 Int. J. Communications, Network and System Sciences, 2013, 6, 415-442 http://dx.doi.org/10.4236/ijcns.2013.610045 Published Online October 2013 (http://www.scirp.org/journal/ijcns) Copyright © 2013 SciRes. IJCNS Alloy Gene Gibbs Energy Partition Function and Equilibrium Holographic Network Phase Diagrams of AuCu-Type Sublattice System Youqing Xie1,2,3*, Xiaobo Li4, Xinbi Liu1,2,3, Yaozhuang Nie5, Hongjian Peng6 1School of Materials Science and Engineering, Central South University, Changsha, China 2Powder Metallurgy Research Institute, Central South University, Changsha, China 3State Key Laboratory of Powder Metallurgy, Central South University, Changsha, China 4College of Mechanical Engineering, Xiangtan University, Xiangtan, China 5School of Physical Science and Technique, Central South University, Changsha, China 6School of Chemistry and Chemical Engineering, Central South University, Changsha, China Email: *xieyouq8088@163.com Received August 1, 2013; revised September 3, 2013; accepted September 14, 2013 Copyright © 2013 Youqing Xie et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Taking AuCu-sublattice system as an example, we present two discoveries and a method. First, the alloy gene se- quences are the central characteristic atom sequences in the basic coordination cluster sequences. Second, the transmis- sion mode of the information about structures and properties of the alloy genes is described by the alloy gene Gibbs energy partition function. The most valuable method in the system sciences is “the whole obtained from a few parts”. We have established the alloy gene database and holographic alloy positioning system of the Au-Cu system, as well as alloy gene Gibbs energy partition function and equilibrium holographic network phase diagrams of the AuCu-type sublattice system. It means that a standard way for researchers to share predictive algorithms and computational meth- ods may be produced during designing advanced alloys. Keywords: Alloy Gene Gibbs Energy Partition Function; Holographic Alloy Positioning System; Equilibrium Holographic Network Phase Diagrams; Systematic Metal Materials Science; Au-Cu System 1. Introduction There are 81 kinds of metal elements accounting for 79% in the Element Periodic Table, of which several alloy system groups can be composed: 2 81 3240C kinds of binary alloy systems, 3 81 85320C kinds of ternary al- loy systems, 4 81 1663740C kinds of quaternary alloy systems, and so on. In order to quickly and efficiently discover advanced alloys, we have to establish the Sys- tematic Metal Materials Science (SMMS) by new think- ing modes and methods of system sciences [1] (Supple- mentary Section 1). “A diversity of structures of a system is attributed to combination and arrangement of structural units in the basic structure unit sequences”. It is the first philosophic proposition of system sciences. Now, we deliver an ex- tensive definition of the gene sequence: the gene se- quence is a basic structure unit sequence carrying a set of transmission information about structures and properties for determining diversity of structures and properties of a system, which may be a biologic or non-biologic system. Therefore, it is our first task to seek alloy gene (AG) se- quences for establishing the SMMS framework. We have discovered that the AG-sequences are the central characteristic atom sequences in the basic coor- dination cluster sequences (Supplementary Figure S1(c)). In the fcc-based lattice Au-Cu system, the basic coordi- nation clusters Au AuAu Cu ii BA Iii and Cu CuAuCu ii BA Iii consist of the central cha- racteristic atoms Au i and Cu i respectively, and the nearest coordinative configuration Au CuIi i . Here, I is the coordinative number and equal to 12; i is the number of Cu-atoms and can change from 0 to 12; (I-i) is the number of Au-atoms. Therefore, the AG-se- quences of the Au-Cu system are the central characteris- tic atom Au i - and Cu i -sequences respectively in the Au i B- and Cu i B-sequences. They may be used to replace *Corresponding author.  