 International Journal of Clinical Medicine, 2013, 4, 459-471 http://dx.doi.org/10.4236/ijcm.2013.410082 Published Online October 2013 (http://www.scirp.org/journal/ijcm) Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis* Teya M. Tietje1,2, Douglas R. Allington1,2, Michael P. Rivey1,2 1Skaggs School of Pharmacy, University of Montana, Missoula, USA; 2Pharmacy Department, Community Medical Center, Mis- soula, USA. Email: teya.tietje@umontana.edu Received August 24th, 2013; revised September 23rd, 2013; accepted October 4th, 2013 Copyright © 2013 Teya M. Tietje et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Alemtuzumab is a humanized mononclonal antibody known to cause rapid depletion of B- and T-cell lymphocytes. Subsequent repletion of these lymphocytes leads to changes in adaptive immunity. Alemtuzumab is approved by the United States Food and Drug Administration (FDA) for the treatment of B-cell lymphocytic leukemia but has been in- vestigated off-label in recent years for treatment of autoimmune diseases, including multiple sclerosis (MS). In MS treatment, alemtuzumab is administered as pulsed therapy, given once daily initially for 5 consecutive days and then for 3 consecutive days at 12-month intervals. Alemtuzumab has recently been compared to interferon beta 1-a in one phase II and two phase III trials in patients with relapsing-remitting MS. Results from the studies show alemtuzumab com- pared to interferon beta 1-a is associated with a greater reduction in the risk of sustained accumulation of disability and is more effective in reducing disease relapse rates. The treatment of MS continues to be a healthcare challenge due to the modest clinical benefit and adverse effect profiles of available disease modifying treatment options. Available data suggest alemtuzumab may offer better efficacy outcomes compared to traditional disease modifying therapies in pa- tients with MS. However, the agent has not been compared to other new disease modifying medications that have been recently introduced. Keywords: Alemtuzumab; Multiple Sclerosis; Monoclonal Antibody; Lymphocyte Depletion 1. Introduction Multiple Sclerosis (MS) is a chronic, progressive in- flammatory disease affecting the central nervous system (CNS), resulting in demyelination and irreversible axonal damage of nerves in the CNS. Abnormal activation of the immune system against antigens results in disruption of the blood-brain barrier (BBB) allowing inflammatory mediators access to the CNS, leading to widespread in- flammation and subsequent neurodegeneration. A result of the destruction of myelin and central neurons is nerve conduction abnormalities. The clinical presentation of MS varies widely between patients as well as during the disease course in individual patients. Depending on the area of the brain and spinal cord damage, symptomatic disease presentation consists of a wide range of symptoms including weakness, fatigue, paresthesias, ataxia, speech dysfunction, and cognitive changes. Magnetic resonance imaging (MRI) evidence of CNS plaques and dissemination of brain lesions is an objective sign of possible MS [1,2]. The diagnosis of MS incorporates a combination of clinical symptomatology that cannot be attributed to another disease state or illness, as well as MRI evidence. However, a symptomatic dis- ease episode or a positive MRI scan is not independently diagnostic and must be evaluated with other evidence after consideration of differential diagnoses, thus making MS a diagnosis of exclusion. There are several subtypes of MS classified based on clinical disease course, with the preponderance (ap- proximately 85%) of patients experiencing a MS disease course characterized by symptomatic episodes lasting at least twenty-four hours followed by remission, or a symptom-free period, lasting at least thirty days. This clinical disease course is referred to as relapsing-remit- ting multiple sclerosis (RRMS). Another MS subtype is secondary progressive (SPMS) described as accrual of disability with or without relapses; many patients with RRMS eventually develop SPMS. Other disease subtypes *The authors report no conflicts of interest in this work. Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 460 are primary progressive (PPMS), described as a slowly progressive disease that begins at disease onset without defined relapses, and progressive relapsing (PRMS) de- scribed as a progressive disease starting at onset but with relapses [1,3]. An optimal pharmacotherapeutic treatment plan has not yet been determined for MS, and there is no cure. Current MS pharmacotherapy is targeted at disease modi- fication while incorporating acute and chronic treatment of symptoms. Disease modifying therapies (DMT) used in MS include interferon beta 1-a, immune modulators, immunosuppressants, and monoclonal antibodies [4]. Monoclonal antibodies used in the treatment of MS tar- get specific immune molecules on the surface of cells to interfere with the inflammatory pathophysiological dis- ease process. Epidemiology of MS An increase in prevalence of MS is seen above the 37th latitude in Northern or Southern Hemisphere countries including Australia, Europe, countries of the Mediterra- nean, New Zealand and the United States, with the high- est prevalence seen in Scotland at 145 - 193 cases per 100,000 people. Epidemiologic studies in the United States have not been extensive; however, a north-to- south gradient is seen with MS occurring in >50 indi- viduals and ≤30 individuals per 100,000 people living above and below the 37th parallel, respectively [5,6]. Mi- gration studies have shown that persons who migrate from an area of high to an area of low MS prevalence have a risk intermediate to their habitation origin, whereas persons who migrate from an area of low to high MS prevalence maintain the low risk of their origin [7-9]. Several potential environmental risk factors for MS including infection, physical environment, climate, diet, and stress have been assessed for causality. Viral infec- tions have long been an area of interest as precipitating factors of MS. Studies have evaluated an association with childhood viral infections including measles, mumps, rubella and varicella, but data have failed to demonstrate a link to MS development [10]. Epstein-Barr virus (EBV) has gained the most interest as a potential environmental factor for MS. An increased EBV antibody seropreva- lence has been found to occur in MS patients compared to controls [11,12]. While current evidence is not suffi- ciently robust to confirm an increased risk of developing MS in patients with prior EBV infection, research of the association remains plausible [11,13-15]. More recently Chlamydia pneumonia and viruses such as herpes sim- plex and retroviruseshave drawn attention as potential candidates for increasing the risk of developing MS but conclusive data are lacking [10,16-18]. Studies evaluating exposure to sunlight and related vi- tamin D concentrations have been mostly consistent in their findings and suggest a decreased risk of MS devel- opment in subjects with high levels of sun exposure be- tween the ages of 6 and 15 years [19]. It has been sug- gested that ultraviolet light may have immunosuppres- sive effects and increases production of vitamin D in the skin. Nevertheless, specific environmental risk factors involved in the etiology of MS remain difficult to isolate and quantify due to many other factors that contribute to the development of MS, as well as confounding factors expressed by genetically susceptible individuals [10]. Several genes are suspected to be associated with MS development including human leukocyte antigen (HLA) class I and II alleles, T-cell receptor alpha, and cytotoxic T-lymphocyte antigen 4 (CTLA4). Results of studies have shown an increased risk for first, second, and third degree relatives of MS patients when compared to the 0.1% general population risk [20]. The greatest risk is observed in an offspring of both parents with MS, with 30.5% of individuals developing the disease, compared to 2.49% in offspring of a single parent with MS [21,22]. Additionally, the disease concordance rate is near 30% in monozygotic twins compared to 4% in dizygotic twins. It is interesting that the increased risk of developing MS in the offspring of two MS positive parents is comparable to that observed in monozygotic twins, independent of pa- rental MS status [23,24]. 