 Open Journal of Synthesis Theory and Applications, 2013, 2, 91-96 http://dx.doi.org/10.4236/ojsta.2013.24012 Published Online October 2013 (http://www.scirp.org/journal/ojsta) Regioselective Synthesis and Biological Evaluation of 1-Hydroxyl Modified Ailanthinone Derivatives as Antimalarials Mahendra D. Chordia*, William F. McCalmont, Kirsten S. Smith, Philip L. Smith* Division of Experimental Therapeutics, Walter Reed Army Institute of Research, Silver Spring, USA Email: *mdchordia@gmail.com, *philip.l.smith@us.army.mil Received August 20, 2013; revised September 26, 2013; accepted October 11, 2013 Copyright © 2013 Mahendra D. Chordia et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT The triterpene quassinoid ailanthinone is a structurally intricate natural product possessing highly potent antimalarial activity against multidrug resistance plasmodium parasites. Although the mechanism of action of ailanthinone is not completely understood, it is presumed to disrupt regular ribosomal functions by inhibiting parasite protein synthesis. Natural scarcity and low solubility of many quassinoid s h ave impeded th eir deve lopment as potential clin ical candid ates, but exquisite potency of ailanthinone against Plasmodium remains compelling in the global fight against malaria. Herein, we report the highly selective synthesis of 1-hydroxyl derivatives of ailanthinone, including ester, carbonate, carbamate and sulfonate derivatives. The key feature of the synthesis is a one-step regioselective modification of the 1-hydroxyl grou p in favor of two other hydroxyl groups at C12 and C13. Der ivatives were obtain ed via direct reaction with acyl/sulfonyl chlorides in the presence of a tertiary amine base without any protection-deprotection. In vitro anti- malarial evaluations of these derivatives were compared with chloroquine and mefloquine against the Plasmodium fal- ciparum clones, D6, W2, and TM91C235. The results demonstrate that modification of the 1-hydroxyl group is achiev- able, and the antimalarial activity of these derivatives relative to the paren t product is sign ificantly retained, thus paving the way for synthesis of derivatives with improved biological availability and/or increased potency. Keywords: Antimalarials; Ailanthinone; Triterpenoids; Quassinoid; Plasmodium Falciparum; Multidrug Resistance 1. Introduction Plants of the family Simaroubaceae have long been util- ized as therapeutic material for a number of diseases be- cause of its anti-malarial, antiviral, amoebicidal, insecti- cidal, anti-feedent and leish manicidal properties in Africa, Asia, and South America [1-4]. The remedial properties of these plants materials are thought to be due to presence of a class of compounds called quassinoids, which be- long to the triterpene group of natural products. Several quassinoid derivatives isolated from various plants have exhibited significant in vitro potency against both chlo- roquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum [5,6]. Quassinoids were first iso- lated over 20 years ago and were initially believed to be unsuitable for clinical development owing to their scar- city, and later for their relatively low therapeutic indices when administered in vivo. Recently, however, a number of new studies have demonstrated that semi-synthetic derivatives of quassinoids possess significant