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 416 atomic pair-sequence [2] and atomic cluster-sequences [3,4], as well as other structural unit sequences [5-7] in the current alloy theories, because they have more ana- logous characters to biologic gene sequences: (1) The AG Au i - and Cu i -sequences carry a set of transmis- sion information: coordinative configurations, electronic structures, physical and thermodynamic properties, which have been obtained by the AG-theory (Supplementary Section 1). (2) Based on the AG-information, a Holo- graphic Alloy Positioning (HAP) system of the Au-Cu system has been established by “the Whole Obtained from a few Parts” (WOP) method (Section 2). (3) AG- information may be transmitted to alloy phases by AG- Gibbs energy partition function (Section 3). The Equilib- rium Holographic Network Phase (EHNP) diagrams of the AuCu-type sublattice system have been obtained by the HAP system (Section 4). (4) Alloy genes may be in- terconvertible, the essence of the orderdisorder transi- tion can be explained by the AG-concentrations distribu- tions (Figure 4). (5) Alloy genes will be used to design alloys, which may be called as the AG-arranging design. 2. Holographic Alloy Positioning System The main objective of the HAP system of an alloy sys- tem is to provide a set of EHNP diagrams for designing advanced alloys. The HAP system of the Au-Cu system consists of AG-theory, AG-Gibbs energy partition func- tions of Au3Cu-, AuCu- and AuCu3-type sublattice sys- tems and balance theory between sublattice systems (Figure 1). Its performing procedures are as follows: (1) The first step is to establish AG-database obtained by the AG-theory, only using experimental mixed enthalpies Figure 1. Holographic alloy positioning system of Au-Cu system. exp m T and mixed volumes exp m VT of the AuCu and AuCu3 compounds [8,9] (Supplementary Figures S2 and S5). (2) The second step is respectively to establish EHNP diagrams of the Au3Cu-, AuCu- and AuCu3-type sublattice systems, according to the AG- Gibbs energy partition functions of Au3Cu-, AuCu- and AuCu3-type sublattice systems and the Bragg-Williams sublattice models [10,11]. (3) The third step is to es- tablish EHNP diagrams of the Au-Cu system based on the balance theory between sublattice systems. So much information is obtained by computing technique and a few experimental data, i.e., WOP method (Supplemen- tary Section 1.5). 3. AG-Gibbs Energy Partition Function “A diversity of properties of a system is attributed to contents and transmission mode of the information of basic structure unit sequences”. It is the second philoso- phic proposition of system sciences. The transmission mode of AG-information is described by AG-Gibbs en- ergy , T-partition function, which consists of the AG-Gibbs energy transmission *,GxT-function and AG-arranging structure Au Cu ,, , ii xxTxxT-func- tion (Supplementary Section 2). An equilibrium orderdisorder transition of the AG-arranging structure of an alloy is defined as that “the AG-Gibbs energy levels Au Cu , ii GTGT and AG- concentrations Au Cu ,, , ii xT xxT occupied at the Au i GT- and Cu i GT-energy levels can respond im- mediately and change synchronously with each small variation in temperature”. It has following general be- haviors: (1) The order → disorder transition upon heating and disorder → order transition upon cooling proceed along the same minimal mixed Gibbs energy min m GT path, i.e., there exists no a so-called hysteresis pheno- menon between both transitions. (2) The min m GT path is continuous and has no jumping phenomenon. 