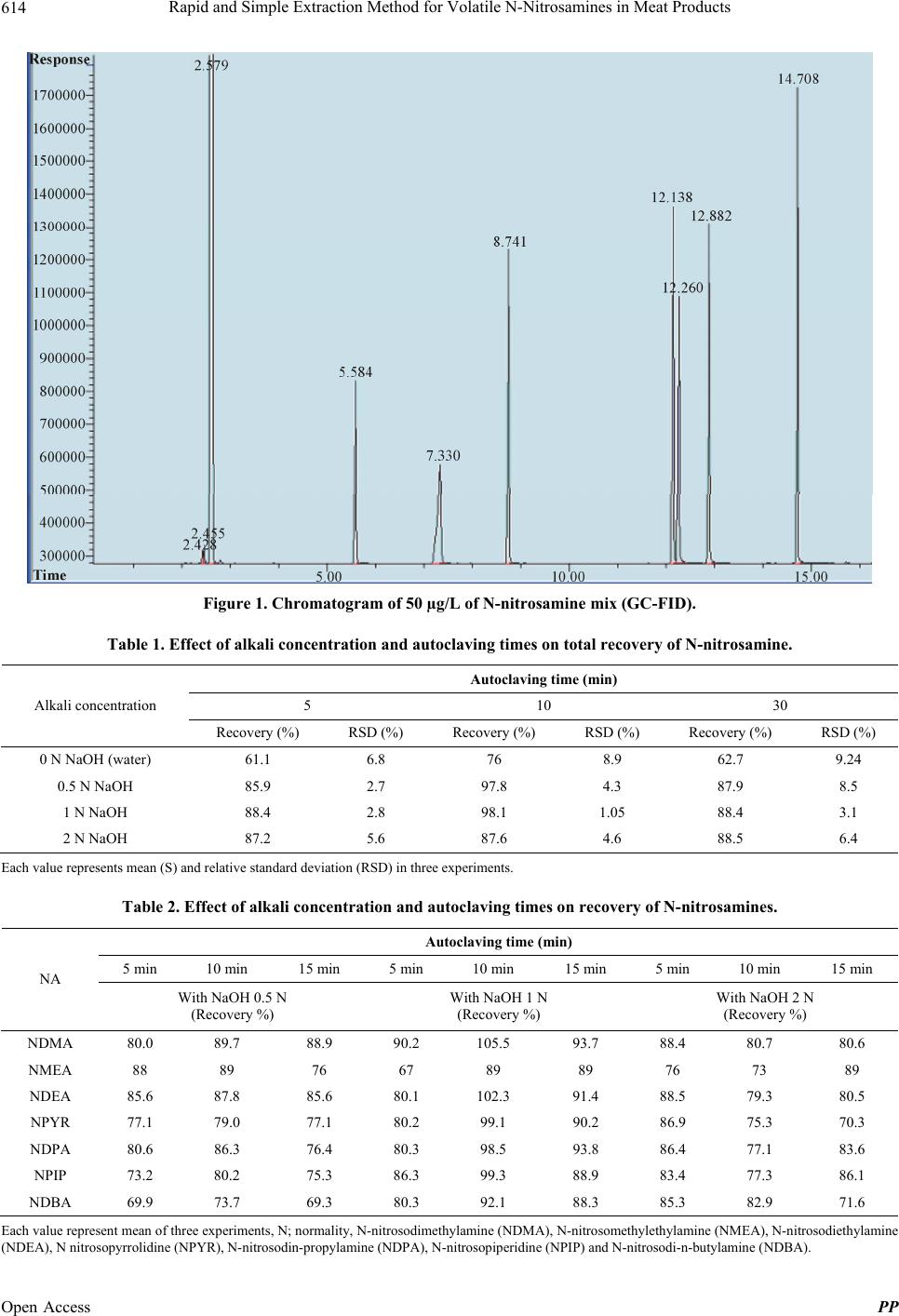
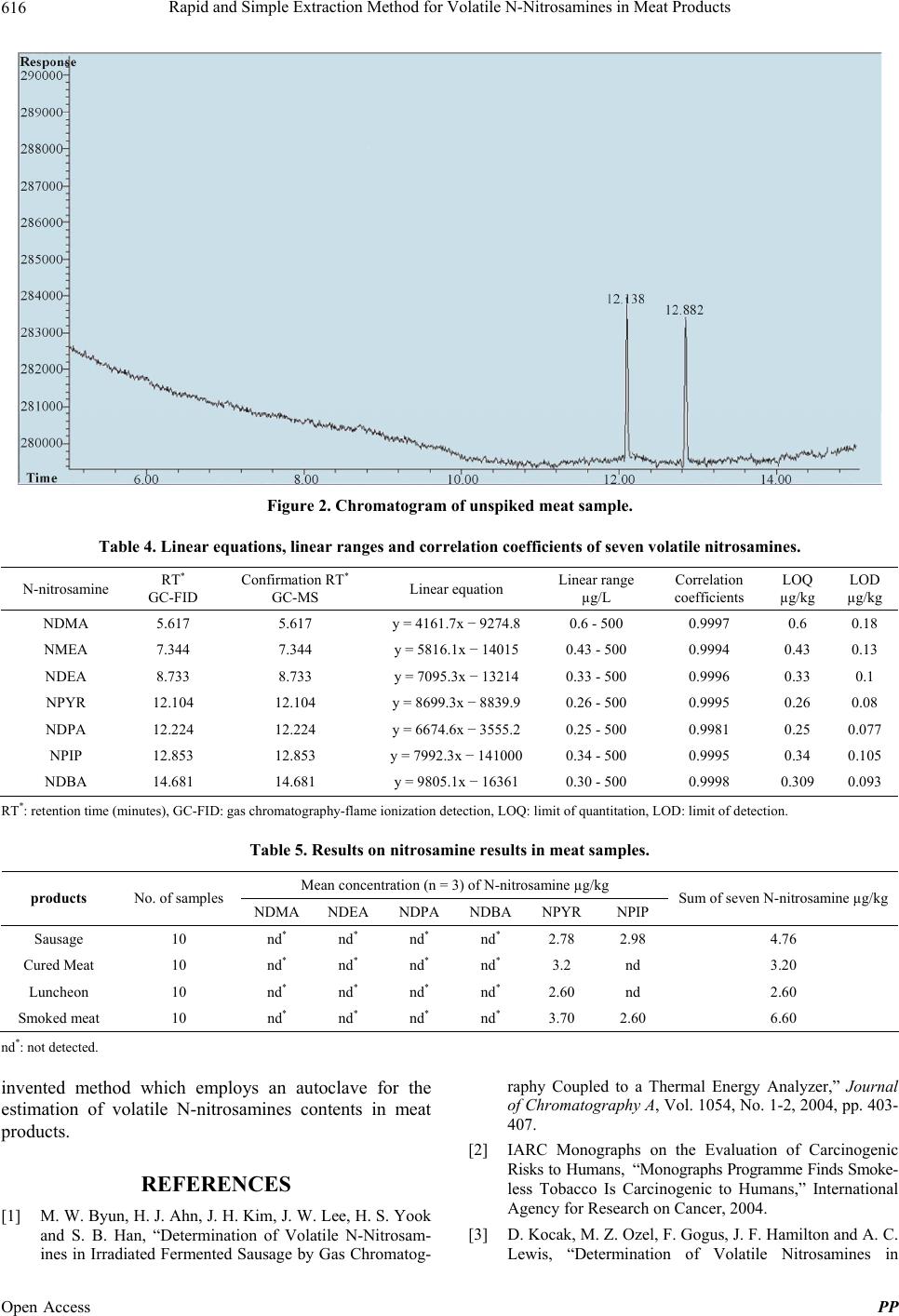
Rapid and Simple Extraction Method for Volatile N-Nitrosamines in Meat Products 617
Grilled Lamb and Vegetables Using Comprehensive Gas
Chromatography—Nitrogen Chemiluminescence Detec-
tion,” Food Chemistry, Vol. 135, No. 4, 2012, pp. 2215-
2220. http://dx.doi.org/10.1016/j.foodchem.2012.07.002
[4] N. Campillo, P. Viñas, N. C. Martinez and M. H. Cór-
doba, “Determination of Volatile Nitrosamines in Meat
Products by Microwave-Assisted Extraction and Disper-
sive Liquid-Liquid Micro-Extraction Coupled to Gas
Chromatography-Mass Spectrometry,” Journal of Chro-
matography A, Vol. 1218, No. 14, 2011, pp. 1815-1821.