2. Pathophysiology of MS Our knowledge regarding the cellular responses and im- munobiology associated with MS has benefitted substan- tially by tissue sample analysis obtained from MS lesions [25] and from over 70 years of animal research, primarily using experimental autoimmune encephalomyelitis (EAE) models [26]. Histologic examination of MS plaques has documented CD4+ T-lymphocytes in perivascular spaces and meninges and CD 8+ T-lymphocytes in the main body of the MS lesion [27]. The specific type and num- ber of cells differs between patients and within a given patient as the MS lesion progresses from an acute to chronic stage [28,29]. B-lymphocytes may act as antigen presenting cells (APC) in the CNS, similar to their known role in the peripheral immune system [30]. Oligo- clonal bands of immunoglobulin (IgG) found in the cere- bral spinal fluid (CSF) of MS patients and directly from MS plaques indicate B-lymphocytes have an important role in the escalated inflammatory response. The demyelinating plaques of MS are dependent upon the recruitment and entry of inflammatory cells into the CNS. Once activated, CD4+ and CD8+ T-lymphocytes and B-lymphocytes from the periphery cross the BBB. Cellular migration from the periphery to the CNS is as- sisted by integrin receptors on lymphocyte membranes Copyright © 2013 SciRes. IJCM 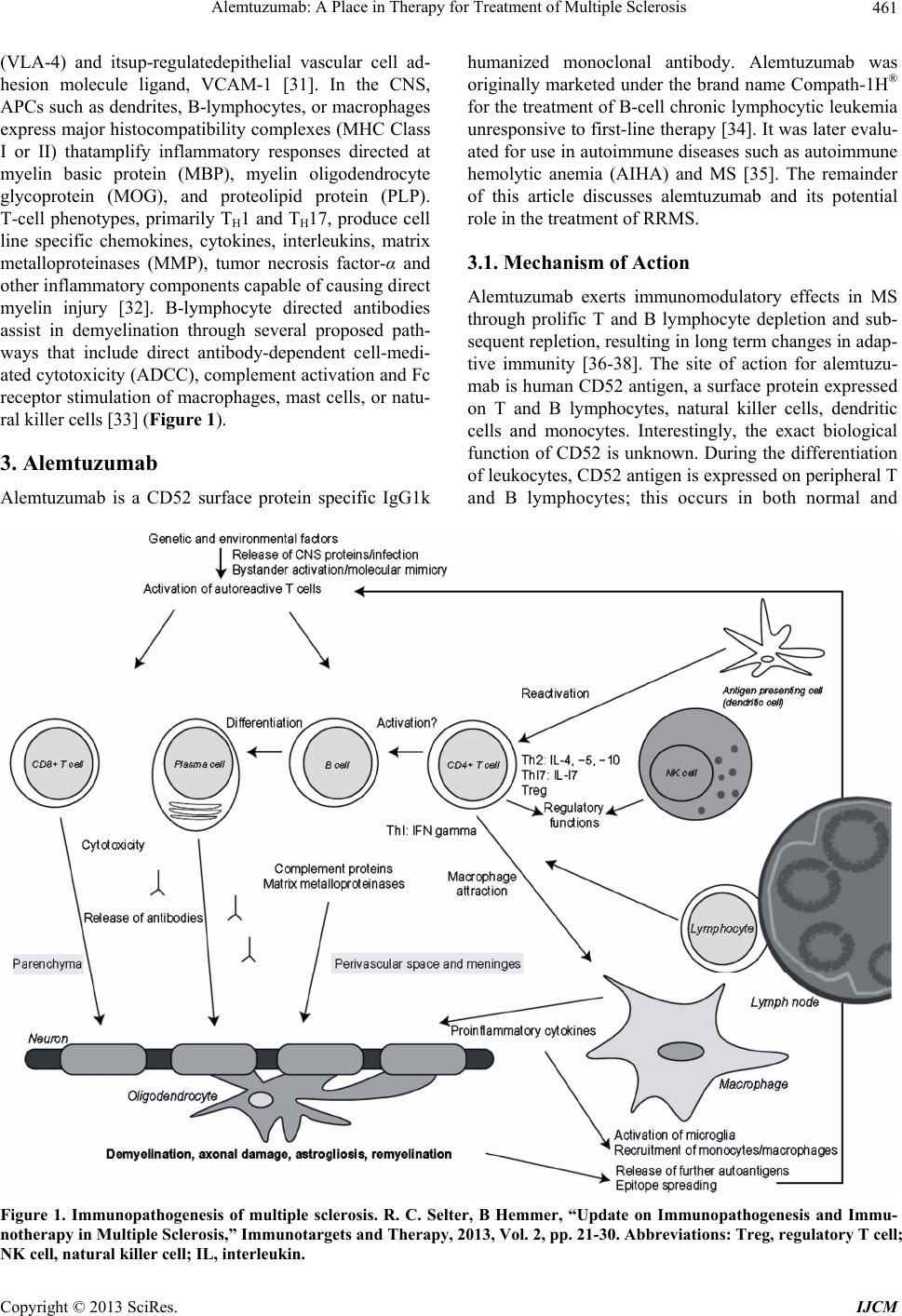 Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis Copyright © 2013 SciRes. IJCM 461 (VLA-4) and itsup-regulatedepithelial vascular cell ad- hesion molecule ligand, VCAM-1 [31]. In the CNS, APCs such as dendrites, B-lymphocytes, or macrophages express major histocompatibility complexes (MHC Class I or II) thatamplify inflammatory responses directed at myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP). T-cell phenotypes, primarily TH1 and TH17, produce cell line specific chemokines, cytokines, interleukins, matrix metalloproteinases (MMP), tumor necrosis factor-α and other inflammatory components capable of causing direct myelin injury [32]. B-lymphocyte directed antibodies assist in demyelination through several proposed path- ways that include direct antibody-dependent cell-medi- ated cytotoxicity (ADCC), complement activation and Fc receptor stimulation of macrophages, mast cells, or natu- ral killer cells [33] (Figure 1). 3. Alemtuzumab Alemtuzumab is a CD52 surface protein specific IgG1k humanized monoclonal antibody. Alemtuzumab was originally marketed under the brand name Compath-1H® for the treatment of B-cell chronic lymphocytic leukemia unresponsive to first-line therapy [34]. It was later evalu- ated for use in autoimmune diseases such as autoimmune hemolytic anemia (AIHA) and MS [35]. The remainder of this article discusses alemtuzumab and its potential role in the treatment of RRMS. 3.1. Mechanism of Action Alemtuzumab exerts immunomodulatory effects in MS through prolific T and B lymphocyte depletion and sub- sequent repletion, resulting in long term changes in adap- tive immunity [36-38]. The site of action for alemtuzu- mab is human CD52 antigen, a surface protein expressed on T and B lymphocytes, natural killer cells, dendritic cells and monocytes. Interestingly, the exact biological function of CD52 is unknown. During the differentiation of leukocytes, CD52 antigen is expressed on peripheral T and B lymphocytes; this occurs in both normal and Figure 1. Immunopathogenesis of multiple sclerosis. R. C. Selter, B Hemmer, “Update on Immunopathogenesis and Immu- notherapy in Multiple Sclerosis,” Immunotargets and Therapy, 2013, Vol. 2, pp. 21-30. Abbreviations: Treg, regulatory T cell; NK cell, natural killer cell; IL, interleukin.  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 462 malignant cells, thereby explaining the development of alemtuzumab as a chemotherapeutic agent [39,40]. Alem- tuzumab adherence to the CD52 protein induces cell lysis via complement deposition and formation of the mem- brane attack complex [40]. Lymphocyte lysis and deple- tion is proposed to promote removal of malignant lym- phocytes and eliminate the involvement of normal lym- phocytes in the inflammatory process. As noted, several subtypes of T lymphocytes have been implicated in the release of inflammatory mediators in MS [41]. Specifically, subtype TH1 lymphocytes re- lease proinflammatory cytokines associated with MS relapses, thereby providing a pharmacotherapeutic target for DMT. Moreover, there is a limited amount of CD52 antigen expressed on the surface of CD34+ hematopoietic cells, which are parent stem cells to CD52+ lymphocytes [40,42]. Decreased CD52 expression on the parent stem cells allows for eradication of mature lymphocytes after alemtuzumab administration without severe depletion of bone marrow cells, allowing for subsequent reconstitu- tion of lymphocytes [40]. 3.2. Pharmacokinetics and Pharmacodynamics The pharmacokinetics of alemtuzumab are intertwined with the pharmacodynamics since pharmacokinetic pa- rameters change as lymphocytes that express the CD52 antigen target of the drug are depleted [43]. Basically, the clearance of the drug is dependent on CD52 availability. This relationship contributes to large intersubject vari- ability in alemtuzumab pharmacokinetics that is common for monoclonal antibody agents that target antigens [44]. Alemtuzumab pharmacokinetics have been character- ized in B-cell chronic lymphocytic leukemia and post stem-cell transplant patients receiving the drug for cancer chemotherapy, usually in combination with other anti- neoplastic drugs [43-45]. No similar data are available in the MS population and caution should be used when ex- trapolating existing information. On the other hand, alemtuzumab in some chemotherapy studies was given by intravenous infusion in doses and regimens similar to those utilized in the MS studies. Limited pharmacoki- netic information is available from late 1990s studies in rheumatoid arthritis patients, although doses used in those studies were typically much larger than MS doses [46]. It is believed that alemtuzumab serum concentrations of 1 - 10 mcg/ml are associated with cellular processes that mediate lymphocyte lysis. Results of studies in bone marrow transplant patients have shown that 10 gm doses given over 5 or 10 consecutive days resulted in mean peak alemtuzumab concentrations of 2.5 mcg/ml and 6.1 mcg/ml, respectively; the drug could still be detected 11 - 23 days after the last dose [45]. Mould et al. [44] deter- mined that the best pharmacokinetic model to describe alemtuzumab was a two-compartment model with zero order input and non-linear Michaelis-Menten elimination strongly influenced by the covariate of the white blood cell count [44]. While the half-life of a drug varies with concentration and time in nonlinear kinetics (i.e. elimination slows with subsequent doses), the elimination of alemtuzumab after repeated doses is clearly prolonged. A half-life range of 5 - 9 days was determined in rheumatoid arthritis patients receiving 100, 250, or 400 mg doses divided over 5 - 10 days [46]. A more prolonged half-life range of 15 - 21 days was measured after alemtuzumab 10 mg given for 5 or 10 days to patients in preparation for stem-cell trans- plant [45]. Large interpatient variability has also been seen for the volume of distribution (approximately 0.2 L/kg) of alemtuzumab