V. Pandey et al. / Case Reports in Clinical Medicine 2 (2013) 454-4 56
Copyright © 2013 SciRes. OPEN ACCESS
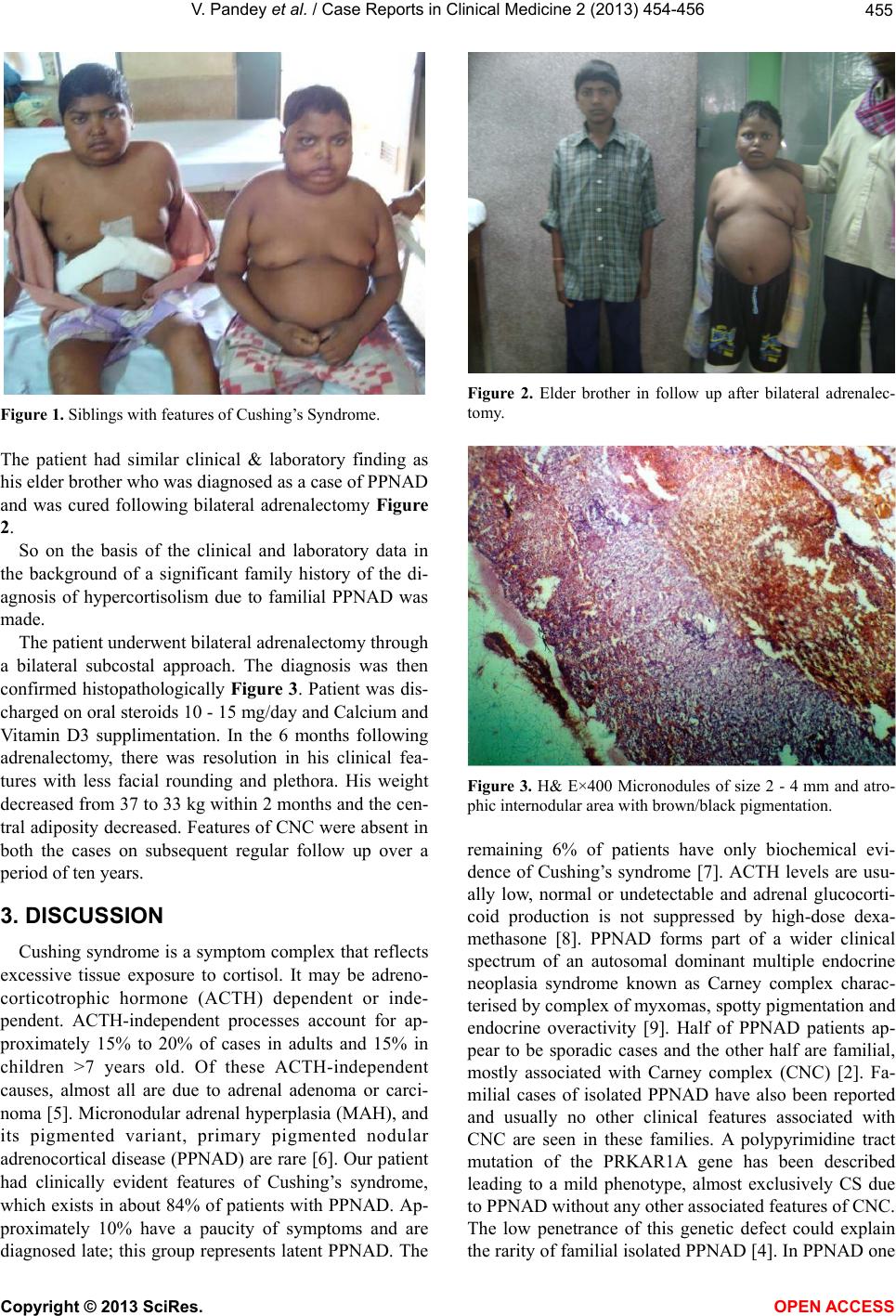
456
or more macronodules (>1 cm in diameter) can be pre-
sent unilaterally or bilaterally, making the differential
diagnosis from ACTH independent macronodular adrenal
hyperplasia (AIMAH) very difficult [10]. In patients with
PPNAD there is delayed paradoxical increase of urinary
free cortisol by 100% or more using the sequential
LDDST and HDDST which forms the basis of Liddle’s
test, differentiating PPNAD from AIMNH [11]. In
PPNAD, the characteristic macroscopic findings usually
described include small brown-black nodules separated
by an atrophic adrenal cortex. The cut surface is yellow,
often with small brown foci. Histologically, the nodules
consist of clear lipid-laden zona-fasciculata-type cells
corresponding to the yellow areas on gross examination,
while the brown foci consist of compact lipid-sparse
zona-reticularis type cells [12]. Bilateral adrenalectomy
is the treatment of choice for CS due to PPNAD. The
laparoscopic approach is associated with a lower mor-
bidity rate compared with the open technique, less post-
operative pain, shorter hospitalization time and lower
overall cost.