http://dx.doi.org/10.1016/j.chroma.2011.02.010
[5] W. G. Lin, F. Wei, F. N. Gu, X. Dong, L. Gao, T. T.
Zhuang, M. B. Yue and J.H. Zhu, “Adsorption of Ni-
trosamines by Mesoporous Zeolite,” Journal of Colloid
and Interface Science, Vol. 348, No. 2, 2010, pp. 621-627.
http://dx.doi.org/10.1016/j.jcis.2010.05.012
[6] R. Andrade, F. G. Reyes and S. Rath, “A Method for the
Determination of Volatile N-Nitrosamines in Food by
HS-SPME-GC-TEA,” Food Chemistry, Vol. 91, No. 1,
2005, pp. 173-179.
http://dx.doi.org/10.1016/j.foodchem.2004.08.015
[7] B. J. Sanchez, E. Ballesteros and M. Gallego, “Automatic
Screening Method for the Pre-Concentration and Deter-
mination of N-Nitrosamines in Water,” Talanta, Vol. 73,
No. 3, 2007, pp. 498-504.
http://dx.doi.org/10.1016/j.talanta.2007.04.007
[8] K. O. Honikel, “The Use and Control of Nitrate and Ni-
trite for the Processing of Meat Products,” Meat Science,
Vol. 78, No. 1-2, 2008, pp. 68-76.
http://dx.doi.org/10.1016/j.meatsci.2007.05.030
[9] R. A. Scanlan, “Encyclopedia of Food and Nutrition,”
Elsevier, 2003, pp. 4142-4147.
http://dx.doi.org/10.1016/B0-12-227055-X/00831-2
[10] M. J. Dennis, P. E. Key, T. Papworth, M. Pointer and R.
C. Massey, “The Determination of Nitrate and Nitrite in
Cured Meat by HPLC/UV,” Food Additives and Con-
taminants, Vol. 7, No. 4, 1990, pp. 455-461.
http://dx.doi.org/10.1080/02652039009373908
[11] F. E. Okieimana, E. O. Akintola, T. C. A. Anucha and M.
M. Ajibola, “Determination of Total Level of Nitrosa-
mine Contamination of Some Consumer Products in Ni-
geria,” International Journal of Environmental Analytical
Chemistry, Vol. 21, No. 4, 1985, pp. 261-266.
http://dx.doi.org/10.1080/03067318508077067
[12] M. T. Matyska, J. J. Pesek and L. Yang, “Screening
method for Determining the Presence of N-Nitrosodie-
thanolamine in Cosmetics by Open-Tubular Capillary Elec-
trochromatography,” Journal of Chromatography A, Vol.
887, No. 1-2, 2000, pp. 487-503.
http://dx.doi.org/10.1016/S0021-9673(00)00451-9
[13] M. Qiang, X. H. Wei, W. Chao, B. Hua, X. G. Cheng, S.
Ning, X. L. Yan and W. J. Bing, “Determination of Ten
Volatile Nitrosamines in Cosmetics by Gas Chromatog-
raphy Tandem Mass Spectrometry,” Chinese Journal of
Analytical Chemistry, Vol. 39, No. 8, 2011, pp. 1201-
1207. http://dx.doi.org/10.1016/S1872-2040(10)60466-5
[14] J. E. Grebel and H. I. Suffet, “Nitrogen-Phosphorus De-
tection and Nitrogen Chemiluminescence Detection of
Volatile Nitrosamines in Water Matrices: Optimization
and Performance Comparison,” Journal of Chromatog-
raphy A, Vol. 1175, No. 1, 2007, pp. 141-144.
http://dx.doi.org/10.1016/j.chroma.2007.09.073
[15] M. Z. Ozel, F. Gongus, S. Yagci, J. F. Hamilton and A. C.