in B-cell chronic leukemia pa- tients; this is likely attributable to patient status but also is caused by changes in CD52 antigen availability [44]. Since the mechanism of clearance of alemtuzumab from the body is not completely understood, dosage adjust- ments based on gender, age, or hepatic or renal function are not available [43]. Furthermore, the clearance of alemtuzumab has been correlated with clinical outcome by Hale et al. [47] who found that chronic lymphocytic leukemia patients who responded to treatment also had slower clearance of the drug [47]. The pharmacodynamics of alemtuzumab are primarily centered on lymphocytes that express the CD52 antigen. Interindividual variability of pharmacodynamic proper- ties of alemtuzumab is also great since baseline white blood cell counts vary between patients and a wide range of plasma concentrations of CD52 have been determined in patients [43,44]. Lymphocyte depletion & reconstitu- tion have been investigated in the MS studies. Consistent with cancer studies, lymphocytes were rapidly depleted days after and slowly reconstituted only months after alemtuzumab treatment in the MS studies [45]. Hill-Cawthorne et al. [48] recently described lympho- cyte reconstitution in 36 SPMS or PPMS patients treated with alemtuzumab between1991-1997 and followed for 384 total person-years. The mean recovery time to the lower end of the normal range was 7.1 months for B-cell and 12.7 months for total lymphocyte counts. T-cell sub- sets CD4+ and CD8+ counts had median recovery times of 20 and 35 months, respectively, but did not return to baseline levels in approximately 70% of patients. In the Phase II and III trials, B-cell lymphocytes reconstituted within 6 months, whereas T-cell lymphocyte recovery was slower and only approached the lower limit of nor- mal after one year [49-51]. Finally, alemtuzumab-binding antibodies can be de- tected in about 30% of treated MS patients before the second course of therapy and increases to over 80% one Copyright © 2013 SciRes. IJCM 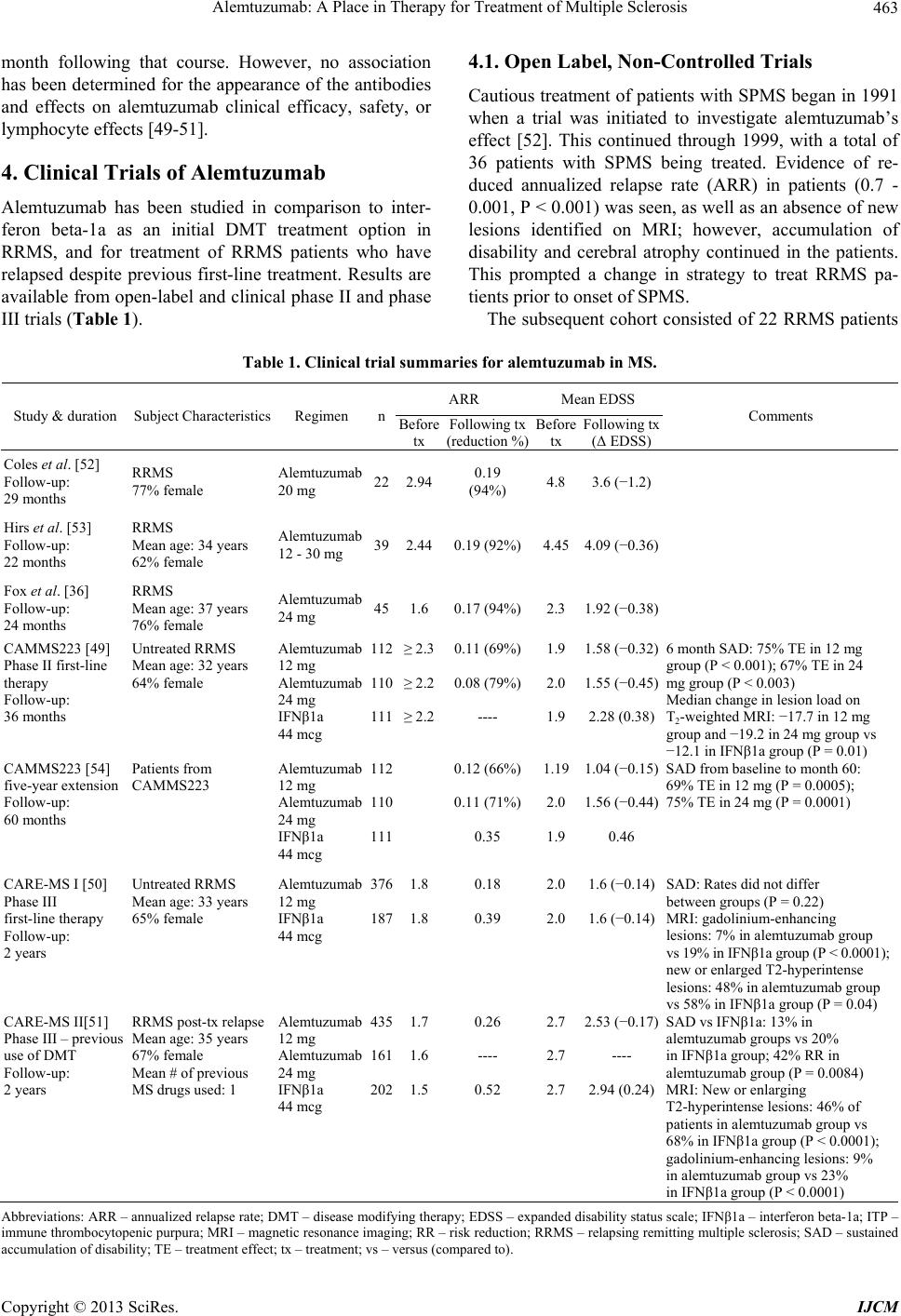 Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 463 month following that course. However, no association has been determined for the appearance of the antibodies and effects on alemtuzumab clinical efficacy, safety, or lymphocyte effects [49-51]. 4. Clinical Trials of Alemtuzumab Alemtuzumab has been studied in comparison to inter- feron beta-1a as an initial DMT treatment option in RRMS, and for treatment of RRMS patients who have relapsed despite previous first-line treatment. Results are available from open-label and clinical phase II and phase III trials (Table 1). 4.1. Open Label, Non-Controlled Trials Cautious treatment of patients with SPMS began in 1991 when a trial was initiated to investigate alemtuzumab’s effect [52]. This continued through 1999, with a total of 36 patients with SPMS being treated. Evidence of re- duced annualized relapse rate (ARR) in patients (0.7 - 0.001, P < 0.001) was seen, as well as an absence of new lesions identified on MRI; however, accumulation of disability and cerebral atrophy continued in the patients. This prompted a change in strategy to treat RRMS pa- tients prior to onset of SPMS. The subsequent cohort consisted of 22 RRMS patients Table 1. Clinical trial summaries for alemtuzumab in MS. ARR Mean EDSS Study & duration Subject Characteristics Regimen nBefore tx Following tx (reduction %) Before tx Following tx (Δ EDSS) Comments Coles et al. [52] Follow-up: 29 months RRMS 77% female Alemtuzumab 20 mg 22 2.940.19 (94%) 4.83.6 (−1.2) Hirs et al. [53] Follow-up: 22 months RRMS Mean age: 34 years 62% female Alemtuzumab 12 - 30 mg 392.440.19 (92%)4.454.09 (−0.36) Fox et al. [36] Follow-up: 24 months RRMS Mean age: 37 years 76% female Alemtuzumab 24 mg 451.60.17 (94%)2.31.92 (−0.38) CAMMS223 [49] Phase II first-line therapy Follow-up: 36 months Untreated RRMS Mean age: 32 years 64% female Alemtuzumab 12 mg Alemtuzumab 24 mg IFNβ1a 44 mcg 112 110 111 ≥ 2.3 ≥ 2.2 ≥ 2.2 0.11 (69%) 0.08 (79%) ---- 1.9 2.0 1.9 1.58 (−0.32) 1.55 (−0.45) 2.28 (0.38) 6 month SAD: 75% TE in 12 mg group (P < 0.001); 67% TE in 24 mg group (P < 0.003) Median change in lesion load on T2-weighted MRI: −17.7 in 12 mg group and −19.2 in 24 mg group vs −12.1 in IFNβ1a group (P = 0.01) CAMMS223 [54] five-year extension Follow-up: 60 months Patients from CAMMS223 Alemtuzumab 12 mg Alemtuzumab 24 mg IFNβ1a 44 mcg 112 110 111 0.12 (66%) 0.11 (71%) 0.35 1.19 2.0 1.9 1.04 (−0.15) 1.56 (−0.44) 0.46 SAD from baseline to month 60: 69% TE in 12 mg (P = 0.0005); 75% TE in 24 mg (P = 0.0001) CARE-MS I [50] Phase III first-line therapy Follow-up: 2 years Untreated RRMS Mean age: 33 years 65% female Alemtuzumab 12 mg IFNβ1a 44 mcg 376 187 1.8 1.8 0.18 0.39 2.0 2.0 1.6 (−0.14) 1.6 (−0.14) SAD: Rates did not differ between groups (P = 0.22) MRI: gadolinium-enhancing lesions: 7% in alemtuzumab group vs 19% in IFNβ1a group (P < 0.0001); new or enlarged T2-hyperintense lesions: 48% in alemtuzumab group vs 58% in IFNβ1a group (P = 0.04) CARE-MS II[51] Phase III – previous use of DMT Follow-up: 2 years RRMS post-tx relapse Mean age: 35 years 67% female Mean # of previous MS drugs used: 1 Alemtuzumab 12 mg Alemtuzumab 24 mg IFNβ1a 44 mcg 435 161 202 1.7 1.6 1.5 0.26 ---- 0.52 2.7 2.7 2.7 2.53 (−0.17) ---- 2.94 (0.24) SAD vs IFNβ1a: 13% in alemtuzumab groups vs 20% in IFNβ1a group; 42% RR in alemtuzumab group (P = 0.0084) MRI: New or enlarging T2-hyperintense lesions: 46% of patients in alemtuzumab group vs 68% in IFNβ1a group (P < 0.0001); gadolinium-enhancing lesions: 9% in alemtuzumab group vs 23% in IFNβ1a group (P < 0.0001) Abbreviations: ARR – annualized relapse rate; DMT – disease modifying therapy; EDSS – expanded disability status scale; IFNβ1a – interferon beta-1a; ITP – immune thrombocytopenic purpura; MRI – magnetic resonance imaging; RR – risk reduction; RRMS – relapsing remitting multiple sclerosis; SAD – sustained ccumulation of disability; TE – treatment effect; tx – treatment; vs – versus (compared to). a Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 464 who had previously failed treatment or had high relapse rates, indicating rapidly progressing disease and poor prognosis. Patients received 20 mg of intravenous alem- tuzumab daily for five consecutive days, with the option of a three-day re-treatment after 12 - 18 months and again after 12 - 30 months. Nineteen (86%) patients re- ceived a second course, and three (14%) patients re- ceived a third course of alemtuzumab following disease relapses. Relapse rates were compared using ARR, and changes in expanded disability status scale (EDSS) scores were used to assess accumulation of disability. After a mean 29-month follow-up, a 94% improvement in ARR (P < 0.001) was seen and EDSS scores had im- proved by an average of 1.2 points. The difference in disability outcomes between the SPMS and RRMS pa- tients suggested the optimal time for alemtuzumab treat- ment was earlier in the disease process, prior to progres- sion to SPMS [52]. The promising results in the Coles et al. [52] RRMS trial prompted another trial of alemtuzumab in 39 pa- tients with aggressive RRMS and a poor prognosis [53]. Thirty-two (82%) patients were treatment-naïve and seven (18%) had previously failed DMT. Alemtuzumab doses were not consistent for all trial subjects due to changesas new information became available. However, all subjects received five consecutive days of 12 - 30 mg alemtuzumab and three consecutive days of 1 g methyl- prednisolone at the beginning of treatment. Retreatment occurred in 13 (33%) patients an average of 17 months after the first treatment, and three (8%) patients received a third treatment an average of 19 months later. Com- parison of pre- and post-treatment ARRs demonstrated a mean difference of 2.27 (P < 0.0001), representing a 92% overall reduction in ARR. The mean change in EDSS after 23 months of follow-up was 0.2 for patients with stable EDSS scores prior to treatment and 0.6 in patients with unstable baseline EDSS scores [53]. In 2012, Fox et al. [36] published results of a study conducted in 45 RRMS patients who previously received interferon beta (IFNβ) for at least six months within two years prior to the study and had two confirmed relapses during IFNβ treatment. All patients received an initial five consecutive days of alemtuzumab therapy and a three-day retreatment 12 months later. At the 24-month follow-up, a 94% reduction in ARR (P < 0.0001) and a mean improvement in EDSS of 0.38 (P = 0.0542) were determined for the alemtuzumab compared to prior treat- ment period [36]. 