Paper Menu >>
Journal Menu >>
 Advances in Bioscience and Biotechnology, 2011, 2, 13-19 ABB doi:10.4236/abb.2011.21003 Published Online February 2011 (http://www.SciRP.org/journal/abb/). Published Online February 2011 in SciRes. http://www.scirp.org/journal/ABB A versatile vector system for generating recombinant EGFP-tagged proteins in yeast Francesco Palma, Laura Chiarantini Dipartimento di Scienze Biomolecolari, Sezione di Biochimica e Biologia Molecolare “Giorgio Fornaini”. Università degli Studi di Urbino “Carlo Bo”, 2–61029, Via Saffi, Urbino, Italy. Email: francesco.palma@uniurb.it Received 5 November 2010; revised 20 November 2010; accepted 26 November 2010. ABSTRACT This paper reports a versatile egfp-tagged pFL61- based expression vector system which allows the production on yeast of homo- and heterologous pro- teins fused with the Enhanced Green Fluorescent Protein (EGFP) at the C-terminus. This expression system, which involves a fluorescent protein, readily allows both to verify the expression and to localize the protein in the yeast cell. The vector carries a Not I site upstream the first codon of the egfp gene. The yeast cells harbouring this plasmid emit a feeble emission compared to the fluorescence expected. Was then investigated the effect of the Not I site, located very close to the start codon, on the expression of the reporter egfp gene using northern and western blot- ting, fluorescence microscopy and flow cytometry. Data indicated that this palindromic site could hide the start codon so as to negatively affect translation. This aspect confers to the proposed expression system an advantage in distinguishing clones after transfor- mation. Keywords: Enhanced GFP; Protein Localization; Tuber borchii Vittad; Saccharomyces cerevisiae 1. INTRODUCTION Green fluorescent protein (GFP), a 238-amino acid polypeptide, is intrinsically fluorescent, thus not requir- ing substrates or co-factors to produce a green emission when excited with near UV light or blue light [1]. GFP has been used as a reporter gene in various organisms, including Escherichia coli, Caernorhabditis elegans, Drosophila melanogaster, Saccharomyces cerevisiae, Aedes aegypti, fish, viruses, mammals and plants [1-7]. Unlike the bacterial β-galactosidase and β-glucuro- nidase, which are widely used reporters in fungi, GFP does not rely on exogenous substrates or co-factors other than oxygen [8]. Therefore, GFP can be used as a fusion tag “in vivo” to localize proteins, to follow their move- ment, or to study the dynamics of the sub-cellular com- partments which target these proteins [1,9]. Since wild-type GFP performs inefficiently in various cellular contexts, efforts were focused upon the improvement of GFP expression and/or fluorescence levels. The enhanced GFP (EGFP) is a red-shifted variant of the wild-type [1,8,10] optimised for brighter green fluo- rescence: the emission spectrum (emission maximum = 507 nm) is 35 times more intense than that of GFP on excitation with blue light (excitation maximum = 488 nm) [11,12].This variant contains the double-amino-acid substitution of Phe-64 to Leu, Ser-65 to Thr and several silent base changes which optimize codon usage for mammals [13], other variants optimised for fungi YEGFP, plant SGFP [5] and green alga CGFP [14] have been reported. EGFP expression in yeast cells produces a diffuse cy- toplasmic and nuclear fluorescence pattern which can be observed in both fixed and living transformed cells [15]. Complementation