4. EHNP-Diagrams of AuCu-Type Sublattice System “Properties are determined by structures; properties should be suitable for environments; environments change struc- tures”. It is the fourth philosophic proposition of system sciences (the third one is presented in Supplementary Section 1.2). The man’s knowledge of relationships of structures, properties and temperature for alloys has been changed from single causality to systematic correlativity, due to discoveries of alloy gene sequences and their in- formation transmission mode, and establishments of the AG-Gibbs energy partition function and HAP system. This systematic correlativity may be described by a set of HNP diagrams. An EHNP diagram consists of curves linked by the  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 417 network ,,qxT -points as functions of the state q, composition x and temperature T. Each position inside the phase region and on the phase boundary (PB) curve may be marked by the ,,qxT -point, where the alloy state q denotes the AG-concentrations (Au i and Cu i ), mixed Gibbs energy m G, order degree , configura- tional entropy c S, mixed characteristic Gibbs energy *m G, mixed enthalpy m , mixed potential energy m E, mixed volume m V, generalized vibration free energy v , generalized vibration energy v U, general- ized vibration entropy v S, mixed heat capacity m C, mixed thermal expansion coefficient m and activi- ties (Au a and Cu a). Each kind of the q-EHNP diagram includes four diagrams: three-dimension qxT phase diagram, two-dimension x qT , q Tx and T qx path phase diagrams. In the text the , m GxT, , T , , c SxT, Au , i xT and Cu , i xT EHNP diagrams are presented. The other EHNP diagrams are shown in Supplementary Figure 6. The calculations of all diagrams are performed by compositional step x = 0.5at.% and temperature step 1KT . These diagrams are interconnected. Therefore, a set of information about structures, properties and their variations with tempera- ture of a designed alloy in the equilibrium state can be pre- dicated by interlinking method and without calculation. 4.1. Mixed Gibbs Energy and Order Degree EHNP Diagrams Using the minimal mixed Gibbs energy path method, we have obtained the three-dimension m GxT and T EHNP diagrams, and two-dimension m x GT , xT , m G Tx , Tx , m T Gx and T path phase diagrams. From these diagrams, we have obtained following main understandings: (1) From Figures 2(a) and (a’), it can be known that there are or- dered phase region (denoted by the symbol “O”), disor- dered phase region (denoted by the symbol “D”), AuCu compound consisted of Au 8 and Cu 4 genes (denoted by the symbol “C”), and the , m PB GxT- and , PB T -phase boundary curves in the each diagram. (2) From Figures 2(b) and (b’ ), it can be known that the Au47.5%Cu52.5% alloy has the highest critical temperature 836 K c T, which is higher than the critical temperature (833K) of the Au50%Cu50% alloy , that agrees with the experimental phenomenon (see Figure 2(c’)). (3) From Figures 2(c) and (c’), it can be known that the phase diagrams can be respectively divided into the several regions by some m G Tx and Tx curves. It is very useful to enrich the EHNP diagrams. (4) From Figure 2(c’), we have discovered that all experimental jumping temperatures T, which are denoted by symbols “*”[12] and “○” [13], approach to the iso-order 0.65 Tx curve. The experimental j Tx curve is very analogous to the equilibrium PB Tx curve, i.e., c Tx curve. However, the experimental jumping long- range order degree R j of the AuCu compound is about 0.8, and the corresponding short-range order de- grees SR is about 0.40. These phenomena show that the experimental order → disorder transition belongs in metastable and non-equilibrium. (5) The isocomposi- tional m x GT and xT path diagrams (Figures 2(d) and (d’)) will be used to establish EHNP diagrams of the Au-Cu system. 