4.2. Active Comparator Trials 4.2.1. CAM MS223 The CAMMS223 study was a randomized, blinded, phase II trial to compare the effects alemtuzumab and interferon beta 1-a on ARR and sustained accumulation of disability (SAD) in previously untreated RRMS pa- tients. This study enrolled patients from December 2002 to July 2004. Patients assigned to receive alemtuzumab were administered either 12 mg (n = 113) or 24 mg (n = 110) intravenously per day for five consecutive days during the first month of therapy, then subsequently for three consecutive days at 12 and 24 months. The inter- feron beta 1-a patients (n = 111) were titrated up to a dose of 44 mcg subcutaneously three times a week dur- ing the study. A disease relapse was defined as the ap- pearance of new or worsening MS symptoms accompa- nied by a change in the neurologic examination lasting at least 48 hours and having been preceded by at least 30 days of clinical stability. The EDSS was used to assess disability, with SAD being defined as an EDSS score increase of ≥1.5 points for patients with a baseline score of 0 and ≥1 for patients with a baseline score of 1 or above. SAD was recorded from the date of qualifying increase in EDSS, and confirmed by EDSS scores twice during a six-month period. Patients had a mean follow-up of 36 months [49]. Alemtuzumab ARRs compared to the interferon beta 1-a group were significantly reduced by 69% and 79% (P < 0.001 with a NNT of 3.1 and 3.9) in the 12 mg and 24 mg groups, respectively. At 36 months, the ARR in the pooled alemtuzumab groups was 0.10 compared to 0.36 for the interferon beta 1-a group. Compared to interferon beta 1-a, alemtuzumab therapy was associated with re- ductions in risk of SAD after six months of 75% (P < 0.001) and 67% (P < 0.001) in the 12 mg and 24 mg groups, respectively. The number needed to treat (NNT) with alemtuzumab compared to interferon beta 1-a to avoid one patient progression to SAD in a 36-month study period was 5.6 in the 12 mg and 6.0 in the 24 mg alemtuzumab groups. The mean EDSS scores signifi- cantly improved by 0.32 and 0.45 in the alemtuzumab groups, compared to a worsening of 0.38 in the interferon beta 1-a group. Finally a reduction in the volume of le- sions on T2-weighted MRI was seen in all study groups, but was significantly more marked after alemtuzumab compared to interferon beta 1-a treatment (P = 0.005). Two notable aspects regarding alemtuzumab dosage emerged in the CAMMS223. First, outcomes for the two alemtuzumab dosage groups were not significantly dif- ferent. Second, in September 2005 during the study, alemtuzumab dosing was temporarily suspended after three subjects were diagnosed with immune thrombocy- topenia (ITP), an antibody and cell mediated suppression and destruction of platelets. The suspension caused two (1%) patients in the trial to not receive alemtuzumab therapy at month 12 and 155 (75%) patients to not re- ceive alemtuzumab at the 24-month dosing time. The study protocol was amended to include formal monitor- Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 465 ing for ITP, and alemtuzumab was resumed. But notably, no significant differences in treatment effect on disability or safety were seen between patient subgroups who re- ceived two cycles and those who received three cycles of alemtuzumab. Adverse events occurred in >99% of all study partici- pants with the most common being infusion-related reac- tions in all groups. Serious infusion reactions occurred in three alemtuzumab patients with one patient discontinu- ing the drug as a result, whereas two interferon beta 1-a patients discontinued treatment due to infusion reactions. Mild to moderate infections and thyroid dysfunction were more frequent in patients receiving alemtuzumab than those receiving interferon beta 1-a. Infection rates were highest during the month following an infusion, but no infections were life-threatening or fatal. Thyroid dys- function occurred up to 30 months after the last dose of study medication, with serious thyroid events occurring in three alemtuzumab-treated patients. Six alemtuzumab and 1 interferon beta 1-a patients developed ITP over a mean follow-up period of 4.5 years; the difference was not statistically significant. The overall incidence rate of ITP for all patients treated with alemtuzumab was 6.2 per 1000 person-years, with 4.2 per 1000 person-years in the 12 mg group and 8.0 per 1000 person-years in the 24 mg group, respectively. In comparison, the rate of ITP in the interferon beta 1-a treated patients was 2.7 per 1000 per- son-years. Study dropouts occurred not only due to ad- verse events but also due to lack of DMT efficacy; 83% of all alemtuzumab and 59% of interferon beta 1-a pa- tients completed the 36-month study.[49] An extension to the CAMMS223 study was initiated in August 2006, four years after initial enrollment into the first study. [54] The extension study was composed of 198 of the original 334 participants and included patients from each of the three treatment (alemtuzumab 12 mg and 24 mg and interferon beta 1-a 44 mcg) groups. Of the original study groups, 47 patients (42%) who contin- ued interferon beta 1-a and 151 patients (68%) who re- ceived alemtuzumab 36 to 48 months earlier were as- sessed in the extension study. Outcomes were assessed from baseline of the original trial period to 60 months. During the extension period, all patients were permitted to use other DMTs for MS including interferon beta 1-a. In 2008, patients in the alemtuzumab groups were given the option to receive additional alemtuzumab 12 mg per day for 3 consecutive days, but the majority of patients did not receive additional alemtuzumab. Results of the extension study showed that, compared with interferon beta 1-a, alemtuzumab decreased the risk of SAD by 69% (p < 0.0001) in the 12 mg group and 75% in the 24 mg group (p < 0.0001) and decreased the ARR by 66% (p < 0.0001) in the 12 mg group and 71% (p < 0.0001) in the 24 mg group [54]. The ARR in the pooled alemtuzumab groups compared to the interferon beta 1-a group was significantly (P < 0.0001) lower for the baseline to the 5-year assessment (0.11 versus 0.35) and was lower, although not statistically significant for the 3-year to 5-year time period (0.14 versus 0.28, P = 0.072). The mean EDSS score decreased in both alemtu- zumab groups while an increased mean EDSS score was experienced in the interferon beta 1-a group [54]. Adverse effects of alemtuzumab were similar to those seen in the original study. Infections due to alemtuzumab decreased in frequency as the extension study progressed and no life-threatening or fatal infections occurred; only herpes zoster infections occurred more commonly in the alemtuzumab compared to interferon beta 1-a patients at the end of the extension study. Similarly, autoimmune adverse effects decreased during the extension study, with thyroid autoimmunity being the most common event [54]. 