in vivo ef- ficacy against P. berghei infections in mice [7]. Fur- thermore, some of these compounds displayed no ob- servable toxicity when administered at therapeutic doses [8]. Although the mechanism of action of the quassinoid natural products as antimalarials is not completely un- derstood, a number of studies suggest that they act as inhibitors of protein synthesis by disrupting normal ri- bosomal function of the parasite. Evidence for this has been demonstrated in cultures of P. falciparum [9]. Quassinoids, therefore, appear to function as antimalari- als through a fundamentally different mechanism from that of chloroqu ine and other current antimalarials. Thus, they represent a new class of potential therapeutics that may be less prone to the development of drug-resistance by the parasite. Studies of quassinoids thus may offer attractive clinical alternative to more relevant quinine based drugs. As part of a continuing program that encompasses the *Corresponding authors. C opyright © 2013 SciRes. OJSTA  M. D. CHORDIA ET AL. 92 collaborative efforts of the Natural Products Group of the Division of Experimental Therapeutics, WRAIR, the Bioresources Development and Conservation Program (BDCP), Silverspring, MD, we selected a highly promis- ing lead quassinoid natural product, ailanthinone, that displays exception al in vitro antimalarial activity with an IC50 of ~2.8 ng/mL against the chlo roquine resistant W2 strain (unpublished results from in ho use screening ). This IC50 exhibited by ailanthinone is comparable to that ob- served for chloroquine used in controls. Furthermore, ailanthinone retains potent anti-malarial activity (IC50 = 4.3 ng/mL) against the multidrug resistant strain of P. falciparum TM91C235, thus providing a new window of opportunity to develop its derivatives as potential thera- peutic agents against malaria. However, development of ailanthinone as an antimalarial is hampered by its natural scarcity and poor solubility in aqueous media. With an aim to develop structure activity relationship repertoire and to alleviate challenges with natural availability by improving its potency/bio-availability, we initiated stud- ies to understand the chemical reactivity of ailanthinone. Our initial studies focused on chemo-selective reactivity of the various hydroxyl groups. Results o btained from th e studies are presented here along with in vitro biological data. 2. Experimental Section 2.1. General Methods Ailanthinone samples used in the current studies were kindly provided by the International Center for Ethno medicine and Drug Development (InterCEDD) program. All other chemicals and solvents were procured from standard sources such as Aldrich (St. Louise, MO) and/or Acros Organics (Morris Plains, NJ). All chemicals pur- chased were of the highest quality available and used without further purification. Preparative silica gel TLC plates were purchased from Analtech Inc. (Newark, De). The TLC and preparative plates were monitored for de- velopment with UV light (short wavelength 254 nm). 