of yeast mutation used to clone genes from heterologous species has been a general ap- proach in molecular biology. For example, the yeast mutant rtf1-1, defective in cell cycle progression and arrests before mitosis was functionally complemented by human p53 [16]. Furthermore, the yeast two-hybrid sys- tem has been widely used to study protein-protein inter- action and EGFP evaluated as an alternative reporter gene for detection by flow cytometry [17]. The PFL61 is a shuttle vector used in complementa- tion of Saccharomyces cerevisiae auxotropic mutant by heterologous cDNAs. This vector contains the phosphor- glycerate kinase promoter (PGK) separated by its termi- nator by a Not I site [18]. We have engineered this vector to obtain a versatile expression system which allows C-terminal fusion of the egfp gene to a hexogenous gene of interest to generate fusion protein whose subcellular localisation can be fol- lowed with fluorescence microscopy in living yeast  F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 14 cells. In this study, we demonstrate the successful expres- sion, using the proposed egfp-tagged pFL61-based ex- pression vector, of two genes from Tuber borchii Vittad. The lectin TBFL-1 is the main soluble secreted pro- tein present in the fruiting body of Tuber borchii Vittad, it is able to selectively bind the exopolysaccharides pro- duced by the ascoma-associated Rhizobium spp [19]. The lectin gene, tbfl-1, encodes for a 12-amino acid N-terminal non-canonical signal peptide, whose se- quence does not match any homologous signal se- quences stored in data banks, and drives the TBFL-1 to the classical secretory pathway, via a non-conventional route, when the gene is expressed in yeast [20]. The other gene, a hexose transporter involved in sugar me- tabolisms, known as Tbhxt1, encodes for a protein which functionally complements the hxt-null mutant Sac- charomyces cerevisiae EBYVW4000 [21]. The hexose transporter has a strong preference for D-glucose over D-fructose and also imports D-mannose. It catalyses the transport via a proton-symport mechanism. The regula- tion of Tbhxt1 gene expression is consistent with its role as a high-affinity D-glucose transporter [21]. Analyses of the expression, in live yeast cells, of the two EGFP- tagged proteins revealed a localization patterns agree with those of previous studies [20-22]. 2. MATERIALS AND METHODS 2.1. Strains Maintenance The Saccharomyces cerevisiae strain INVSc1 (Invitro- gen, Life Technologies, USA) was propagated on YPD agar. Transformation of yeast was performed using the yeast transformation kit (Sigma-Aldrich, USA) [23,24]. The recombinant yeast strain was grown, at 30°C, in a minimal medium composed of “yeast nitrogen base without amino acids” (Sigma-Aldrich, USA), 2% w/w D-glucose and supplemented with “yeast synthetic drop- out medium supplement, without uracil” (Sigma-Aldrich, USA). For the experiments described in this paper a single colony of yeast was grown overnight, in liquidin an or- bital shaking dry incubator. One ml was added to 50 ml of the same fresh medium. After 2 h aliquots were used for flow cytometry analysis, while for the other experi- ments the culture was grown until OD600 reached 1.0. The construction of recombinant plasmids were carried out in Escherichia coli strain BL21 by standard proto- cols. 2.2. Preparation of Recombinant Vectors Construction of the recombinant plasmids pBS(GFP) was performed as followed. Five micrograms of the