4.2.2. C AR E-MS I Results of the CARE-MS I study, a phase III trial com- paring alemtuzumab to interferon beta 1-a as first line therapy, were released in 2012 [50]. The study enrolled 563 patients with confirmed, active, and untreated RRMS who had the disease less than five years and had suffered at least two relapses in the previous two years, with the most recent relapse being within the previous year. Pa- tients also had an EDSS score of no greater than 3. Pa- tients were randomized in a 2:1 ratio to receive a 12 mg of alemtuzumab intravenous infusion for five days at study initiation and three days at 12 months, or were ti- trated to interferon beta 1-a 44 mcg administered subcu- taneously three times a week. All patients also received methylprednisolone 1 g daily by the intravenous route for 3 consecutive days at study enrollment and at 12 months. Patients were excluded if they previously received MS DMT, including immunosuppressive, investigational, or monoclonal antibody therapy, or if they had a progres- sive MS disease course or other clinically significant autoimmune disorder. Because of concern for an in- creased herpes virus infection rate, the study protocol was amended and alemtuzumab patients received oral acyclovir 200 mg twice daily as prophylaxis. Acyclovir was given on alemtuzumab infusion days and for the next 28 days [50]. Primary efficacy outcomes assessed were MS RR and time to SAD confirmed over a 6-month period. MS re- lapse and SAD were defined the same as in CAMM223. Patients who did not experience a MS relapse or suffer SAD were defined as free of clinical disease activity. Secondary study outcomes included aspects of changes in the primary efficacy outcomes, brain MRI changes including the presence of both gadolinium-enhancing lesions and new or enlarging T2-hyperintense lesions, Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 466 and safety reflected by adverse events [50]. Alemtuzumab reduced the MS RR at 2 years when the study concluded, with 77.6% of patients who received alemtuzumab relapse-free, compared to 58.7% of inter- feron beta 1-a patients (P < 0.0001). However, there was no difference between treatment groups for SAD rates, observed in 8% of alemtuzumab compared to 11% of interferon beta 1-a patients (P = 0.22). Interestingly, the SAD rate in the interferon beta 1-a group was less than half the rate that had been determined in the CAMMS223 study. A significant difference was seen for the secon- dary outcome of 2-year clinically disease-free prevalence, occurring in 279 (74%) alemtuzumab compared to 104 (56%) interferon beta 1-a patients (P < 0.0001). Mixed results were observed for the MRI radiological outcomes. There was no difference (P = 0.31) in the me- dian change in volume of T2-hyperintense lesions be- tween groups, although alemtuzumab compared to inter- feron beta 1-a was associated with a decreased propor- tion of patients with new or enlarging T2-hyperintense lesions(48% versus 58%, p = 0.04). Moreover, there were significantly fewer patients in the alemtuzumab group with gadolinium-enhancing lesions at 24 months (7% versus 19%, P < 0.0001), as well as a significantly smaller median change in brain parenchymal fractions (−0.867% alemtuzumab and −1.488% interferon beta 1-a, P < 0.0001). When the clinical and MRI disease-free results were combined, the joint outcome was found in 139/360 (39%) alemtuzumab and 46/172 (27%) inter- feron beta 1-a patients, respectively (P = 0.006) [50]. At least one adverse event occurred in 96% of patients receiving alemtuzumab and 92% of patients receiving interferon beta 1-a. As seen in the CAMMS223 study, the most frequently observed adverse events in alemtu- zumab patients were infusion-related reactions (90%) and of these, 3% were serious reactions. Infections (67% versus 45% of patients) and thyroid dysfunction (18% versus 6% of patients) occurred more frequently in alemtuzumab compared to interferon beta 1-a treated patients, but the occurrences were predominantly mild to moderate in severity. Acyclovir prophylaxis was not found to lessen the risk of herpetic infection in alemtu- zumab patients. Three alemtuzumab patients developed ITP, of which two were successfully treated and one re- solved spontaneously [50]. 4.2.3. C ARE-MS II Nearly concurrent with CARE-MS I, the phase III CARE-MS II study was conducted in RRMS patients who had MS relapse despite treatment with first line DMT [51]. Trial design mirrored that of the CARE-MS I study, but patients admitted to the CARE-MS II study had MS no longer than 10 years, an EDSS score less than 5, and must have suffered a MS relapse after at least 6 months of treatment with interferon beta 1-a or glati- ramer. Approximately 70% of patients had previously used a single MS medication, 23% had used two MS medications, and less than 7% had used more than two MS medications [51]. Patients were randomized in a 2:2:1 ratio to receive alemtuzumab 12 mg or 24 mg or interferon beta 1-a 44 mcg given in the same regimen used for other studies. In December of 2008 after 14 months of subject recruitment, randomization to the alemtuzumab 24 mg arm was ter- minated to facilitate more rapid recruitment into the other two groups. As such, final subject groups were composed of 436 patients in the alemtuzumab 12 mg, 173 patients in the alemtuzumab 24 mg, and 231 patients in the inter- feron beta 1-a groups. Follow-up for the study was 24 months, and 755 (90%) of participants completed the study. Primary and secondary outcomes assessed were the same as those in the MS-CARE I study, using the RR and time to 6-month SAD as primary endpoints [51]. Results of the CARE-MS II demonstrated superiority of alemtuzumab compared to interferon beta 1-a for pri- mary study outcomes [see Table 1]. Due to the ran- domization change, only the alemtuzumab 12 mg group was included for the primary endpoint comparisons with interferon beta 1-a. Alemtuzumab reduced the ARR 49.4% (P < 0.0001) over that observed in the interferon beta 1-a group, with relapses occurring in 35% of alem- tuzumab and 53% of interferon beta 1-a patients, respec- tively. A 42% (P = 0.0084) reduced risk of SAD was observed with alemtuzumab, affecting 13% alemtuzumab versus 20% interferon beta 1-a patients. Significant benefits associated with alemtuzumab were also found for the mean change in EDSS score during the study and the percentages of patients who were relapse-free, had sustained reduction in disability for 6 months, had new or enlarging lesions on MRI, and were clinically plus MRI disease-free. When the alemtuzumab 12 mg and 24 mg groups were compared for clinical outcomes, only new MRI lesion formation was improved with the larger dos- age [51]. A subgroup analysis was conducted in patients with highly active RRMS (≥2 relapses in the year prior to randomization and ≥1 gadolinium enhancing lesion at baseline) who had relapsed while receiving DMT. The subgroup consisted of 101 patients (23.7%) in the alem- tuzumab 12 mg group and 42 patients (20.8%) in the interferon beta 1-a group. After two years, 24.2% of 101 patients treated with alemtuzumab were disease-activity free compared to 0% of 42 patients treated with inter- feron beta 1-a (P = 0.0002); 35.8% of patients in the alemtuzumab subgroup had relapses compared to 60% in the interferon beta 1-a subgroup, 7.4% had SAD com- pared to 17.5% in the alemtuzumab and interferon beta 1-a subgroups, respectively [55]. Copyright © 2013 SciRes. IJCM 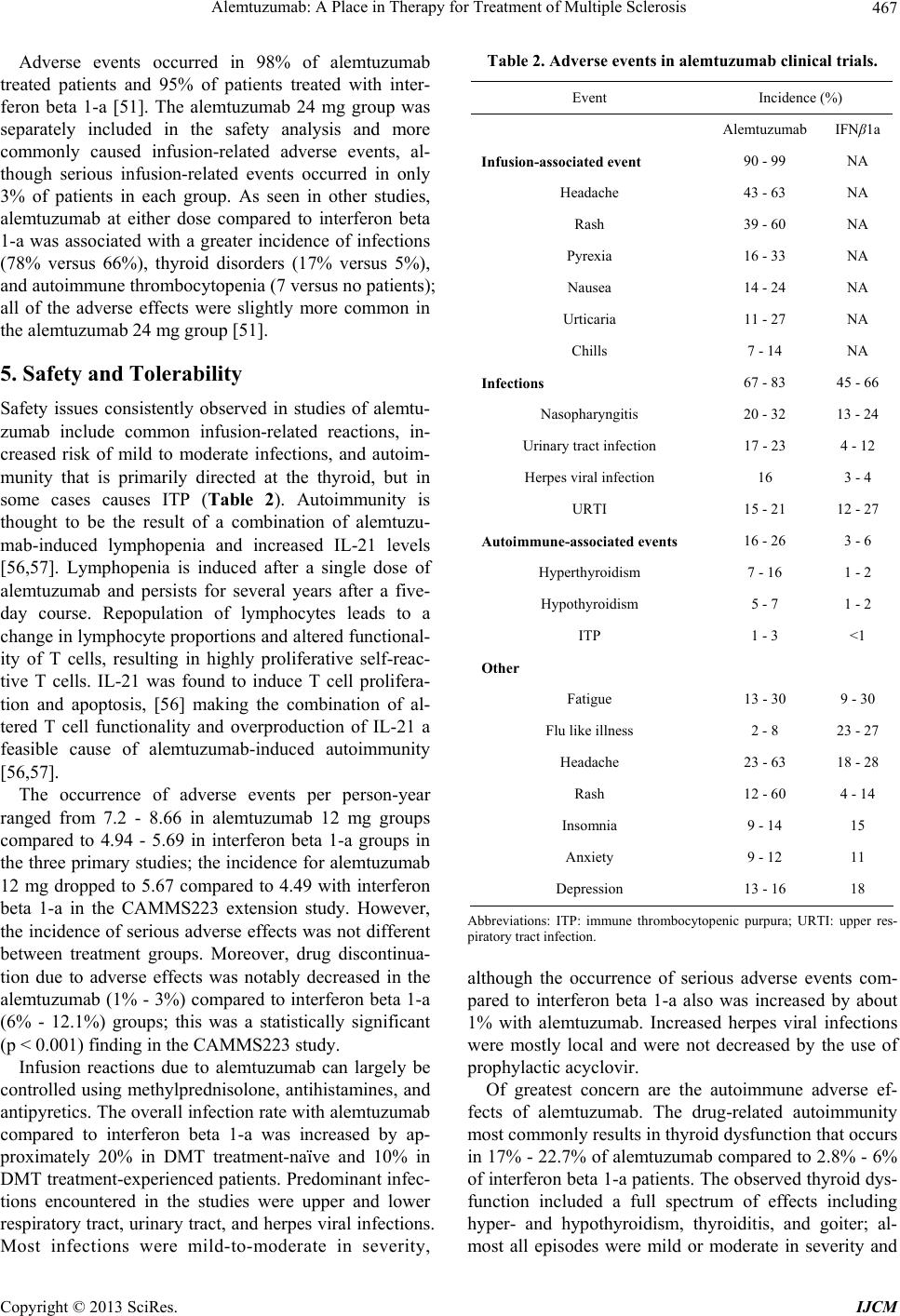 Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 467 Adverse events occurred in 98% of alemtuzumab treated patients and 95% of patients treated with inter- feron beta 1-a [51]. The alemtuzumab 24 mg group was separately included in the safety analysis and more commonly caused infusion-related adverse events, al- though serious infusion-related events occurred in only 3% of patients in each group. As seen in other studies, alemtuzumab at either dose compared to interferon beta 1-a was associated with a greater incidence of infections (78% versus 66%), thyroid disorders (17% versus 5%), and autoimmune thrombocytopenia (7 versus no patients); all of the adverse effects were slightly more common in the alemtuzumab 24 mg group [51]. 