4.2. Configuration Entropy and AG- Concentration EHNP Diagrams The configuration entropy , c SxT and AG-con- centration Au Cu ,, , ii xT xxT EHNP diagrams have been established, based on the AG-arranging structure Au Cu ,, , ii xxTxxT-function in the , T-func- tion. From Figures 3 and 4, it can be known that each kind of q-EHNP diagram includes not only the 4-type diagrams indicated above, but also other–type diagrams. From Figure 3, we have obtained following main under- standings: (1) The configuration entropy of each ordered alloy can change continually from the configuration en- tropy of the maximum order degree max state to one of the ideal disordered state, that need not to induct any parameter. It means that we should take the ideal disor- dered state as the standard. (2) The structural units used for calculating configuration entropy should be in agree- ment with the structural units used for calculating corre- sponding energy levels. These are two rules to establish partition function. However, these rules are often ne- glected in the currently used thermodynamic models of alloy phases [14]. From Figure 4, we have obtained following main un- derstandings: (1) The AG-concentration EHNP diagram may be described by two modes: the Au i xT and Cu i xT EHNP diagrams (Figures 4(a) and (a’)) in the AG-arranging crystallography [15], where the Au i and Cu i x are the probabilities occupied at the lattice points; the Au i xi and Cu i xi EHNP diagrams (Figures 4(a), (a’), (c) and (c’)) in the AG-arranging band theory, where the Au i and Cu i are the probabili- ties occupied at the Au i GT and Cu i GT energy lev- els. (2) There exists a probability of AG-arranging struc- tures in the alloy phases, which is described by the AG- concentration Au Cu ,, , ii xT xxT functions. This probability leads to atom-scale heterogeneity of proper- ties of alloys. (3) There exists an emergent phenomenon of AG-arranging structures in the ordered alloy phases, which is defined as that some AG-concentrations of the alloy in ordered state are larger than one in the disor- dered state, such as Au 9 , Au 8 , Au 7 , Cu 3 , Cu 4 and Cu 5 (Figures 4(b)-(c’)). (4) Alloy genes may be inter-  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 418 (a) (a’) (b) (b’) (c) (c’) (d) (d’) Figure 2. Mixed Gibbs energy and order degree EHNP diagrams. (a), (a’), Three-dimension m GxT and xT diagrams with phase boundary curves m PB GxT, and PB xT ,; (b), (b’), Two-dimension isocompositional m x GT and xT path diagrams with phase boundary curves m PB GT and PB T ; (c), (c’), Two-dimension iso-mixed Gibbs energy m G Tx and iso-order degree Tx diagrams with phase boundary curves PB Tx ; (d), (d’), Two-dimension isothermal m T Gx and Tx diagrams with phase boundary curves m PB Gx and PB x .  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 419 (a) (b) (c) (d) Figure 3. Configuration entropy EHNP diagrams of the AuCu-type sublattice system. a) Three-dimension c xT phase diagram with phase boundary curve c PB xT,; b) Three-dimension c xT .Au phase diagram with phase boundary curve ., cAu PB xT of the Au-component; c) Three-dimension .cCu xT diagram with phase boundary curve ., cCu PB xT of the Cu-component; d) Three-dimension ..cAu cCu SxT combinational phase diagrams with phase boundary curves ., cAu PB xT and ., cCu PB xT of the Au- and Cu-components. convertible. The essence of the order → disorder tran- sition of the stoichiometric AuCu compound consisting of Au 8 - and Cu 4 -alloy genes is that the Au 8 - and Cu 4 -alloy genes are split into the Au i - and Cu i -se- quences, respectively; otherwise the essence of the dis- order → order transition of the stoichiometric disordered AuCu alloy is that the Au i - and Cu i -sequences are degenerated into the Au 8 - and Cu 4 -alloy genes (Fig- ures 4(d) and (d’)). 