ret- roviral plasmid PINCO [25], carrying the EGFP cDNA, were digested with Hind III and Not I overnight. The EGFP cDNA was separated by agarose gel electrophore- sis and purified using the “Qiaquik Gel Extraction Kit”. At the same time, 5 micrograms of plasmid pBlueScript II KS (Stratagene, USA) were digested with the same restriction enzyme, dephosphorylated with calf intestinal phosphatase (CIP) and purified using the “Qiaquik PCR Purification Kit”. One hundred micrograms of plasmid and 50 micrograms of cDNA were used in 10 µl of a ligation reaction. The obtained plasmid, pBS(GFP), was used for the preparation of yeast expression vectors. The yeast expression vectors, pFL61(GFP) and pFL61(GFPmut), were constructed as follows. Five micrograms of the plasmid pBS(GFP) were di- gested with Nco I and Not I overnight. The insert was filled with T4 DNA polymerase in the presence of dNTPs. Meanwhile, 5 micrograms of the plasmid pFL61 [18], was incubated overnight with Not I, filled with T4 and dNTPs and dephosphorylated using calf intestinal phosphatase (CIP, Boehringer Mannheim GmbH, Ger- many). After purification, both plasmid and insert were used in a ligation reaction to produce the expression vector pFL61(GFP). The plasmid DNA was used as a template for mis-matched primer mutagenesis [26] to produce the pFL61(GFPmut) vector carrying a Not I site upstream to the EGFP start codon. The primers utilized are reported in Table 1. The production of pFL61(TBFL1-EGFP) and pFL61 (TBHXT1-EGFP) was performed as described below. The cDNA inserts of both tbhxt and tbfl-1 genes were obtained by PCR, using a proofreading polymerase, from total fruiting body cDNA, using the primers re- ported in Table 1. The PCR products were incubated Ta b le 1 . Primer sequences employed in the mutagenesis and in the gene amplification. The Not I sites are underlined and the start codons are typed on bold letter. Gene Forward Primer Reverse Primer Primers mutagenesis CAACAAATATAAAACCAGCGG CCGCAATGGTGAGCAAGG CCTTGCTCACCATTGCGG CCGCTGGTTTTATATTTGTTG tbhxt1 GenBank = AY956320 ATGGGTTTCATCATCAAG GCGGCCGCAACCTCGCCGTGCC tbfl-1 GenBank = U83996 ATGTCTTCTAAAATTGTTCGTGAGCC CAGAAGGCGGCCGCAACCTGGACC  F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 15 with Taq DNA polymerase at 72°C for 5 minutes and then were cloned using the pGEM®-T Vector System (Promega, USA). The recombinant pGEM vectors was digested with Not I and the insert cloned in the dephos- phorylated Not I-linearised pFL61(GFPmut). 2.3. Analysis of Gene Expression by Northern and We stern Blotting Northern blot analyses were performed using total RNA extracted from 1.5 milligrams of wet weight cells har- vested from log-phase yeast-grown cultures by the RNeasy Plant Mini Kit (Qiagen GmbH, Germany). Twenty micrograms of total RNA were used in a stan- dard northern blot analysis. A hybridization probe was purified from the pBS-EGFP digested with Nco I and Not I. The cDNA fragment was P-labeled using the RediPrime kit (Amersham Biosciences, UK) and the filter exposed in a Phospho Imager apparatus. The ACT1 probe was used as housekeeping gene The western blot analyses of yeast total proteins were conducted by processing 50 ml of log-phase minimal medium yeast-grown cultures. After cell harvesting by centrifugation, 0.5 g of each wet weight yeast pellet, was frozen in liquid nitrogen and ground into a powder with mortar and pestle. The powder was then suspended in 1 ml of 10 mM Tris-HCl, pH 8.0 and 0.1% w/v SDS. The