5. Safety and Tolerability Safety issues consistently observed in studies of alemtu- zumab include common infusion-related reactions, in- creased risk of mild to moderate infections, and autoim- munity that is primarily directed at the thyroid, but in some cases causes ITP (Table 2). Autoimmunity is thought to be the result of a combination of alemtuzu- mab-induced lymphopenia and increased IL-21 levels [56,57]. Lymphopenia is induced after a single dose of alemtuzumab and persists for several years after a five- day course. Repopulation of lymphocytes leads to a change in lymphocyte proportions and altered functional- ity of T cells, resulting in highly proliferative self-reac- tive T cells. IL-21 was found to induce T cell prolifera- tion and apoptosis, [56] making the combination of al- tered T cell functionality and overproduction of IL-21 a feasible cause of alemtuzumab-induced autoimmunity [56,57]. The occurrence of adverse events per person-year ranged from 7.2 - 8.66 in alemtuzumab 12 mg groups compared to 4.94 - 5.69 in interferon beta 1-a groups in the three primary studies; the incidence for alemtuzumab 12 mg dropped to 5.67 compared to 4.49 with interferon beta 1-a in the CAMMS223 extension study. However, the incidence of serious adverse effects was not different between treatment groups. Moreover, drug discontinua- tion due to adverse effects was notably decreased in the alemtuzumab (1% - 3%) compared to interferon beta 1-a (6% - 12.1%) groups; this was a statistically significant (p < 0.001) finding in the CAMMS223 study. Infusion reactions due to alemtuzumab can largely be controlled using methylprednisolone, antihistamines, and antipyretics. The overall infection rate with alemtuzumab compared to interferon beta 1-a was increased by ap- proximately 20% in DMT treatment-naïve and 10% in DMT treatment-experienced patients. Predominant infec- tions encountered in the studies were upper and lower respiratory tract, urinary tract, and herpes viral infections. Most infections were mild-to-moderate in severity, Table 2. Adverse events in alemtuzumab clinical trials. Event Incidence (%) Alemtuzumab IFNβ1a Infusion-associated event 90 - 99 NA Headache 43 - 63 NA Rash 39 - 60 NA Pyrexia 16 - 33 NA Nausea 14 - 24 NA Urticaria 11 - 27 NA Chills 7 - 14 NA Infections 67 - 83 45 - 66 Nasopharyngitis 20 - 32 13 - 24 Urinary tract infection 17 - 23 4 - 12 Herpes viral infection 16 3 - 4 URTI 15 - 21 12 - 27 Autoimmune-associated events 16 - 26 3 - 6 Hyperthyroidism 7 - 16 1 - 2 Hypothyroidism 5 - 7 1 - 2 ITP 1 - 3 <1 Other Fatigue 13 - 30 9 - 30 Flu like illness 2 - 8 23 - 27 Headache 23 - 63 18 - 28 Rash 12 - 60 4 - 14 Insomnia 9 - 14 15 Anxiety 9 - 12 11 Depression 13 - 16 18 Abbreviations: ITP: immune thrombocytopenic purpura; URTI: upper res- piratory tract infection. although the occurrence of serious adverse events com- pared to interferon beta 1-a also was increased by about 1% with alemtuzumab. Increased herpes viral infections were mostly local and were not decreased by the use of prophylactic acyclovir. Of greatest concern are the autoimmune adverse ef- fects of alemtuzumab. The drug-related autoimmunity most commonly results in thyroid dysfunction that occurs in 17% - 22.7% of alemtuzumab compared to 2.8% - 6% of interferon beta 1-a patients. The observed thyroid dys- function included a full spectrum of effects including hyper- and hypothyroidism, thyroiditis, and goiter; al- most all episodes were mild or moderate in severity and Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 468 managed with conventional therapy. Comprehensive mo- nitoring uncovered many of the thyroid adverse events and also provided for early detection and treatment of ITP. ITP occurred in 0.8% - 2.8% of alemtuzumab pa- tients but most were classified as a serious adverse event [49-51]. Alemtuzumab currently carries a black box warning for fatal cytopenias, infusion reactions and in- fections and is classified as pregnancy category C by the FDA [58]. 6. Conclusions MS is a lifelong illness and as such requires lifelong treatment. Current first-line DMTs for MS offer modest clinical benefit and have considerable adverse effects associated with their use. Previous parenteral therapies for treatment of MS require at least weekly administra- tion, whereas alemtuzumab administration is necessary only five days in the first 12 months and three days in the following 12 months, with evidence of sustained clinical benefit up to 60 months and possibly beyond. When available alemtuzumab studies are evaluated together, consistent benefit from the investigational agent compared to conventional therapy has been seen in both previously untreated RRMS patients and those who have relapsed on first-line conventional therapy. Significant decreases in the primary outcome of MS RR have been associated with alemtuzumab in each study and a reduced risk of SAD is observed in both the CAMMS223 and the CARE-MS II trials. Safety outcomes for both the 12 mg and 24 mg alemtuzumab doses were similar, and efficacy of the 24 mg dose was evaluated only in the CAMMS223 trial, with no differences seen between doses for clinical or MRI efficacy outcomes. Adverse effects associated with alemtuzumab infu- sions usually occur within one month of administration and can be screened for and effectively managed or treated. Nevertheless, the drug has been associated with an increased frequency of adverse effects compared to interferon beta 1-a-based therapy. Specifically, increased vigilance for infections and autoimmune diseases, in- cluding mainly thyroid dysfunction but also immune thrombocytopenia, appears to be required for alemtuzu- mab therapy. Current trials have shown sustained benefit up to 60 months when alemtuzumab therapy is administered at 0, 12, and 24 months. However, continued dosing of alem- tuzumab beyond the 24th month has not been evaluated. This is an area for continued research and will require an extended duration of study. Alemtuzumab, marketed under the trade name Lem- trada™, gained support from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) in June 2013 [59] and was approved for the treatment of RRMS by the European Commission in September 2013 [60]. Alemtuzumab is currently under review by the US FDA for the treatment of RRMS [59]. REFERENCES [1] R. L. Talbert, J. T. DiPiro, G. R. Matzke, L. M. Posey, B. G. Wells and G. C. Yee, “Multiple Sclerosis,” In: R. L Talbert, J. T. DiPiro, G. R. Matzke, L. M. Posey, B. G. Wells and G. C. Yee (Eds.), Pharmacotherapy: A Patho- physiologic Approach, 8e. http://nv-ezproxy.roseman.edu:2062/content.aspx?aID=7 984977 [2] J. Noseworthy, C. Lucchinetti, M. Rodriguez and B. Weinshenker, “Multiple Sclerosis,” The New England Journal of Medicine, Vol. 343, No. 13, 2000, pp. 938-952. http://dx.doi.org/10.1056/NEJM200009283431307 [3] C. Polman, S. Reingold, B. Banwell, M. Clanet, J. A. Co- hen, M. Filippi, K. Fujihara, E. Havrdova, M. Hutchinson, L. Kappos, F. D. Lublin, X. Montalban, P. O’Connor, M. Sandberg-Wollheim, A. J. Thompson, E. Waubant, B. Weinshenker and J. S. Wolinsky, “Diagnostic Criteria for Multiple Sclerosis: 2010 Revisions to the Mcdonald Cri- teria,” Annals of Neurology, Vol. 69, No. 2, 2011, pp. 292-302. http://dx.doi.org/10.1002/ana.22366 [4] E. Zintzaras, C. Doxani, T. Mprotsis, C. Schmid and G. Hadjigeorgiou, “Network Analysis of Randomized Con- trolled Trials in Multiple Sclerosis,” Clinical Therapeu- tics, Vol. 34, No. 4, 2012, pp. 857-869. http://dx.doi.org/10.1016/j.clinthera.2012.02.018 [5] M. Pugliatti, S. Sotgiu and G. Rosati, “The Worldwide Prevalence of Multiple Sclerosis,” Clinical Neurology and Neurosurgery, Vol. 104, No. 3, 2002, pp. 182-191. http://dx.doi.org/10.1016/S0303-8467(02)00036-7 [6] G. Rosati, “The