4. CONCLUSION
Isolated familial PPNAD is a rare entity and has got a
better prognosis than familial PPNAD associated with
Carney Complex. This observation has important conse-
quences for clinical management, follow-up and genetic
counselling of such patients.
REFERENCES
[1] Carney, J.A. and Young, W.F. (1992) Primary pigmented
nodular adrenocortical disease and its associated condi-
tions. Endocrinologist, 2, 6-21.
http://dx.doi.org/10.1097/00019616-199201000-00003
[2] Stratakis, C.A. and Kirschner, L.S. (1998) Clinical and
genetic analysis of primary bilateral adrenal diseases (mi-
cro- and macronodular disease) leading to Cushing syn-
drome. Hormone and Metabolic Research, 30, 456-63.
http://dx.doi.org/10.1055/s-2007-978914
[3] Stratakis, C.A., Kirschner, L.S. and Carney, J.A. (2001)
Clinical and molecular features of the Carney complex:
Diagnostic criteria and recommendations for patient evalu-
ation. The Journal of Clinical Endocrinology & Meta-
bolism, 86, 4041-4046.
http://dx.doi.org/10.1210/jc.86.9.4041
[4] Groussin, L., Horvath, A., Jullian, E., Boikos, S., Rene-
Corail, F., Lefebvre, H., et al. (2006) A PRKAR1A muta-
tion associated with primary pigmented nodular adreno-
cortical disease in 12 kindreds. The Journal of Clinical
Endocrinology & Metabolism, 91, 1943-1949.
http://dx.doi.org/10.1210/jc.2005-2708
[5] Arnaldi, G., Angeli, A., Atkinson, A.B., et al. (2003) Di-
agnosis and complications of Cushing’s sy ndrome: A
consensus statement. The Journal of Clinical Endocri-
nology & Metabolism, 88, 5593-5602.
http://dx.doi.org/10.1210/jc.2003-030871
[6] Stratakis, C.A. (2007) Adrenocortical tumors, primary
pigmented adrenocortical disease (PPNAD)/Carney com-
plex, and other bilateral hyperplasias: The NIH studies.
Hormone and Metabolic Research, 39, 467-473.
http://dx.doi.org/10.1055/s-2007-981477
[7] Carney, J.A., Gordon, H., Carpenter, P.C., Shenoy, B.V.
and Go, V.L. (1985) The complex of myxomas, spotty
pigmentation, and endocrine overactivity. Medicine, 64,
270-283.
http://dx.doi.org/10.1097/00005792-198507000-00007
[8] Sarlis, N.J., Chrousos, G.P., Doppman, J.L., Carney, J.A.
and Stratakis, C.A. (1997) Primary pigmented nodular
adrenocortical disease: Reevaluation of a patient with
Carney complex 27 years after unilateral adrenalectomy.
The Journal of Clinical Endocrinology & Metabolism, 82,
1274-1278. http://dx.doi.org/10.1210/jc.82.4.1274
[9] Groussin, L., Jullian, E., Perlemoine, K., Louvel, A., Le-
heup, B., Luton, J.P., et al. (2002) Mutations of the
PRKAR1A gene in Cushing’s syndrome due to sporadic
primary pigmented adrenocortical disease. The Journal of
Clinical Endocrinology & Metabolism, 87, 4324-4329.
http://dx.doi.org/10.1210/jc.2002-020592
[10] Doppman, J.L., Travis, W.D., Nieman, L., Chrousos, G.P.,
Gomez, M.T., Cutler, G.B., et al. (1989) Cushing syn-
drome due to primary pigmented nodular adrenocortical
disease: Findings at CT and MR imaging. Radiology, 172,
415-420.
[11] Stratakis, C.A., Sarlis, N., Kirschner, L.S., Carney, J.A.,
Doppman, J.L., Nieman, L.K., Chrousos, G.P. and Pa-
panicolaou, D.A. (1999) Paradoxical response to dexa-
methasone in the diagnosis of primary pigmented nodular
adrenocortical disease. Annals of Internal Medicine, 131,
585-591.
http://dx.doi.org/10.7326/0003-4819-131-8-199910190-0
0006
[12] Neville, A.M., McGee, J.O’D., Isaacson, P.G. and Wright,
N.A. (1992) Oxford Textbook of pathology. Vol. 2b. In:
Pathology of systems. Oxford medical publications. Ox-
ford University Press, New York, 1968-1986.