cell lysate was clarified by centrifugation in 1.5-ml tubes at 12,000 g for 15 min. Protein content was assayed by the BioRad method. Equal amount of protein extract (20µg) were resolved on a 15% SDS-polyacrilamide gels [27], transferred to a nitrocellulose membrane (Hy- bond-C Extra, Amersham Biosciences, UK.) and then detected with a mouse monoclonal antiGFP antibody (Chemicon International, USA). The secondary antibody was horseradish peroxidase-conjugated goat anti-mouse (Amersham Biosciences, UK). The immune complexes were visualized with the ECL detection system (Amer- sham Biosciencesces, UK) accordingly to the manufac- turer’s instructions. 2.4. Microscopic Study and FACS Analysis Microscopic evaluation of EGFP expression in the re- combinant yeast clones was performed by direct cell observation using a Leica DMLB fluorescence micro- scope, equipped with a FITC filter and a DC300 F cam- era. The early log-phase yeast cultures in minimal medium were analyzed using a FACScan flow cytometer and CellQuest software (Becton Dickinson, San Jose, CA, USA) with a standard excitation wavelength of 488 nm. 3. RESULTS AND DISCUSSION. Our objective was to develop of a pFL61-based versatile plasmid construction system suitable for the efficient preparation of yeast expression vectors for producing C-terminal EGFP-tagged proteins. The pFL61 vector [18] allows an efficient gene expression under the control of the constitutive PGK promoter. We chose to localize the fluorescent domain at the C-terminus to allow the pro- teins, which have an N-terminal signal peptide, to follow their own localization pathway [28-30]. The EGFP open reading frame was obtained from the PINCO retroviral plasmid [25]. Its Hind-Not fragment was first subcloned the into the pBlueScript-KS vector to obtain an easier to handle plasmid, named pBS(GFP), which carrying an Nco I unique site. The digestion with Nco I and Not I of the pBS(GFP), followed by a treat- ment with T4 DNA polymerase and dNTPs, produce an EGFP cDNA with blunt ends. For generation of the pFL61(EGFP) plasmid, the blunt fragment was cloned in the linearised pFL61. The re- sulting plasmid was further engineered, using the site-specific mutagenesis procedure, to generate a unique Not I site upstream the EGFP start codon [26]. The novel vector was named pFL61(EGFPmut). The recovery of the Not I site, destroyed by insertion of EGFP cDNA, made the vector suitable for cloning employments. Furthermore the mutation preserved the integrity of the Kozak sequence [31]. The pFL61(EGFPmut) vector was used to express two Tuber borchii proteins in yeast: the TBFL-1 lectin [19] and TBHXT1 hexose transporter [21]. A reproducible cloning protocol was developed in or- der to obtain the versatile construction system. The coding regions (ORF) of the target genes, the tbfl-1 or the tbhxt1, were amplified by PCR using the primers shown in Ta b le 1 . The forward primer was de- signed to start from ATG codon while the reverse primer contained a Not I site localized close to stop codon so to make the coding region in frame to the EGFP sequence. The amplified-fragment was subcloned in the pGEM-T vector system. This step permitted to add to the cDNA a further Not I site, came from the pGEM polylinker, up- stream the 5’ ATG. The Not I digestion of the recombi- nant pGEM(ORF) plasmid generated a cDNA fragment compatible with the expression vector pFL61(EGFPmut). The schematic diagram in Figure 1 summarizes the steps previously