Prevalence of Multiple Sclerosis in the World: An Update,” Neurological Sciences, Vol. 22, No. 2, 2001, pp. 117-139. http://dx.doi.org/10.1007/s100720170011 [7] G. Dean, H. McLoughlin, R. Brady, A. Adelstein and J. Tallett-William, “Multiple Sclerosis among Immigrants in Greater London,” British Medical Journal, Vol. 1, No. 6014, 1976, pp. 861-864. http://dx.doi.org/10.1136/bmj.1.6014.861 [8] B. Visscher, R. Detels, A. Coulson, R. Malmgren and J. Dudley, “Latitude, Migration and the Prevalence of Mul- tiple Sclerosis,” American Journal of Epidemiology, Vol. 106, No. 6, 1977, pp. 470-475. [9] C. Gale and C. Martyn, “Migrant Studies in Multiple Sclerosis,” Progress in Neurobiology, Vol. 47, No. 4-5, 1995, pp. 425-448. [10] R. Marrie, “Environmental Risk Factors in Multiple Scle- rosis Aetiology,” Lancet, Vol. 3, No. 12, 2004, pp. 709- 718. http://dx.doi.org/10.1016/S1474-4422(04)00933-0 [11] A. Ascherio, K. Munger, E. Lenette, D. Spiegelman, M. A. Hernán, M. J. Olek, S. E. Hankinson and D. J. Hunter, “Epstein-Barr Virus Antibodies and Risk of Multiple Sclerosis: A Prospective Study,” Journal of the American Medical Association, Vol. 286, No. 24, 2001, pp. 3083- 3088. http://dx.doi.org/10.1001/jama.286.24.3083 Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 469 [12] R. Marrie and C. Wolfson, “Multiple Sclerosis and Ep- stein-Barr Virus,” Canadian Journal of Infectious Dis- eases, Vol. 13, No. 2, 2002, pp. 111-118. [13] B. Serafini, L. Muzio, B. Rosicarelli and A. Francesca, “Radioactive in Situ Hybridization for Epstein-Barr Vi- rus-Encoded Small RNA Supports Presence of Epstein- Barr Virus in the Multiple Sclerosis Brain,” Brain, Vol. 136, No. 7, 2013, p. e233. http://dx.doi.org/10.1093/brain/aws315 [14] R. Magliozzi, B. Serafini, B. Rosicarelli, B. Chiappetta, C. Veroni, R. Reynolds and F. Alosi, “B-Cell Enrichment and Epstein-Barr Virus Infection in Inflammatory Corti- cal Lesions in Secondary Progressive Multiple Sclerosis,” Journal of Neuropathology & Experimental Neurology, Vol. 72, No. 1, 2013, pp. 29-41. http://dx.doi.org/10.1097/NEN.0b013e31827bfc62 [15] L. Levin, K. Munger, M. Rubertone, C. A. Peck, E. T. Lennette, D. Spiegelman and A. Ascherio, “Temporal Relationship between Elevation of Epstein-Barr Virus Antibody Titers and Initial Onset of Neurological Symp- toms in Multiple Sclerosis,” Journal of the American Medical Association, Vol. 239, No. 20, 2005, pp. 2496- 2500. http://dx.doi.org/10.1001/jama.293.20.2496 [16] P. Challoner, K. Smith, J. Parker, D. L. MacLeod, S. N. Coulter, T. M. Rose, E. R. Schultz, J. L. Bennett, R. L. Garber and M. Chang, “Plaque-Associated Expression of Human Herpesvirus 6 in Multiple Sclerosis,” Proceedings of the National Academy of Sciences, Vol. 92, No. 16, 1995, pp. 7440-7444. http://dx.doi.org/10.1073/pnas.92.16.7440 [17] J. Pietiläinen, J. Virtanen, L. Uotila, L. Saloned, M. Ko- skiniemi and M. Färkkilä, “HHV-6 Infection in Multiple Sclerosis. A Clinical and Laboratory Analysis,” European Journal of Neurology, Vol. 17, No. 3, 2010, pp. 506-509. http://dx.doi.org/10.1111/j.1468-1331.2009.02718.x [18] S. Simpson, B. Taylor, D. Dwyer, J. Taylor, L. Blizzard, A. L. Ponsonby, F. Pittas, T. Dwyer and l. van der Mei, “Anti-HHV-6 IgG Titer Significantly Predicts Subsequent Relapse Risk in Multiple Sclerosis,” Multiple Sclerosis Journal, Vol. 18, No. 6, 2012, pp. 799-806. http://dx.doi.org/10.1177/1352458511428081 [19] N. Summerday, S. Brown, D. Allington and M. Rivey, “Vitamin D and Multiple Sclerosis: Review of a Possible Association,” Journal of Pharmacy Practice, Vol. 25, No. 1, 2011, pp. 75-84. http://dx.doi.org/10.1177/0897190011421839 [20] G. Ebers, “Environmental Factors and Multiple Sclero- sis,” Lancet Neurology, Vol. 7, No. 3, 2008, pp. 268-277. http://dx.doi.org/10.1016/S1474-4422(08)70042-5 [21] N. Robertson, J. O’Riordan, J. Chataway, D. P. Kingsley, D. H. Miller, D. Clayton and D. A. Compston, “Offspring Recurrence Rates and Clinical Characteristics of Conjugal Multiple Sclerosis,” Lancet, Vol. 349, No. 9065, 1997, pp. 1587-1590. http://dx.doi.org/10.1016/S0140-6736(96)07317-5 [22] G. Ebers, I. Yee, A. Sadavnick, P. Duquette and the Ca- nadian Collaborative Study Group, “Conjugal Multiple Sclerosis Population-Based Prevalence and Recurrence Risks in Offspring,” Annals of Neurology, Vol. 48. No. 6, 2000, pp. 927-931. http://dx.doi.org/10.1002/1531-8249(200012)48:6<927:: AID-ANA14>3.0.CO;2-F [23] G. Ebers, D. Bulman, A. Sadovnick, D. W. Paty, S. War- ren, W. Hader, T. J. Murray, T. P. Seland, P. Duquette, T. Grey, R. Nelson, M. Nicolle and D. Brunet, “A Popula- tion-Based Study of Multiple Sclerosis in Twins,” The New England Journal of Medicine, Vol. 315, No. 26 1986, pp. 1638-1642. http://dx.doi.org/10.1056/NEJM198612253152603 [24] A. Sadovnick, H. Armstrong, G. Rice, D. Bulman, L. Ha- shimoto, D. W. Paty, S. A. Hashimoto, S. Warren, W. Hader, T. J. Murray, et al., “A Population-Based Study of Multiple Sclerosis in Twins: Update,” Annals of Neurol- ogy, Vol. 33, No. 3, 1993, pp. 281-285. http://dx.doi.org/10.1002/ana.410330309 [25] M. H. Barnett and J. W. Prineas, “Relapsing and Remit- ting Multiple Sclerosis: Pathology of the Newly Forming Lesion,” Annals of Neurology, Vol. 55, No. 4, 2004, pp. 458-468. http://dx.doi.org/10.1002/ana.20016 [26] R. Gold, C. Linington and H. Lassmann, “Understanding Pathogenesis and Therapy of Multiple Sclerosis via Ani- mal Models: 70 Years of Merits and Culprits in Experi- mental Autoimmune Encephalomyelitis Research,” Brain, Vol. 129, No. 8, 2006, pp. 1953-1971. http://dx.doi.org/10.1093/brain/awl075 [27] F. W. Gay, T. J. Drye, G. W. Dick and M. M. Esiri, “The Application of Multifactorial Cluster Analysis in the Staging of Plaques in Early Multiple Sclerosis. Identifica- tion and Characterization of the Primary Demyelinating Lesion,” Brain, Vol. 120, No. 8, 1997, pp. 1461-1483. http://dx.doi.org/10.1093/brain/120.8.1461 [28] H. Lassmann, W. Brück and C. F. Lucchinetti, “The Im- munopathology of Multiple Sclerosis: An Overview,” Brain Pathology, Vol. 17, No. 2, 2007, pp. 210-218. http://dx.doi.org/10.1111/j.1750-3639.2007.00064.x [29] C. F. Luchinetti, W. Brück, J. Parisi, B. Scheithauer, M. Rodriquez and H. Lassman, “Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination,” Annals of Neurology, Vol. 47, No. 6, 2000, pp. 707-717. http://dx.doi.org/10.1002/1531-8249(200006)47:6<707:: AID-ANA3>3.0.CO;2-Q [30] R. C. Selter and B. Hemmer, “Update on Immunopatho- genesis and Immunotherapy in Multiple Sclerosis,” Im- munotargets and Therapy, Vol. 2, 2013, pp. 21-30. http://www.dovepress.com/update-on-immunopathogenes is-and-immunotherapy-in-multiple-sclerosis-peer-reviewe d-article-ITT-recommendation1 [31] J. W. Peterson, L. Bö, S. Mörk, A. Chang, R. M. Ranso- hoff and B. D. Trapp,“VCAM-1 Positive Microglia Tar- get Oligodendrocytes at the Border of Multiple Sclerosis Lesions,” Journal of Neuropathology & Experimental Neurology, Vol. 61, No. 6, 2002, pp. 539-546. [32] A. Minagar and J. S. Alexander, “Blood-Brain Barrier Disruption in Multiple Sclerosis,” Multiple Sclerosis, Vol. 9, No. 3, 2003, pp. 540-549. http://dx.doi.org/10.1191/1352458503ms965oa [33] D. M. Wingerchuk, C. F. Lucchinetti and J. H. Nosewor- Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis 470 thy, “Multiple Sclerosis: Current Pathophysiological Con- cepts,” Laboratory Investigation, Vol. 81, No. 3, 2001, pp. 263-281. http://dx.doi.org/10.1038/labinvest.3780235 [34] H. Waldmann and G. Hale, “CAMPATH: From Concept to Clinic,” Philosophical Transactions of the Royal Soci- ety B: Biological Sciences, Vol. 360, No. 1461, 2005, pp. 1707-1711. http://dx.doi.org/10.1098/rstb.2005.1702 [35] D. Gómez-Almagues, M. Solano-Genesta, L. Tarín-Ar- zaga, J. L. Herrera-Garza, O. G. Cantú-Rodríguez, C. H. Gutiérrez-Aguirre and J. C. Jaime-Pérez, “Low-Dose Ri- tuximab and Alemtuzumab Combination Therapy for Pa- tients with Steroid-Refractory