1H-NMR analyses were performed on Bruker Avance 300 MHz or Varian Unity 300 MHz spectrophotometers. The chemical shifts are noted in ppm as δ values and coupling constant are reported as J in Hz. The ESI mass spectral data for compounds were performed on LCQ (Thermo Finnigan) ion trap LCMS instrument equipped with waters HPLC. Samples are prepared in HPLC grade acetonitrile and injected in LCMS using H2O:CH3CN formic acid (1%) solvent system in isocratic at flow rate of 0.3 mL/min. The mass spectra were recorded in posi- tive ion mode using following parameters: capillary tem- perature 250˚C. ESI spay voltage 4.5 kV, multiplyer 1500 V at WRAIR’s mass-spectrometry facilities. The in vitro antimalarial biological data was collected at in house facilities. 2.2. Experimental Procedures Acylation of ailanthinone with acetic anhydride: A solu- tion of acetic anhydride (0.1 M, 300 L, 0.03 mmol) in dry CH2Cl2 was added to premixed cold solution (4˚C) of ailanthinone (7.2 mg, 0.015 mmol) and triethyl amine (100.0 L, 0.72 mmol) in dry CH2Cl2 (1.0 mL). The mix- ture was stirred for 1 hr and allowed to warm room tem- perature and further stirred for 4 hrs. The TLC analysis of the reaction mixture revealed formation of two new non-polar products. The reaction mixture was diluted with ethyl acetate (10 mL) and washed with water (5 mL) followed by 10 % NaHCO3 (5 mL) solution. The organic layer was finally washed with brine and dried over Na2SO4 and concentrated under reduced pr essure to yield crude residue. Purification of crude material by prepara- tive TLC (Silica gel GF, 20 × 20, 250 micron) yielded two compounds polar 1-acetyl ailanthinone (5.1 mg, 65%) and a non-polar ring opened 1, 20-diacety seco-ailan- thinone ( 2.2 mg, 26%) . 1H-NMR of 1-acetyl-ailanthione (4) (CDCl3): δ 0.96 (t, J = 7.8 Hz, 3H, CH3), 0.97 (q, J = 7.2 Hz, 3H, CH3), 1.09 (d, J = 6.9 Hz, 3H, CH3), 1.18 (d, J = 6.0 Hz, 3H, CH3), 1.35 (s, 3H, C10-CH3) 1.45 (m, 1H), 1.60 (m, 1H), 1.96 (m, 1H), 1.99 (brs, 3H), 2.26 (s, 3H, COCH3), 2.35 (m, 2H), 2.42 (q, J = 6.9 Hz, 1H, CHCO), 2.62 (s, 1H), 3.16 (brd, J = 12.0 Hz, 1H), 3.56 (brd, J = 4.5 Hz, 1H), 3.68 (d, J = 9.0 Hz, 1H), 3.96 (d, J = 9.0 Hz, 1H), 4 .31 (dq, J = Hz, 2H, OCH2) 4.63 (dd, J = 1.2, 4.8 Hz, 1H), 5.28 (s, 1H), 5.49 (brd, J = 6.0 Hz, 1H), 5.70 (s, 1H), 6.05 (q, J = 1.8 Hz, 1H, C3-H). LCMS: observed ESI m/z 543 [M+Na]+; calculated for C27H35O10Na 543. 1H-NMR of 1,20-diacetyl seco-ailanthinone (5) (CDCl3): δ 0.94 (t, J = 7.8 Hz, 3H, CH3), 0.98 (q, J = 7.2 Hz, 3H, CH3), 1.24 (d, J = 6.9 Hz, 3H, CH3), 1.28 (s, 3H, C10-CH3) 1.51 (m, 1H), 1.65 (m, 1H), 1.96 (m, 1H), 1.97 (brs, 3H), 2.05 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 2.27 (m, 1H), 2.49 (m, 1H), 3.21 (brd, J = 12.3 Hz, 1H), 3.31 (s, 1H), 3.80 (d, J = 12.9 Hz, 1H, C-20H), 3.84 (d, J = 2.4 Hz, 1H), 3.94 (s, 1H), 4.66 (d, J = 12.6 Hz, 1H, C-20H), 4.97 (t, J = 1.8 Hz, 1H), 5.19 (s, 1H), 5.98 (d, J = 9.0 Hz, 1H), 6.05 (q, J = 1.2 Hz, 1H, C3-H). LCMS: observed ESI m/z 585 [M+Na]+; calculated for C29H38O11Na 585. General procedure for modification of 1-hydroxyl group: Ailanthinone (3) (5.0 mg, 0.0105 mmol) was dissolved in CH2Cl2 (1.0 mL) and to it was added triethylamine (50.0 L, 0.36 mmol) and cooled in ice bath. Subsequently the acyl chloride reagent (0.01 mmol, from freshly made 1 mM solution in CH2Cl2) was added. The mixture was stirred at 5˚C for 1hr and then allowed to warm to room temperature (4 - 16 hrs). The reaction was followed by Copyright © 2013 SciRes. OJSTA  M. D. CHORDIA ET AL. 93 TLC analysis. For most of the reactions a very small amount of ailanthinone remained unreacted (~5% - 10%) as a polar spot and new non-polar