Lewis, “Determination of Volatile Nitrosamines in Vari-
ous Meat Products Using Comprehensive Gas Chroma-
tography-Nitrogen Chemiluminescence Detection,” Food
and Chemical Toxicology, Vol. 48, No. 11, 2010, pp.
3268-3273. http://dx.doi.org/10.1016/j.fct.2010.08.036
[16] B. Hfiger and R. Niessner, “Determination of N-Nitroso-
methylaniline and Methylaniline in the Gas Phase,” Mi-
crochimica Acta, Vol. 122, No. 1-2, 1996, pp. 35-44.
http://dx.doi.org/10.1007/BF01252403
[17] J. A. Incavo and M. A. Schafer, “Simplified Method for
the Determination of N-Nitrosamines in Rubber Vulcani-
zates,” Analytica Chimica Acta, Vol. 557, No. 1-2, 2006,
pp. 256-261. http://dx.doi.org/10.1016/j.aca.2005.10.018
[18] D. Tsikas, “Methods of Quantitative Analysis of the Ni-
tric Oxide Metabolites Nitrite and Nitrate in Human Bio-
logical Fluids,” Free Radical Research, Vol. 39, No. 8,
2005, pp. 797-815.
http://dx.doi.org/10.1080/10715760500053651
[19] J. W. Dallinga, D. M. Pachen, A. H. Lousberg, J. A. Van
Geel, G. M. Houben, R. W. Stockburgger, J. M. Maanen
and J. C. Kleinjans, “Volatile N-Nitrosamines in Gastric
Juice of Patients with Various Conditions of the Gastro-
intestinal Tract Determined by Gas Chromatography-
Mass Spectrometry and Related to Intragastic pH and Ni-
trate and Nitrite Levels,” Cancer Letters, Vol. 124, No. 2,
1998, pp. 119-125.
http://dx.doi.org/10.1016/S0304-3835(97)00467-9
[20] Y. Xia, J. E. McGuffey, S. Bhattacharyya, B. Sellergren,
E. Yilmaz and L. Wang, “Analysis of the Tobacco-Speci-
fic Nitrosamine 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-
Butanol in Urine by Extraction on a Molecularly Im-
printed Polymer Column and Liquid Chromatography/
Atmospheric Pressure Ionization Tandem Mass Spec-
trometry,” Analytical Chemistry, Vol. 77, No. 23, 2005,
pp. 7639-7645. http://dx.doi.org/10.1021/ac058027u
[21] G. Bellec, M. C. Cauvin, K. L. Calve, Y. Dreano, H.
Gouerou, J. F. Menez and F. Berthou, “Analysis of
N-Nitrosamines by High-Performance Liquid Chroma-
tography with Post-Column Photohydrolysis and Colori-
metric Detection,” Journal of Chromatography A, Vol.
727, No. 1, 1996, pp. 83-92.
http://dx.doi.org/10.1016/0021-9673(95)01073-4
[22] N. V. Komarova and A. A. Velikanov, “Determination of
Volatile N-Nitrosamines in Food by High-Performance
Liquid Chromatography with Fluorescence Detection,”
Journal of Analytical Chemistry, Vol. 56, No. 4, 2001, pp.
359-363. http://dx.doi.org/10.1023/A:1016652213062
[23] Association of Official Analytical Chemists, “N-Nitrosa-
mines (Volatile) in Fried Bacon. Mineral Oil Vacuum
Distillation—Thermal Energy Analyzer Method,” No.
982.22, Official Methods of Analysis, 15th Edition, 1990.
[24] E. L. Greenfield, W. J. Smith and A. J. Malanovski,
“Mineral Oil Vacuum Distillation for Nitrosamines in
Fried Bacon, with Thermal Energy Analyser,” Journal of
the Association Official Analytical Chemists, Vol. 65, No.
6, 1982, pp. 1319-1332.
Open Access PP