5. Conclusions In this paper, these creative achievements are of broad significances: (1) Four philosophic propositions of sys- tem sciences will prove stimulating to researchers work- ing on general system sciences, matter systems and social systems, and who may well find value in using them as a philosophic thinking logic for establishing various sys- tem science frameworks. (2) the AG-sequence instead of compositional atoms, compositional atom pairs and compositional atom clusters sucessively proposed during last one hundred years will prove stimulating to re- searchers working on physics, chemistry and material science, and who may well find value in using it as basic structure unit sequence for establish SMMS framework. (3) The AG-Gibbs energy partition function composed of AG-Gibbs energy level model of chemical potential and AG-statistic model of entropy replaces two current func- tions: one is composed of atom pair energy level model  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 420 Figure 4. AG-concentration EHNP diagrams. (a), (a’), Three-dimension i xxT Au and i xxT Cu diagrams; (b), (b’), Three-dimension i xxi Au and i xxi Cu diagrams of the ordered Au(1-x)Cux alloys with maximum order degree max at 0K; (c), (c’), Three-dimension i xxi Au and i xxi Cu diagrams of the perfectly disordered Au(1-x)Cux alloys; (d), (d’), Three-dimension i xTi Au and i xTi Cu path diagrams of disordering for the stoichiometric AuCu compound.  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 421 of chemical potential and compositional atom statistic model of entropy; another one is composed of electron energy level model of chemical potential and composi- tional atom cluster model of entropy. That will prove stimulating to researchers working on physics, chemistry and materials science, and who may well find value in using it as a theoretic foundation for establishing alloy system sciences of binary and ternary alloy systems. (4) The HAP system based on the AG-Gibbs energy parti- tion function and the WOP method will prove stimulat- ing to researchers working on physics, chemistry and materials science, and who may well find value in using it as a platform for establishing EHNP diagrams of alloy systems. (5) The great holographic alloy database con- sisting of AG-database and EHNP diagrams of alloy sys- tems will prove stimulating to others working on materi- als engineering, and who may well find value in using it as a tool for materials discovery, design, manufacture and application, which may be called as “AG-Arranging (AGA) Engineering”. The investigation in AuCu-type sublattice system may be analogous to Mendel’s investi- gation in genetic characters of peas. 6. Acknowledgements The work was supported by the National Natural Science Foundation of China (51071181); and by the Natural Science Foundation of Hunan province (2012FJ4044), China. REFERENCES [1] Y. Q. Xie, “Systematic Science of Alloys,” Central South University Press, Changsha, China, 2012. [2] H. A. Bethe, “Statistical Theory of Superlattices,” Pro- ceedings of the Royal Society of London, Vol. 150, No. 871, 1935, pp. 552-575. http://dx.doi.org/10.1098/rspa.1935.0122 [3] R. Kikuchi, D. De Fontaine, M. Murakami and T. Naka- mura, “Ternary Phase Diagram Calculations—II Exam- ples of Clustering and Ordering Systems,” Acta Metallur- gica, Vol. 25, 1997, pp. 207-219. http://dx.doi.org/10.1016/0001-6160(77)90124-9 [4] M. Asta and D. De Fontaine, “First-Principles Study of Phase Stability of Ti-Al Intermetallic Compounds,” Journal of Materials Research, Vol. 8, No. 10, 1993, pp. 2554-2568. http://dx.doi.org/10.1557/JMR.1993.2554 [5] V. Ozolins, C. Wolverton and A. Zunger, “Cu-Au, Ag-Au, Cu-Ag, and Ni-Au Intermetallics: First-Principles Study of Temperature-Composition Phase Diagrams and Struc- tures,” Physical Review B, Vol. 57, No. 11, 1998, pp. 6427-6443. http://dx.doi.org/10.1103/PhysRevB.57.6427 [6] B. Sundman, S. G. Fries and W. A. Oates, “A Thermo- dynamic Assessment of the Au-Cu System,” Calphad, Vol. 22, No. 2, 1998, pp. 335-354. http://dx.doi.org/10.1016/S0364-5916(98)00034-0 [7] Hillert, M., “The compound energy formalism,” Journal of Alloys and Compounds, Vol. 320, No. 2, 2001, pp. 161-176. http://dx.doi.org/10.1016/S0925-8388(00)01481-X [8] Y. Q. Xie, X. B. Liu, X. B. Li, H. J. Peng and Y. Z. Nie, “Potential Energies of Characteristic Atoms on Basis of Experimental Heats of Formation of AuCu and AuCu3 Compounds,” Transactions of Nonferrous