compound as major spot was appeared. The reaction mixture was then diluted with CH2Cl2 and concentrated under reduced pressure; the residue was re-dissolved in chloroform (0.2 mL) and loaded on preparative TLC plate (Silica gel GF, 20 × 20, 500 micron). The preparative TLC purification was per- formed first by running the plate in 1:3 (EtOAc:hexanes) and then 1:1 (EtOAc:hexanes). The non-polar UV active band was collected and desorbed with ethyl acetate (3 × 2 mL). The combined ethyl acetate layer evaporated un- der reduced pressure to yield sufficiently pure 1-hydroxyl modified ailanthinone derivatives (70% - 90%). The pu- rity of sample was primarily accessed by 1H-NMR ana- lysis and compared with parent ailanthinone. The newly derived compounds were submitted without further puri- fication for their in vitro biological evaluation as anti- malarials. 1H-NMR of 1-methyloxycarbonyl- ailanthion e (6) (CDCl3): δ 0.95 (t, J = 7.5 Hz, 3H, CH3), 0.97 (q, J = 7.8 Hz, 3H , CH 3), 1.09 (brd, J = 6.9 Hz, 3H, CH3), 1.17 (d, J = 6.0 Hz, 3H, CH3), 1.34 (s, 3H, C10-CH 3) 1.51 (m, 1H), 1.76 (m, 1H), 1.98 (brs, 3H), 2.24-2.39 (m, 2H), 2.42 (q, J = 6.9 Hz, 1H, CHCO), 2.64 (s, 1H), 3.11 (brd, J = 12.0 Hz, 1H), 3.56 (brs, 1H), 3.68 (d, J = 9.0 Hz, 1H), 3.88 (s, 3H, OCH3), 3.96 (d, J = 9.0 Hz, 1H), 4.63 (dd, J = 1.2, 4.8 Hz, 1H), 5.01 (s, 1H), 5.48 (brd, J = 6.0 Hz, 1H), 5.54 (s, 1H), 6.09 (t, J = 1.8 Hz, 1H, C3-H). LCMS: ob- served ESI m/z 559 [M+Na]+; calculated for C27H36O11Na 559. 1H-NMR of 1-ethyloxycarbonyl-ailanth inone (7) (CDCl3): δ 0.95 (t, J = 7.5 Hz, 3H, CH3), 0.96 (q, J = 7.8 Hz, 3H , CH 3), 1.09 (brd, J = 6.9 Hz, 3H, CH3), 1.17 (d, J = 6.0 Hz, 3H, CH3), 1.33 (s, 3H, C10-CH3), 1.34 (t, J = 7.5 Hz, CH3) 1.50 (m, 1H), 1.77 (m, 1H), 1.98 (brs, 3H, CH3), 2.03 (m, 1H), 2.23-2.38 (m, 2H), 2.42 (q, J = 6.0 Hz, 1H), 2.63 (s, 1H), 3.11 (brd, J = 12.0 Hz, 1H), 3.56 (brd, J = 4.5 Hz, 1H), 3.67 (d, J = 9.0 Hz, 1H ), 3.96 (d, J = 9.0 Hz, 1H), 4.30 (dq, J = 9.0 Hz, 2H, OCH2) 4.62 (dd, J = 1.2, 4.8 Hz, 1H), 5.00 (s, 1H), 5.48 (brd, J = 9.0 Hz, 1H), 5.60 (brs, 1H), 6.08 (q, J = 1.8 Hz, 1H, C3-H). LCMS: observed ESI m/z 573 [M+Na]+; calculated for C28H38O11Na 573. 1H-NMR of 1-isobutyloxycarbonyl-ailanthinone (8) (CDCl3): δ 0.95 (t, J = 7.5 Hz, 3H, CH3), 0.96 (d, J = 7.5 Hz, 6H, CH3 × 2), 0.96 (q, J = 7.8 Hz, 3H, CH3), 1.09 (brd, J = 6.9 Hz, 3H, CH3), 1.18 (d, J = 6.0 Hz, 3H, CH3), 1.34 (s, 3H, C10-CH3), 1.50 (m, 1H), 1.77 (m, 1H), 1.98 (brs, 3H, CH3), 2.03 (m, 1H), 2.23-2.38 (m, 2H), 2.42 (q, J = 6.0 Hz, 1H), 2.63 (s, 1H), 3.11 (brd, J = 12.0 Hz, 1H), 3.56 (brd, J = 4.5 Hz, 1H), 3.68 (d, J = 9.0 Hz, 1H), 3.96 (d, J = 9.0 Hz, 1H), 4.03 (dq, J = 9.0 Hz, 2H, OCH2) 4.62 (dd, J = 1.2, 4.8 Hz, 1H), 5.00 (s, 1H), 5.48 (brd, J = 9.0 Hz, 1H), 5.64 (s, 1H), 6.08 (q, J = 1.8 Hz, 1H, C3-H). LCMS: observed ESI m/z 601 [M+Na]+; calculated for C30H42O11Na 601. 1-Benzoyl-ailanthione (9) (CDCl3): δ 0.96 (t, J = 7.5 Hz, 3H, CH3), 0.97 (q, J = 7.5 Hz, 3H, CH3), 1.05 (d, J = 6.9 Hz, 3H, CH3), 1.18 (d, J = 6.0 Hz, 3H, CH3), 1.35 (s, 3H, C10-CH3) 1.45 (m, 1H), 1.60 (m, 1H), 1.96 (m, 1H), 1.99 (brs, 3H), 2.80 (dt, J = 1.8, 9.0 Hz, 1H), 2.35 (m, 2H), 2.42 (q, J = 6.9 Hz, 1H, CHCO), 2.70 (s, 1H), 3.21 (brd, J = 12.0 Hz, 1H), 3.38 (t, J = 4.5 Hz, 1H), 3.67 (d, J = 9.0 Hz, 1H), 3.97 (d, J = 9.0 Hz, 1H), 4.64 (dd, J = 1.2, 4.8 Hz, 1H), 5.42 (s, 1H), 5.45 (brd, J = 6.0 Hz, 1H), 5.55 (s, 1H), 6.10 (q, J = 1.8 Hz, 1H, C3-H), 7.46 (t, J = Hz, 2H, ArH), 7.61 (t, J = Hz, 1H, ArH), 8.06 (d, J = Hz, 2H, ArH). 