described. Following this procedure we were able to obtain the vectors pFL61(TBFL1-EGFP) and pFL61(TBHXT1- EGFP) carrying the two Tuber borchii genes [19,21]. We used a complementation study, exploiting the fluo- rescence properties of EGFP, to demonstrate the correct expression of recombinant chimeric gene. In fact, in fluorescence microscopy the cells expressing the EGFP should show a diffuse cytoplasmic fluorescence [15], due to the soluble nature of the protein, while, the yeast 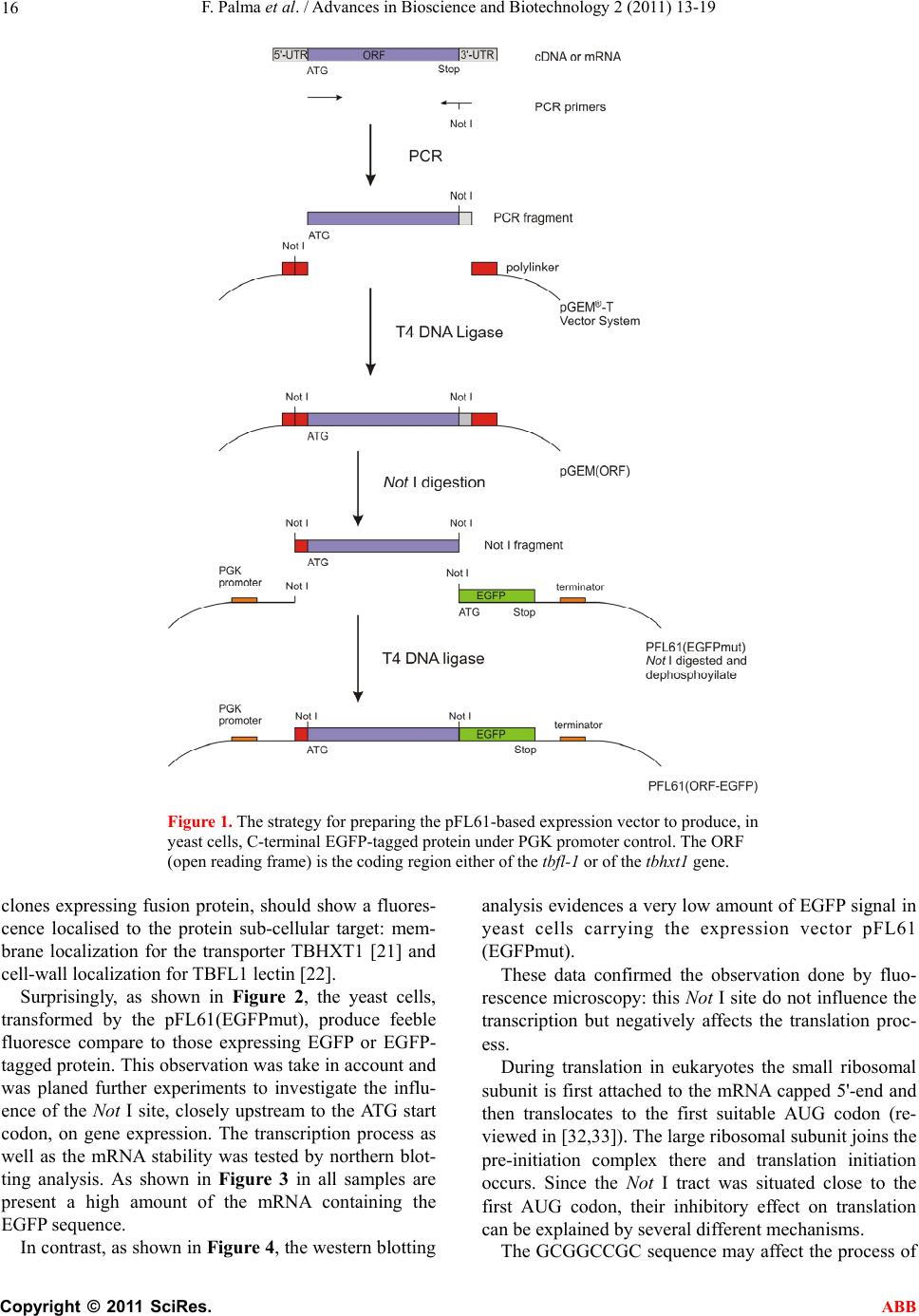 F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 16 Figure 1. The strategy for preparing the pFL61-based expression vector to produce, in yeast cells, C-terminal EGFP-tagged protein under PGK promoter control. The ORF (open reading frame) is the coding region either of the tbfl-1 or of the tbhxt1 gene. clones expressing fusion protein, should show a fluores- cence localised to the protein sub-cellular target: mem- brane localization for the transporter TBHXT1 [21] and cell-wall localization for TBFL1 lectin [22]. Surprisingly, as shown in Figure 2, the yeast cells, transformed by the pFL61(EGFPmut), produce feeble fluoresce compare to those expressing EGFP or EGFP- tagged protein. This observation was take in account and was planed further experiments to investigate the influ- ence of the Not I site, closely upstream to the ATG start codon, on gene expression. The transcription process as well as the mRNA stability was tested by northern blot- ting analysis. As shown in Figure 3 in all samples are present a high amount of the mRNA containing the EGFP sequence. In contrast, as shown in Figure 4, the western blotting analysis evidences a very low amount of EGFP signal in yeast cells carrying the expression vector pFL61 (EGFPmut). These data confirmed the observation done by fluo- rescence microscopy: this Not I site do not influence the transcription but negatively affects the translation proc- ess. During translation in eukaryotes the small ribosomal subunit is first attached to the mRNA capped 5'-end and then translocates to the first suitable AUG codon (re- viewed in [32,33]). The large ribosomal subunit joins the pre-initiation complex there and translation initiation occurs. Since the Not I tract was situated close to the first AUG codon, their inhibitory effect on translation can be explained by several different mechanisms. The GCGGCCGC sequence may affect the process of  F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 17 (a) (b) (c) (d) Figure 2. Fluorescence microscopy analyses of the expression of GFP fusion proteins in recom- binant yeast cells harbouring different expression vectors. The Saccharomyces cerevisiae cells transformed with the following expression vec- tors: a) pFL61(EGFP); b) pFL61(EGFPmut); c) pFL61 (TBFL1-EGFP); d) pFL61(TBHXT1- EGFP) are shown. The pictures were obtained by the fluorescent microscopy analysing live cells and were token by a camera setting the exposition time to 1 sec. Figure 3. Northern blot of 20 µg of total RNA extracted from 1.5 mg of wet weight yeast cells log-phase harvested harbouring different expres- sion vectors. Lane 1, pFL61(EGFP); lane 2, pFL61(EGFPmut); lane 3, pFL61(TBFL1-EGFP); lane 4, pFL61(TBHXT1-EGFP). In panel A the hybridization was performed with 32P labelled EGFP cDNA as specific probe. In panel B the ACT1 probe was 32P labelled and used as internal control. Figure 4. Western blotting analysis of total pro- teins from yeast cells harbouring different