Metals Society of China, Vol. 19, No. 5, 2009, pp.1243-1256. http://dx.doi.org/10.1016/S1003-6326(08)60436-7 [9] Y. Q. Xie, X. B. Liu, X. B. Li, H. J. Peng and Y. Z. Nie, “Volume Sequences of Characteristic Atoms Separated from Experimental Volumes of AuCu and AuCu3 Com- pounds,” Transactions of Nonferrous Metals Society of China, Vol. 19, No. 6, 2009, pp. 1559-1617. http://dx.doi.org/10.1016/S1003-6326(09)60077-7 [10] W. L. Bragg and E. J. Williams, “The Effect of Thermal Agitation on Atomic Arrangement in Alloys,” Proceed- ings the Royal of Society A, Vol. 145, No. 855, 1934, pp. 699-730. http://dx.doi.org/10.1098/rspa.1934.0132 [11] W. L. Bragg and E. J. Williams, “The Effect of Thermal Agitation on Atomic Arrangement in Alloys II,” Pro- ceedings the Royal of Society A, Vol. 151, No. 874, 1935, pp. 540-566. http://dx.doi.org/10.1098/rspa.1935.0165 [12] R. A. Oriani, “Thermodynamics of Order Alloy. II. The Gold-Copper System,” Acta Metallurgica, Vol. 2, No. 4, 1954, pp. 608-615. http://dx.doi.org/10.1016/0001-6160(54)90196-0 [13] F. N. Rhines, W. E. Bond and R. A. Rummel, “Constitu- tion of Order Alloys of the System Copper-Gold,” Transactions of the American Society of Metals, Vol. 47, 1955, pp. 578-597. [14] W. A. Oates, F. Zhang, S.-L .Chen and Y.A. Chang, “Im- proved Cluster-Site Approximation for the Entropy of Mixing in Multicomponent Solid Solutions,” Physical Re- view B, Vol. 59, No. 17, 1999, pp. 11221-11225. http://dx.doi.org/10.1103/PhysRevB.59.11221 [15] Y. Q. Xie, X. B. Liu, H. J. Peng, Y. Z. Nie, X. B. Li and Y. F. Li, “Characteristic Atom Arranging Crystallography of Alloy Phases for Au-Cu System,” Science China: Technological Sciences, Vol. 54, No. 6, 2011, pp. 1560- 1567. http://dx.doi.org/10.1007/s11431-011-4390-4 [16] Y. Q. Xie, “Atomic Energies and Gibbs Energy Functions of Ag-Cu Alloys,” Science in China (Series E), Vol. 41, No. 2, 1998, pp. 146-156. http://dx.doi.org/10.1007/BF02919677 [17] Y. Q. Xie and X. D. Zhang, “Atomic Volume and Vol- ume Functions of Ag-Cu Alloys,” Science in China (Se- ries E), Vol. 41, No. 2, 1998, pp. 157-168. http://dx.doi.org/10.1007/BF02919678 [18] Y. Q. Xie, “A New Potential Function with Many-Atom Interactions in Solid,” Science in China (Series A), Vol. 36, No.1, 1993, pp. 90-99. [19] Y. Q. Xie, “Electronic Structure and Properties of Pure Iron,” Acta Metallurgica et Materialia, Vol. 42, No. 11, 1994, pp. 3705-3715. http://dx.doi.org/10.1016/0956-7151(94)90436-7 [20] Y. Q. Xie, “Relationship between Partial and Average  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 422 Atomic Volumes of Components in Au-Ni Alloys”, Transactions of Nonferrous Metals Society of China, Vol. 21, No. 8, 2011, pp. 1801-1807. http://dx.doi.org/10.1016/S1003-6326(11)60934-5  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 423 Supplementary Information 1. Alloy Gene (AG) Theory of Au-Cu System 1.1. New Thinking Modes of System Sciences ‘A diversity of structures of a system is attributed to combination and arrangement of structure units in the basic structure unit sequence’. It is the first philosophic proposition of system sciences proposed by us. For ex- amples, the diversity of species in the biological system is attributed to splices of some biological genes in the biological gene sequences; the diversity of free atom states in the free atom system is attributed to arrangement of electrons in the electronic orbital (state) sequence de- scribed by four quantum numbers; the diversity of crys- talline species in the geometric crystallography is attrib- uted to combination of symmetric elements in the sym- metric element sequence; the diversity of music scores in the music system is attributed to combination and ar- rangement of notes in the music note sequence, and so on. This philosophic proposition guides us to seek the most important structure units for designing advanced alloys. They are the central characteristic atoms in the basic co- ordination clusters, and