1-Piperdinylcarbonyl-ailanthinone (10) (CDCl3): δ 0.96 (t, J = 7.5 Hz, 3H, CH3), 0.98 (q, J = 7.5 Hz, 3H, CH3), 1.11 (brd, J = 6.9 Hz, 3H, CH3), 1.18 (d, J = 6.0 Hz, 3H, CH3), 1.19 (s, 3H, C10-CH3) 1.53 (m, 1H), 1.77 (m, 1H), 1.96 (m, 1H), 2.01 (brs, 3H), 2.19-2.46 (m, 3H), 2.73 (s, 1H), 2.98 (d, J = 7.8 Hz, 1H), 3.56 (brd, J = 4.5 Hz, 1H), 3.69 (d, J = 9.0 Hz, 1H), 3.96 (d, J = 9.0 Hz, 1H), 4.06 (s, 1H ), 4.63 (dd, J = 1.2, 4.8 Hz, 1H), 5.18 (s, 1H), 5.57 (brd, J = 6.0 Hz, 1H), 6.14 (q, J = 1.8 Hz, 1H, C3-H). LCMS: observed m/z 590.12, calculated [M+H]+ 590. 1-p-Bromophenylsulfonyl-ailanthione (11) (CDCl3): δ 0.96 (t, J = 7.5 Hz, 3H, CH3), 0.97 (q, J = 7.5 Hz, 3H, CH3), 1.17 (d, J = 6.9 Hz, 3H, CH3), 1.20 (d, J = 6.0 Hz, 3H, CH3), 1.30 (s, 3H, C10-CH3) 1.52 (m, 1H), 1.76 (m, 1H), 1.96 (m, 1H), 1.97 (brs, 3H), 2.25-2.49 (m, 3H), 2.83 (s, 1H), 3.02 (brs, 1H), 3.17 (brd, J = 12.0 Hz, 1H), 3.59 (brs, 1H), 3.69 (d, J = 9.0 Hz, 1H), 3.95 (d, J = 9.0 Hz, 1H), 4.55 (s, 1H) 4.67 (dd, J = 1.2, 4.8 Hz, 1H), 5.10 (s, 1H), 5.48 (brd, J = 6.0 Hz, 1H), 5.99 (q, J = 1.8 Hz, 1H, C3 -H), 7.72 (d, J = 7.6 Hz, 2H), 7.90 (d, J = 7.6 Hz, 2H). LCMS: observed ESI m/z 719 and 721 [M+Na]+; calculated for C31H37BrO11SNa 71 9 a nd 721. 2.3. In Vitro An ti malarial Assay The antimalarial activities for all new compounds along with ailanthinone, chloroquine and mefloquine were ob- tained from three strains of parasites, namely D6 (resis- tant to mefloquine but susceptible to chloroquine, qui- nine and pyrimethamine), W2 (resistant to chloroquine, quinine and pyrimethamine but susceptible to mefloquine) and TM91C215 (a multiple drug resistant isolate from Thailand). The assays are based on standard biological screens performed at WRAIR. The detailed procedure is described elsewhere [10,11] and is limited to the assess- ment of the intrinsic activity against plasmodium in the erythrocyte like asexual life cycle (blood schizontocides). In brief, the cultured parasites were incub ated with varied concentration o f diluted drugs in 96 well microtiter plate Copyright © 2013 SciRes. OJSTA  M. D. CHORDIA ET AL. 94 for 48 hrs and then [3H]-hypoxanthine was added and its uptake in parasite was measured after additional incuba- tion of 24 h rs by first lysing and recov ering the DNA on glass fiber filter using Packard Filtermate Cell Harvester. The data were recorded on scintillation counter (Packard Top count microplate reader) and the results recorded as counts per minutes (CPM) per well at each drug concen- tration divided by the arithmetic mean of the CPM ob- tained from control untreated parasite wells. The loss in incorporation of radio-lab eled hypox anthine in parasite is measure of antimalarial activities of the compound tes- ted. 3. Results and Discussion Although ailanthinone possess impressive in vitro anti- malarial activity, further impetus for its development is hampered by its low natural abundance. In addition, little information is available about chemical reactivity of ai- lanthinone, particularly with respect to the hydroxyl moieties, and their relationship to its observed an timalar- ial activity. Ogura et al. [12] reported acylation of ailan- thinone using excess of acetic anhydride/pyridine