ex- pression vectors. Lane 1, pFL61(EGFP); lane 2, pFL61(EGFPmut); lane 3, pFL61(TBFL1- EGFP); lane 4, pFL61(TBHXT1-EGFP). recognition of the start codon after the translocation of the small ribosomal subunit from RNA 5'-end, called scanning (reviewed in [32]). We hypnotized that this Not I sequence, close to ATG, might form a hairpin which reduces the rate of translation. It is well documented that eukaryotic mRNAs with highly structured 5'-UTRs are relatively inefficient translationally [34,35]. This is likely due to the inability of the 40S ribosomal subunit and/or associated RNA helicases to unwind stable sec ondary structures in the 5'-UTR during scanning and AUG recognition [36]. This phenomenon gave an ad- vantage to the proposed yeast expression vector system, in fact a bright florescence were observed only in yeast cells carrying the exogenous gene fused with egfp; in contrast a feeble florescence emission was found in clone harbouring the void vector and no fluorescence was found in the cells containing the vector where the exogenous genes were in a wrong orientation (data not shown). The phenotypic differences among the yeast clones were also analysed by flow cytometer. The yeast cultures were analysed in an early stage of growth to have a bet- ter plasmid retention rate [37]. The Figure 5 reports the FACS graph of the different yeast clones. The filled histogram represents a negative control yeast cells. The line histograms describe the fluorescence distribution of the four yeast clone cells. The green line represents the yeast carrying pFL61 (EGFPmut) that shows lower fluorescence than pFL61(EGFP), magenta line, pFL61(TBF1-EGFP), blue line, and pFL61(TBHXT1-EGFP), orange line. Unfor- tunately, this comparative analysis evidences that the different fluorescence properties among the yeast clones do not permit to sort them by flow cytometry. The versatile expression system based on pFL61, re- (a)  F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 18 (b) Figure 5. Cytometry analyses of transformed yeast cells con- stitutively expressing EGFP-tagged proteins The Panel A re- ports the analysis of a yeast clone. In panel B is shown the fluorescence intensity by yeast cells harbouring the vector pFL61(EGFPmut) in green line, harbouring pFL61(EGFP) in magenta line; pFL61(TBFL1-EGFP) in blue line, and harbour- ing pFL61(TBHXT1-EGFP) in orange line; the filled histo- gram was obtained by untransformed yeast cells. ported in this paper, allows obtaining, in a few steps, a vector for producing proteins fused with EGFP in yeast. We also noted that the palindromic Not I restriction site, close to the start codon, affects the translation process. This phenomenon could be further investigated to create vectors that generate a fluorescent phenotype only if they carry an exogenous gene inserted in the correct orientation. REFERENCES [1] Chalfie, M., Tu, Y., Euskirchen, G., Ward, W.W. and Prasher, D. C. (1994) Green fluorescent protein as a marker for gene expression. Science, 263, 802-805. doi:10.1126/science.8303295 [2] Plautz, J.D., Day, R.N., Dailey, G.M., Welsh, S.B., Hall, J. C., Halpain, S. and Kay, S.A. (1996) Green fluorescent protein and its derivatives as versatile markers for gene expression in living Drosophila melanogaster, plant and mammalian cells. Gene, 173, 83-87. [3] Higgs, S., Traul, D., Davis, B.S., Kamrud, K.I., Wilcox, C.L. and Beaty, B.J. (1996) Green fluorescent protein expressed in living mosquitoes--without the