called as alloy genes (AG). “A diversity of properties of a system is attributed to the contents and transmission mode of the information of the basic structure unit sequences”. It is the second phi- losophic proposition of system sciences proposed by us. It is also essential condition to establish a systematic science theory of a certain system. It is a protracted and tortuous history to research structures, properties, evolu- tions and functions of the biologic gene sequences for establishing biosystematics. It is another good example to research structures, properties, evolutions and func- tions of electronic state sequences for establishing sys- tematic science of free atom system. Through a long- term exploring we have proposed a fictitious structure unit (Supplementary Section 1.3), which is the character- istic crystal consisted of the same alloy genes with known potential energy and volume, and with the same based lattice to the alloy system. Then, we have estab- lished the valence bond theory and thermodynamics of characteristic crystals. By both these theories, we can calculate the valence electron structures of characteristic crystals, which may represent ones of alloy genes, and the physical and thermodynamic properties of charac- teristic crystals, which may represent contributions of alloy genes to properties of alloys. Now, we have dis- covered that the transmission mode of the AG-in-forma- tion about structures and properties can be described by the AG-Gibbs energy partition function. 1.2. Structural Levels of Alloy Systems “The complexity and entirety of a complex system are attributed to the multilevel of the structures and proper- ties and to the various correlatives between different structural levels, between different properties and be- tween the structures and properties.” It is the third phi- losophic proposition of system sciences proposed by us. The SMMS framework contains three levels: the elec- tron-structures of atoms, atom- and electron (valence bond)-structures of phases and phase-structures of or- ganizations (Supplementary Figure S1(a)), which are simplified as atom-, phase- and organization-levels, re- spectively. The traditional crystallography and thermo- dynamics of alloys contain phase-level and organization- level. The first-principles electron theory of alloys be- longs in the electron-structures of phases (Supplemen- tary Figure S1(b)). Therefore, traditional alloy theories have no atom-level theory, i.e., AG-theory. 1.3. Structural Unit Sequences of the Au-Cu Al- loy System In order to explain diversities of structures and properties of alloy phases, we proposed three structural unit se- quences. Basic coordination cluster B-sequences: Au Au Au 012 i BBA ; Cu Cu Cu 012 i BBB . They are formed by the central characteristic atoms Au Cu , ii AA and the nearest coordinative configuration 12 iAu iCu . The alloy phase is formed by the Basic Coordination Cluster Overlapping (Simplified as BCO model) (Sup- plementary Figure S1(c)). Alloy genes A-sequences: Au Au Au 012 i AA; Cu Cu Cu 012 i AA. They are the central characteristic atoms of basic coordination clusters. The alloy phase is formed by the Alloy Gene Arranging (Simplified as AGA model) (Supplementary Figure S1(c)). Characteristic crystal C-sequences: Au Au Au 012 i CCC ; Cu Cu Cu 012 i CCC . Each fictitious characteristic crystal consists of the same characteristic atoms. The alloy phase is formed by the Characteristic Crystal Mixing (Simpli- fied as CCM model) (see Supplementary Figure S1(d), Supplementary Section 1.6). Recently, we have discovered that the alloy gene se- quences are very useful to design advanced alloys. 