over- night to afford the 1, 12, 20-triacetate derivative a he- miketal ring opened seco-ailanthinone. In another report glaucarubolone, a parent quassinoid devoid of the C-20 ester, was first protected as the trimethylsilyl ethers and subsequently esterfied at the C-15 hydroxyl group with succinic anhydride by Morré et al. [13]. Additionally, when a controlled acylation reaction of ailanthinone was performed using acetic anhydride and triethylamine at low temperature under mild conditio ns, which afforded a major C-1 acetylated ailanthinone (65%) along with a minor diacetate (C-1 and C-20) derivative (26%) (Sche- me 1). The outcome of the reaction not only indicated that C-1 hydroxyl group of ailanthinone was the more reac- tive when compared with the remaining two other hy- droxyl group at C-12 and C-13 positions, however these results also suggested that the cyclic hemiketal between the C-12 ketone and the C-20 hydroxyl was unstable un- O O HO OH OO OO OH Ac2O(2.0eq)/NEt 3 4oCtoRT O O OH O OO AcO OAcO Minor: 1, 20-diacetate derivative (26%) O O HO OH OO OO O O Major:1-Acetyl derivative (65%) Scheme 1. Acetylation of ailanthione with acetic anhydride. der those reaction conditions. The use of a slight excess of acetic anhydride may have been responsible for pro- moting the hemiketal ring to open followed by acetyla- tion of the resulting C-20 hydroxyl group. Thus, after few attempts, it was observed that using a more reactive acylating reagent in less than stoichiometric amount (~0.95 eq.) in the presence of a tertiary amine base re- sulted in a highly regioselective 1-hydroxyl acyl-pro- tected product that could be isolated along with unre- acted ailanthinone (approximately 5% - 10%). Using these optimized conditions, several 1-hydroxyl protected derivatives, such as the ester, carbonate, carbamate and sulfonate, were synthesized (Scheme 2). It must be noted here that the exclusive formation of 1-hydroxyl deriva- tives clearly demonstrates the higher reactivity of the C-1 hydroxyl over the C-12 and C-13 hydroxyl groups. In addition it can also be inferred from the formation of the C-1 and C-20 diacetate product that the C-13 hydroxyl group is the least reactive functional group. This conclu- sion is borne out by the observation that the hemiacetal ring opening and subsequent acylation of primary al- cohol of C-20 hydroxyl. The hemiacetal ring opening is reversible under the reaction condition but upon ring opening the C-20-hydroxyl reactivity being primary hy- droxy group is pretty high thus resulting in formation of C-20 acylation product. Acylation at C-20 position ren- der the ring-opening and closing process irreversible and resist the reformation the cyclic ketal. The new 1-hy- droxyl protected compounds were primarily character- ized by 1H-NMR and mass spectral data and compared the observed data with the parent ailanthinone to confirm their structures (Figure 1). The derivatives were then evaluated for their in vitro antimalarial activities with three available plasmodium strains: D6 (resistant to me- floquine but susceptible to chloroquine, quinine and pyrimethamine), W2 (resistant to chloroquine, quinine and pyrimethamine but susceptible to mefloquine), and TM91C215 (a multiple drug resistant isolates from Thai- land). A typical dose response curve is shown in Figure 2 obtained for 1 -ethox ycarbon yl ailan th inone. The r esu lts of these assays are presented in Table 1. The IC50 values (ng/mL) obtained from the assays for these compounds were compared with parent ailanthinone and with two well established antimalarial drugs, chloroquine and me- floquine. 