requirement of transformation. Biotechniques, 21, 660-664. [4] Cubitt, A.B., Heim, R., Adams, S.R., Boyd, A.E., Gross, L.A. and Tsien, R.Y. (1995) Understanding, improving and using green fluorescent proteins. Trends in Bio- chem ical Sciences, 20, 448-455. doi:10.1016/S0968-0004(00)89099-4 [5] Chiu, W., Niwa, Y., Zeng, W., Hirano, T., Kobayashi, H. and Sheen, J. (1996) Engineered GFP as a vital reporter in plants. Current Biology, 6, 325-330. doi:10.1016/S0960-9822(02)00483-9 [6] Baulcombe, D.C., Chapman, S. and Santa, C.S. (1995) Jellyfish green fluorescent protein as a reporter for virus infections. Plant Journal, 7, 1045-1053. doi:10.1046/j.1365-313X.1995.07061045.x [7] Amsterdam, A., Lin, S. and Hopkins, N. (1995) The Ae- quorea victoria green fluorescent protein can be used as a reporter in live zebrafish embryos. Developmental Biol- ogy, 171, 123-129. doi:10.1006/dbio.1995.1265 [8] Prasher, D.C., Eckenrode, V.K., Ward, W.W., Prendergast, F.G. and Cormier, M.J. (1992) Primary structure of the Aequorea victoria green-fluorescent protein. Gene, 111, 229-233. [9] Prasher, D.C. (1995) Using GFP to see the light. Trends in Genetics, 11, 320-323. doi:10.1016/S0168-9525(00)89090-3 [10] Inouye, S. and Tsuji, F.I. (1994) Aequorea green fluores- cent protein. Expression of the gene and fluorescence characteristics of the recombinant protein. FEBS Letters, 341, 277-280. doi:10.1016/0014-5793(94)80472-9 [11] Heim, R., Cubitt, A.B. and Tsien, R.Y. (1995) Improved green fluorescence. Nature, 373, 663-664. doi:10.1038/373663b0 [12] Heim, R. and Tsien, R.Y. (1996) Engineering green fluo- rescent protein for improved brightness, longer wave- lengths and fluorescence resonance energy transfer. Cur- rent Biology, 6, 178-182. doi:10.1016/S0960-9822(02)00450-5 [13] Cormack, B.P., Valdivia, R.H. and Falkow, S. (1996) FACS-optimized mutants of the green fluorescent protein (GFP). Gene, 173, 33-38. [14] Fuhrmann, M., Oertel, W. and Hegemann, P. (1999) A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in Chlamydomonas rein- hardtii. Plant Journal, 19, 353-361. doi:10.1046/j.1365-313X.1999.00526.x [15] Rizzuto, R., Brini, M., Pizzo, P., Murgia, M. and Pozzan, T. (1995) Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Current Biology, 635-642. doi:10.1016/S0960-9822(95)00128-X [16] Koerte, A., Chong, T., Li, X., Wahane, K., and Cai, M., "Suppression of the yeast mutation rft1-1 by human p53," Journal of Biological Chemistry, Vol. 270, No. 38, 22-9-1995, pp. 22556-22564. [17] Starling, A.L., Ortega, J.M., Gollob, K.J., Vicente, E.J., ndrade-Nobrega, G. M., and Rodriguez, M. B., "Evalua- tion of alternative reporter genes for the yeast two-hybrid system," Genetics and Molecular Research, Vol. 2, No. 1, 2003, pp. 124-135. [18] Minet, M., Dufour, M.E. and Lacroute, F. (1992) Com- plementation of Saccharomyces cerevisiae auxotrophic mutants by Arabidopsis thaliana cDNAs. Plant Journal, 2, 417-422. [19] Cerigini, E., Palma, F., Barbieri, E., Buffalini, M. and Stocchi, V. (2008) The Tuber borchii fruiting body-specific protein TBF-1, a novel lectin which inter- acts with associated Rhizobium species. FEMS Microbi- ology Letters, 284, 197-203. doi:10.1111/j.1574-6968.2008.01197.x [20] Palma, F., Cerigini, E. and Stocchi, V. (2007) Yeast ex- pression of the Tuber borchii fruiting body specific pro- tein, TBF-1: identification of a noncanonical signal pep- tide. FEMS Microbiology Letters, 272, 114-119. doi:10.1111/j.1574-6968.2007.00748.x [21] Polidori, E., Ceccaroli, P., Saltarelli, R., Guescini, M., Menotta, M., Agostini, D., Palma, F. and Stocchi, V.  