Autoimmune Cytopenias,” Blood, Vol. 116, No. 23, 2010, pp. 4783-4785. http://dx.doi.org/10.1182/blood-2010-06-291831 [36] E. J. Fox, H. C. Sullivan, S. K. Gazda, L. Mayer, L. O’Donnell, K. Melia and S. L. Lake, “A Single-Arm, Open-Label Study of Alemtuzumab in Treatment-Re- fractory Patients With Multiple Sclerosis,” European Jour- nal of Neurology, Vol. 19, No. 2, 2012, pp. 307-311. http://dx.doi.org/10.1111/j.1468-1331.2011.03507.x [37] L. Klotz, S. Meuth and H. Wiendl, “Immune Mechanisms of New Therapeutic Strategies in Multiple Sclerosis—A Focus on Alemtuzumab,” Clinical Immunology, Vol. 142, No. 1, 2012, pp. 24-30. http://dx.doi.org/10.1016/j.clim.2011.04.006 [38] B. Bielekova and B. Becker, “Monoclonal Antibodies in MS: Mechanism of Action,” Neurology, Vol. 74, No. S1, 2010, pp. S31-S40. http://dx.doi.org/10.1212/WNL.0b013e3181c97ed3 [39] S. Rodig, J. Abramson, G. Pinkus, S. P. Treon, D. M. Dor- fman, H. Y. Dong, M. A. Shipp and J. L. Kutok, “Het- erogeneous CD52 Expression among Hematologic Neo- plasm: Implications for the Use of Alemtuzumab (CAM- PATH-1H),” Clinical Cancer Research, Vol. 12, No. 23, 2006, pp. 7174-7179. http://dx.doi.org/10.1158/1078-0432.CCR-06-1275 [40] A. Minagar, S. Alexander, A. Sahraian and R. Zivadinov, “Alemtuzumab and Multiple Sclerosis: Therapeutic Ap- plication,” Expert Opinion on Biological Therapy, Vol. 10, No. 3, 2010, pp. 421-429. http://dx.doi.org/10.1517/14712591003586806 [41] J. L. Bennett and O. Stϋve, “Update on Inflammation, Neurodegeneration, and Immunoregulation in Multiple Sclerosis: Therapeutic Implication,” Clinical Neurophar- macology, Vol. 32, No. 3, 2009, pp. 121-132. http://dx.doi.org/10.1097/WNF.0b013e3181880359 [42] M. Klabusay, V. Sukova, P. Coupek, Y. Brychtova and J. Mayer, “Different Levels of CD52 Antigen Expression Evaluated by Quantitative Fluorescence Cytometry Are Detected on B-lymphocytes, CD34+ Cells and Tumor Cells of Patients With Chronic B-cell Lymphoprolifera- tive Diseases,” Cytometry, Vol. 72, No. 5, 2007, pp. 363- 370. http://dx.doi.org/10.1002/cyto.b.20181 [43] T. Elter, I. Molnar, J. Kuhlmann, M. Hallek and C. Wen- dtner, “Pharmacokinetics of Alemtuzumab and the Rele- vance in Clinical Practice,” Leuk Lymphoma, Vol. 49, No. 12, 2008, pp. 2256-2262. http://dx.doi.org/10.1080/10428190802475303 [44] D. R. Mould, A. Baumann, J. Kuhlmann, M. J. Keating,S. Weitman, P. Hillmen, L. R. Brettman, S. Reif and P. L. Bonate, “Population Pharmacokinetics-Pharmacodynamics of Alemtuzumab (CampathR) in Patients With Chronic Lymphocytic Leukaemia and its Link to Treatment Re- sponse,” British Journal of Clinical Pharmacology, Vol. 64, No. 3, 2007, pp. 278-291. http://dx.doi.org/10.1111/j.1365-2125.2007.02914.x [45] P. Rebello, K. Cwynarski, M. Varughese, A. Eades, J. F. Apperley and G. Hale, “Pharmacokinetics of CAM- PATH-1H in BMT Patients,” Cytotherapy, Vol. 3, No. 4, 2001, pp. 261-267. http://dx.doi.org/10.1080/146532401317070899 [46] J. D. Isaacs, V. K. Manna, N. Rapson, K. J. Bulpitt, B. L. Hazleman, E. L. Matteson, E. W. St.Clair, T. J. Schnitzer and J. M. Johnston, “Campath-1H in Rheumatoid Arthri- tis—An Intravenous Dose-Ranging Study,” British Jour- nal of Rheumatology, Vol. 35, No. 3, 1996, pp. 131-140. http://dx.doi.org/10.1093/rheumatology/35.3.231 [47] G. Hale, P. Rebello, L. R. Brettman, C. Fegan, B. Ken- nedy, E. Kimby, M. Leach, J. Lundin, H. Mellstedt, P. Moreton, A. C. Rawstron, H. Waldmann, A. Osterborg and P. Hillmen, “Blood Concentrations of Alemtuzumab and Antiglobulin Responses in Patients With Chronic Lymphocytic Leukemia Following Intravenous or Sub- cutaneous Routes of Administration,” Blood, Vol. 104, No. 4, 2004, pp. 948-955. http://dx.doi.org/10.1182/blood-2004-02-0593 [48] G. A. Hill-Cawthorne, T. Button, O. Tuohy, J. L. Jones, K. May, J. Somerfield, A. Green, G. Giovannoni, D. A. S. Compston, M. T. Fahey and A. J. Coles, “Long Term Lymphocyte Reconstitution after Alemtuzumab Treat- ment of Multiple Sclerosis,” Journal of Neurology, Neu- rosurgery, and Psychiatry, Vol. 83, No. 3, 2012, pp. 298- 304. http://dx.doi.org/10.1136/jnnp-2011-300826 [49] A. J. Coles, D. A. Compston, K. W. Selmaj, S. L. Lake, S. Moran, D. H. Margolin, K. Norris and P. K. Tandon, “Alemtuzumab vs. Interferon Beta-1a in Early Multiple Sclerosis,” The New England Journal of Medicine, Vol. 359, No. 17, 2008, pp. 1786-1801. http://dx.doi.org/10.1056/NEJMoa0802670 [50] J. A. Cohen, A. J. Coles, D. L. Arnold, C. Confavreux, E. J. Fox, H. P. Hartung, E. Havrdova, K. W. Selmaj, H. L. Weiner, E. Fisher, W. Brinar, G. Giovannoni, M. Stoja- novic, B. I. Ertik, S. L. Lake, D. H. Margolin, M. A. Panzara and D. A. Compston, “Alemtuzumab Versus In- terferon Beta 1a as First-Line Treatment For Patients With Relapsing-Remitting Multiple Sclerosis: A Ran- domized Controlled Phase 3 Trial,” Lancet, Vol. 380, No. 9856, 2012, pp. 1819-1828. http://dx.doi.org/10.1016/S0140-6736(12)61769-3 [51] A. J. Coles,C. L. Twyman, D. L. Arnold, J. A. Cohen, C. Confavreux, E. J. Fox, H. P. Hartung, E. Havrdova, K. W. Selmaj, H. L. Weiner, T. Miller, E. Fisher, R. Sandbrink, S. L. Lake, D. H. Margolin, P. Oyuela, M. A. Panzara and D. A. Compston, “Alemtuzumab for Patients With Rela- psing Multiple Sclerosis after Disease-Modifying Ther- apy: A Randomized Controlled Phase 3 Trial,” Lancet, Vol. 380, No. 9856, 2012, pp. 1829-1839. http://dx.doi.org/10.1016/S0140-6736(12)61768-1 [52] A. J. Coles, A. Cox, E. Le Page, J. Jones, S. A. Trip, J. Copyright © 2013 SciRes. IJCM  Alemtuzumab: A Place in Therapy for Treatment of Multiple Sclerosis Copyright © 2013 SciRes. IJCM 471 Deans, S. Seaman, D. H. Miller, G. Hale, H. Waldmann and D. A. Compston, “The Window of Therapeutic Op- portunity in Multiple Sclerosis: Evidence From Mono- clonal Antibody Therapy,” Journal of Neurology, Vol. 253, No. 1, 2006, pp. 98-108. http://dx.doi.org/10.1007/s00415-005-0934-5 [53] C. Hirst, A. Pace, T. Pickersgill, R. Jones, B. N. McLean, J. P. Zajicek, N. J. Scolding and N. P. Robetrson, “Cam- path 1-H Treatment in Patients With Aggressive Relaps- ing Remitting Multiple Sclerosis,” Journal of Neurology, Vol. 255, No. 2, 2008, pp. 231-238. http://dx.doi.org/10.1007/s00415-008-0696-y [54] A. J. Coles, E. Fox, A. Vladic,S. K. Gazda, V. Brinar, K. W. Selmaj, A. Skoromets, I. Stolyarov, A. Bass, H. Sul- livan, D. H. Margolin, S. L. Lake, S. Moran, J. Palmer, M. S. Smith and D. A. Compston, “Alemtuzumab More Ef- fective Than Interferon β-1a at 5-Year Follow-Up of CAMMS223 Clinical Trial,” Journal of Neurology, Vol. 78, No. 14, 2012, pp. 1069-1078. http://dx.doi.org/10.1212/WNL.0b013e31824e8ee7 [55] S. Krieger, D. Arnold, J. Cohen, A. J. Coles, C. Con- favreux, E. J. Fox, H. Hartung, E. Havrdova, K. Selmaj, H. L. Weiner, T. A.Miller, C. L. Twyman, S. L. Lake, D. H. Margolin, M. A. Panzara and A. Compston, “Alemtu- zumab is Efficacious in Highly-Active RRMS Patients in CARE-MS II,” 2013. https://cmscactrims.confex.com/cmscactrims/2013/webpr ogram/Paper1191.html [56] J. L. Jones, C. Phuah, A. L. Cox, S. A. Thompson, M. Ban, J. Shawcross, A. Walton, S. J. Sawcer, A. Compston and A. J. Coles, “IL-21 Drives Secondary Autoimmunity in Patients With Multiple Sclerosis, Following Therapeu- tic Lymphocyte Depletion With Alemtuzumab (Campath— 1H),” Journal of Clinical Investigation, Vol. 119, No. 7, 2009, pp. 2052-2061. [57] L. Costelloe, J. Jones and A. Coles, “Secondary Autoim- mune Diseases Following Alemtuzumab Therapy for Multiple Sclerosis,” Expert Review of Neurotherapeutics, Vol. 12, No. 3, 2012, pp. 333-341. http://dx.doi.org/10.1586/ern.12.5 [58] Alemtuzumab, “Millennium and ILEX Partners,” 2001. [59] S. Jeffrey, “Lemtrada Gets Positive Opinion from Euro- pean CHMP in MS,” 2013. http://www.medscape.com/viewarticle/807066 [60] “European Commission Approves Genzyme’s Multiple Sclerosis Treatment Lemtrada™ (alemtuzumab),” 2013. http://online.wsj.com/article/PR-CO-20130917-900101.ht ml
|