0.95 eq. electrophile NEt3,CH 2Cl2, 0oCtoRT 75-90% after Prep TLCO O HO OH OO OO RO Ailanthinone 1-Hydroxy modified ailanthionones Ailanthi no ne Scheme 2. Synthesis of 1-Hydroxy modified ailanthinones. Copyright © 2013 SciRes. OJSTA 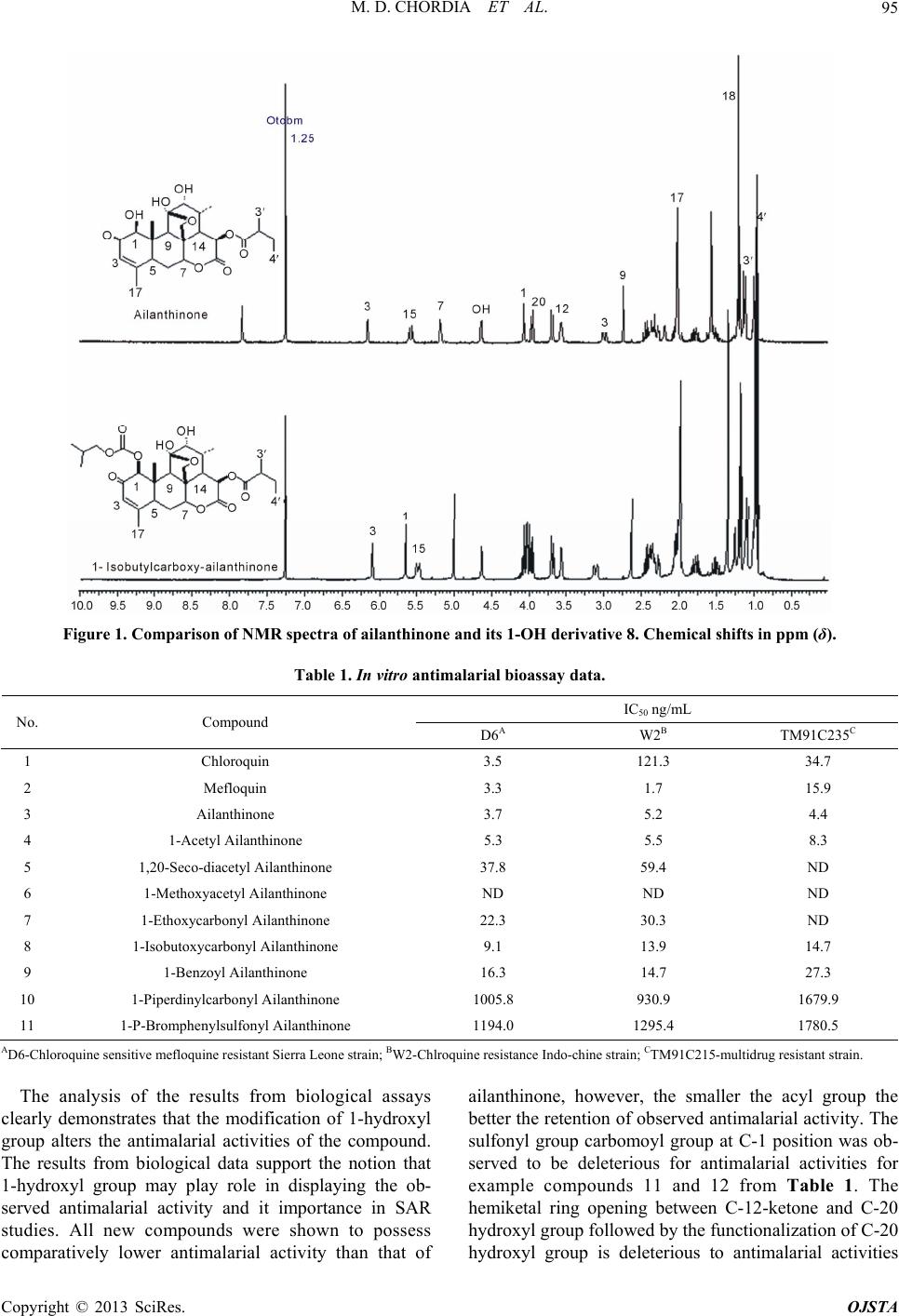 M. D. CHORDIA ET AL. Copyright © 2013 SciRes. OJSTA 95 Figure 1. Comparison of NMR spectra of ailanthinone and its 1-OH derivative 8. Chemical shifts in ppm (δ). Table 1. In vitro antimalarial bioassay data. IC50 ng/mL No. Compound D6A W2B TM91C235C 1 Chloroquin 3.5 121.3 34.7 2 Mefloquin 3.3 1.7 15.9 3 Ailanthinone 3.7 5.2 4.4 4 1-Acetyl Ailanthinone 5.3 5.5 8.3 5 1,20-Seco-diacetyl Ailanthinone 37.8 59.4 ND 6 1-Methoxyacetyl Ailanthinone ND ND ND 7 1-Ethoxycarbonyl Ailanthinone 22.3 30.3 ND 8 1-Isobutoxycarbonyl Ailanthinone 9.1 13.9 14.7 9 1-Benzoyl Ailanthinone 16.3 14.7 27.3 10 1-Piperdinylcarbonyl Ailanthinone 1005.8 930.9 1679.9 11 1-P-Bromphenylsulfonyl Ailanthinone 1194.0 1295.4 1780.5 AD6-Chloroquine sensitive mefloquine resistant S i erra Leone strain; BW2-Chlroquine resistance Indo-chine strain ; CTM9 1C2 15 -multidrug r es ista nt stra in. The analysis of the results from biological assays clearly demonstrates that the modification of 1-hydroxyl group alters the antimalarial activities of the compound. The results from biological data support the notion that 1-hydroxyl group may play role in displaying the ob- served antimalarial activity and it importance in SAR studies. All new compounds were shown to possess comparatively lower antimalarial activity than that of ailanthinone, however, the smaller the acyl group the better the retention of observ ed antimalarial activity. The sulfonyl group carbomoyl group at C-1 position was ob- served to be deleterious for antimalarial activities for example compounds 11 and 12 from Table 1. The hemiketal ring opening between C-12-ketone and C-20 hydroxyl group followed by the functionalization of C-20 hydroxyl group is deleterious to antimalarial activities  M. D. CHORDIA ET AL. 96 Figure 2. Representative dose-response curve (1-ethylcar- boxy-ailanthinone). (example 5). This decrease in biological activity may stem from the loss of rigid cyclic conformation of the ailanthinone skeleton, but further studies are needed to confirm these observations. The results presented here also suggest that proper modification of C-1 hydroxyl group may improve bioavailability. In conclusion, the highly selective modification of C-1 hydroxyl group of ailanthinone is possible without pro- tection/deprotection of the other hydroxyl groups. Such modification may be accomplished without negatively impacting antimalarial activity, and thus may be signify- cant to develop ailanthinone for clinical use in future. In addition, the cyclic hemiacetal formed between the C-12 ketone and C-20 hydroxyl is likely an essential featu re of ailanthinone to hold proper conformation of overall structure that is necessary for its potent antimalarial ac- tivity. 