F. Palma et al. / Advances in Bioscience and Biotechnology 2 (2011) 13-19 Copyright © 2011 SciRes. ABB 19 (2007) Hexose uptake in the plant symbiotic ascomycete Tuber borchii Vittadini: biochemical features and expres- sion pattern of the transporter TBHXT1. Fungal Genetics and Biology, 44, 187-198. doi:10.1016/j.fgb.2006.08.001 [22] De Bellis, R., Agostini, D., Piccoli, G., Vallorani, L., Potenza, L., Polidori, E., Sisti, D., Amoresano, A., Pucci, P., Arpaia, G., Macino, G., Balestrini, R., Bonfante, P. and Stocchi, V. (1988) The tbf-1 gene from the white truffle Tuber borchii codes for a structural cell wall protein spe- cifically expressed in fruitbody. Fungal Genetics and Bi- ology, 25, 87-99. doi:10.1006/fgbi.1998.1092 [23] Ito, H., Fukuda, Y., Murata, K. and Kimura, A. (1983) Transformation of intact yeast cells treated with alkali cations. Journal of Bacteriology, 153, 163-168. [24] Hill, J., Donald, K.A. and Griffiths, D.E. (1991) DMSO- enhanced whole cell yeast transformation. Nucleic Ac- ids Research, 19, 5791. doi:10.1093/nar/19.20.5791 [25] Grignani, F., Kinsella, T., Mencarelli, A., Valtieri, M., Riganelli, D., Grignani, F., Lanfrancone, L., Peschle, C., Nolan, G. P. and Pelicci, P. G. (1998) High-efficiency gene transfer and selection of human hematopoietic pro- genitor cells with a hybrid EBV/retroviral vector ex- pressing the green fluorescence protein. Cancer Research, 58, 14-19. [26] R.Higuchi "Recombinant PCR," In: Innis, M.A., Gelfand, D.H., Sninsky, J.J. and White T.J. Ed., (1990) PCR Pro- tocols: A Guide to Methods and Applications, Academic Press, Inc., San Diego. [27] Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680-685. doi:10.1038/227680a0 [28] Rothman, J.E. (1994) Mechanisms of intracellular pro- tein transport. Nature, 372, 55-63. doi:10.1038/372055a0 [29] Palade, G. (1975) Intracellular Aspects of the Process of Protein Synthesis. Science, 189, 867. doi:10.1126/science.189.4206.867-b [30] Nombela, C., Gil, C., and Chaffin, W.L. (2006) Non-conventional protein secretion in yeast. Trends in Microbiology, 14, 15-21. doi:10.1016/j.tim.2005.11.009 [31] Kozak, M. (1987) An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Research, 15, 8125-8148. doi:10.1093/nar/15.20.8125 [32] Kozak, M. (1999) Initiation of translation in prokaryotes and eukaryotes. Gene, 234, 187-208. [33] Gingras, A.C., Raught, B. and Sonenberg, N. (1999) eIF4 initiation factors: effectors of mRNA recruitment to ri- bosomes and regulators of translation. Annual Review of Biochemistry, 68, 913-963. doi:10.1146/annurev.biochem.68.1.913 [34] Kozak, M. (1980) Influence of mRNA secondary struc- ture on binding and migration of 40S ribosomal subunits. Cell, 19, 79-90. [35] Kozak, M. (1986) Influences of mRNA secondary struc- ture on initiation by eukaryotic ribosomes. Proceedings of the National Academy of Sciences of the United States of America, 83, 2850-2854. doi:10.1073/pnas.83.9.2850 [36] Kozak, M. (1989) Circumstances and mechanisms of inhibition of translation by secondary structure in eu- caryotic mRNAs. Molecular and Cellular Biology, 9, 5134-5142. [37] Ishii, J., Izawa, K., Matsumura, S., Wakamura, K., Tan- ino, T., Tanaka, T., Ogino, C., Fukuda, H. and Kondo, A. (2009) A simple and immediate method for simultane- ously evaluating expression level and plasmid mainte- nance in yeast. Journal of Biochemistry, 145, 701-708. doi:10.1093/jb/mvp028 |