4. Acknowledgements We highly appreciate Dr. Maurice M. Iwu and Dr. Chris O. Okunji for providing us ailanthinone sample. Thanks are due to Norma Roncal and Lucia Gerena for collecting in vitro antimalarial data. REFERENCES [1] B. C. Joshi, R. P. Sharma and A. Khare, “Ailanthus Quas- sinoids and Their Biological Activity,” Natural Product Communications, Vol. 2, No. 8, 2007, pp. 869-880. [2] Z. Guo, S. Vangapandu, R. W. Sindelar, L. A. Walker and R. D. Sindelar, “Biologically Active Quassinoids and Their Chemistry: Potential Leads for Drug Design,” Cur- rent Medicinal Chemistry, Vol. 12, No. 2, 2005, pp. 173- 190. http://dx.doi.org/10.2174/0929867053363351 [3] S. C. Bhatnagar, A. J. Caruso and J. Polonky, “Biologi- cally-Active Quassinoids-Synthetic Methodology for the Conversion of Chaparrin into Glaucarbolone Esters and Quassinoid Analogs,” Tetrahedron, Vol. 43, No. 15, 1987, pp. 3471-3480. http://dx.doi.org/10.1016/S0040-4020(01)81638-0 [4] J. Polonsky, “Quassinoid Bitter Principles. II,” Progress in the Chemistry of Organic Natural Products, Vol. 47, 1985, pp. 221-264. [5] R. M. Ekong, G. C. Kirby, G. Patel, J. David Phillipson and D. C. Warhurst, “Comparision of the in Vitro Activi- ties of Quassinoids with Activity against Plasmodium- Falciparum, Anisomycine and Some Other Inhibitors of Eukaryotic Protein-Synthesis,” Biochemical Pharmacol- ogy, Vol. 40, No. 2, 1990, pp. 297-301. http://dx.doi.org/10.1016/0006-2952(90)90691-D [6] I. Muhammad and V. Samoylenko, “Antimalarial Quas- sinoids: Past, Present and Future,” Expert Opinion on Drug Discovery, Vol. 2, 2007, pp. 1065-1084. http://dx.doi.org/10.1517/17460441.2.8.1065 [7] H.-S. Kim, Y. Shibata, N. Ko, N. Ikemoto, Y. Ishizuka, et al., “Potent in Vivo Antimalarial Activity of 3,15-di-O- Acetylbruceolide against Plasmodium Berghei Infection in Mice,” Parasitology International, Vol. 48, 2000, pp. 271-274. http://dx.doi.org/10.1016/S1383-5769(99)00023-9 [8] N. Murakami, M. Sugimoto, M. Kawanishi, S. Tamura , H. S. Kim, et al., “New Semisynthetic Quassinoids with in Vivo Antimalarial Activity,” Journal of Medical Chemis- try, Vol. 46, No. 4, 2003, pp. 638-641. http://dx.doi.org/10.1021/jm0201971 [9] G. C. Kirby, M. J. O’Neill, J. D. Phillipson and D. C. Warhurst, “In Vitro Studies on the Mode of Action of Quassinoids with Activity Chloroquine-Resistant Plas- modium Falciparum,” Biochemical Pharmacology, Vol. 38, No. 24, 1989, pp. 4367-4374. http://dx.doi.org/10.1016/0006-2952(89)90644-8 [10] J. D. Chulay, J. D. Haynes and C. L. Diggs, “Plasmo- dium-Falciparum—Assessment of in Vitro Growth by [H-3]-Labeled Hypoxanthine Incorporation,” Experimen- tal Parasitology, Vol. 55, No. 1, 1983, pp. 138-146. http://dx.doi.org/10.1016/0014-4894(83)90007-3 [11] W. K. Milhous, N. F. Weatherly, J. H. Bowdre and R. E. Desjardins, “In Vitro Activities of and Mechanisms of Resistance to Antifol Antimalarial Drugs,” Antimicrobial Agents Chemotherapy, Vol. 27, No. 4, 1985, pp. 525-530. http://dx.doi.org/10.1128/AAC.27.4.525 [12] M. Ogura, G. A. Cordell, D. Kinghorn and N. R. Farns- worth, “Potential Anti-Cancer Agents 6. Constituents of Ailanthus-Excelsa (Simaroubaceae),” Lloydia, Vol. 40, 1977, pp. 579-584. [13] D. J. Morré, P. A. Grieco and D. M. Morré, “Mode of Action of the Anticancer Quassinoids—Inhibition of the Plasma Membrane NADH Oxidase,” Life Sciences, Vol. 63, 1998, pp. 595-604. http://dx.doi.org/10.1016/S0024-3205(98)00310-5 Copyright © 2013 SciRes. OJSTA
|