1.4. The Separated Theory of Potential Energies and Volumes of Alloy Genes The AG-theory consists of three parts (Supplementary Figure S2(a)): separated theory of potential energies and volumes of alloy genes [16,17], valence bond theory [18,19] and thermodynamics of characteristic crystals (see Equations (9) to (24)). The extensive properties ,qxT , , A qxT, , B qxT functions of a given alloy phase and its com- ponents can be obtained by a transmission law of the  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 424 Figure S1. Theoretic levels and three structural unit sequences. a) Theoretic levels in SMMS framework; b) Theoretic levels in the traditional alloy theory; c) Basic coordination cluster sequences and AG-sequences: BBB Au Au Au 0812 , BBB Cu CuCu 12 40 and AAA Au Au Au 0812 , AAA Cu CuCu 12 40 ; d) The characteristic crystal-sequences: CCC Au Au Au 0812 , CCC Cu CuCu 12 40 . 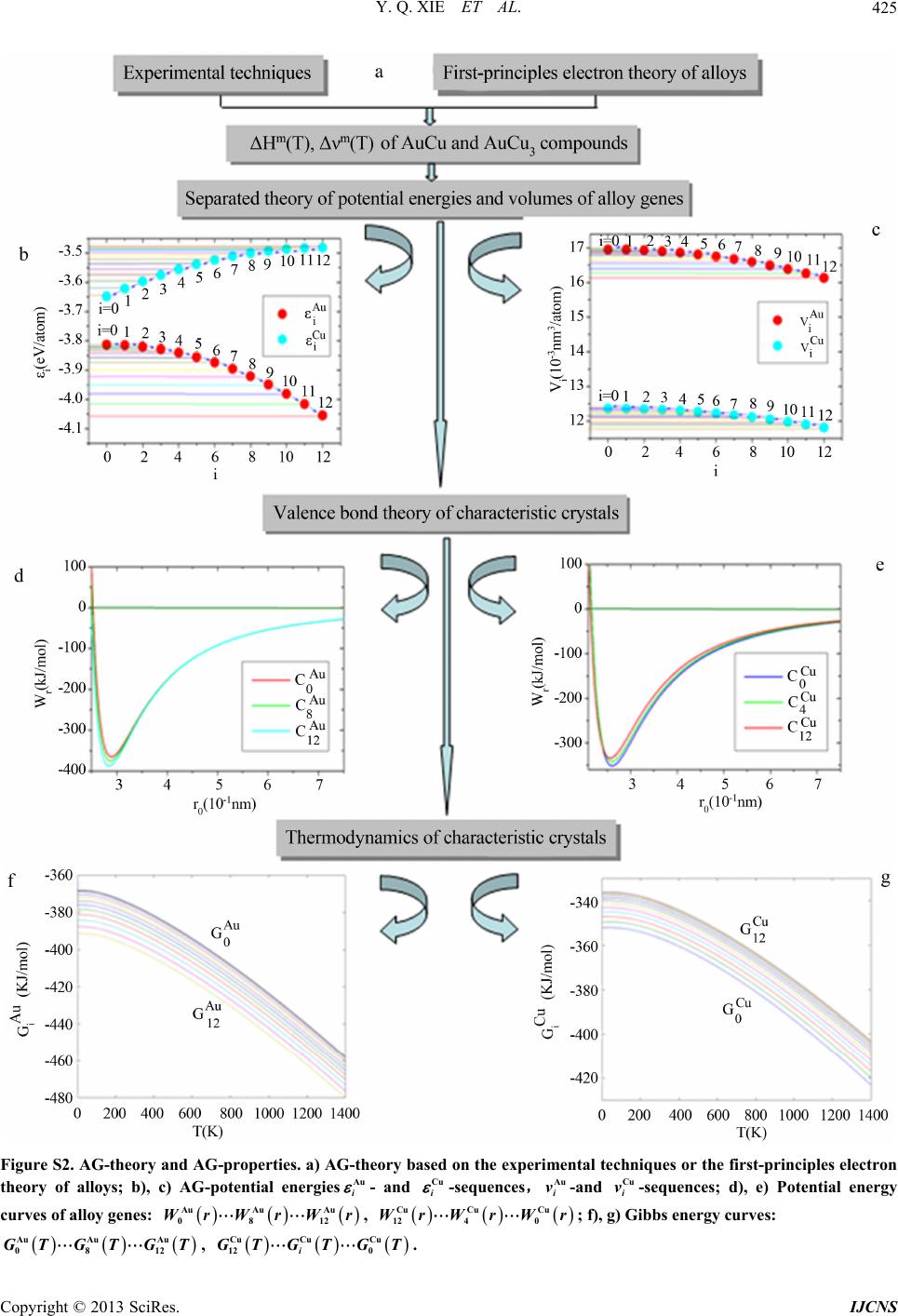 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 425 Figure S2. AG-theory and AG-properties. a) AG-theory based on the experimental techniques or the first-principles electron theory of alloys; b), c) AG-potential energiesi Au - and i Cu -sequences,i vAu -and i vCu -sequences; d), e) Potential energy curves of alloy genes: WrWrWr Au AuAu 0812 , WrWrWr Cu CuCu 12 40 ; f), g) Gibbs energy curves: GT GTGT Au Au Au 0812 , i GT GT GT Cu CuCu 12 0 .  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 426 extensive q-properties of alloy genes, which may be sim- plified as AGT law: 00 0 0 ,, , ,, ,, II AA BB iiii ii IAA AiiA i IBB BiiB i qxTxxTq TxxTq T qxTxxTqTx qxTxxTqTx (1) where, the q denotes potential energy (E) and volume (v), which are the functions as temperature (T); the ),(TxxA i and , B i xT are the concentrations of the alloy genes; which are the functions of the composition (x) and tem- perature (T); A and denote compositions of the A and B components, respectively. In order to make the Equation (1) become the simple, applicable and separable ,,qxT function, three type relations of A i q and i q with i have been designed: (i)Type I of linear relation, 00 0 AA AA iI BBB iI I qq iIqq qq IiIqq (2) (ii)Type II of concave parabola relation, 2 00 2 0 AA AA iI BBB iI I qq qIqq qq IiIqq (3) (iii)Type III of convex parabola relation, 2 00 0 0 2 0 2 2 AA AAAA iI I BB BB iI I BB I qqiI qqiIqq qq IiIqq IiIq q (4) where, the 0 A q and qdenote, respectively, q-proper- ties of the primary state alloy genes; the A q and 0 q denote, respectively, q-properties of the terminal state alloy genes. By combining Equations (2)-(4) and substituting them into Equation (1), nine ,qxT functions can be ob- tained. In Supplementary Table S1 the nine general ,qxT -functions of alloy phases can be used to com- pounds, ordered and disordered alloy phases. In Sup- plementary Table S2, the nine ,qxT -functions can be used for the disordered alloy phases only. 1.5. Methodology The most valuable method in the systematic science is “the whole obtained from a few parts”, which may be simplified as the WOP method. It means that a system can be reproduced from it’s a few parts. Such as the whole of a tree can be reproduced from a seed, a branch or even a leaf of this tree. It means also that the whole information of a system can be obtained from the infor- Table S1. Nine general qxT,-functions of alloy phases at T K*. No. Type A B Functions 1 I I 000 00 , II AB AAABBB ABIiIi I ii iIi qxTxqTxqTxqT qTxqT qT II 2 I Ⅱ 2 000 00 , II AB AAABBB ABIiIi I ii iIi qxTxqTxqTxqT qTxqT qT II 3 Ⅰ Ⅲ 2 000 2 00 2 , II AB AAABBB ABI iIiI ii II iI i i qxTxqTxqTxqT qTxqT qT II 4 Ⅱ Ⅰ 2 000 00 , II AB AAABBB ABIiIi I ii iIi qxTxqTxqTxqT qTxqT qT II 5 Ⅱ Ⅱ 22 000 00 , II AB AAABBB ABIiIi I ii iIi qxTxqTxqTxqT qTxqT qT II 6 Ⅱ Ⅲ 2 2 000 2 00 2 , II AB AAABBB ABI iIiI ii II iI i i qxTxqT xqTxqTqTx qTqT II 7 ⅢⅠ 2 000 2 00 2 , II ABAAA BBB ABIiIi I ii Ii iIi qxTxqT xqTxqT qTxqT qT II 8 ⅢⅡ 2 2 000 2 00 2 , II AB AAABBB ABIiIi I ii Ii iIi qxTxqTxqTxqT qTx qT qT II 9 Ⅲ Ⅲ 2 2 000 22 0 2 2 , I AB AAABBB ABI iIiI i II iI i Ii i qxTxqTxqTxqT qTx qT qT II *: Here, the , AA ii xxT and , BB ii xxT.  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 427 Table S2. qxT,-functions of disordered alloy phases at T K. No. Type A B Functions 1 Ⅰ I 000 ,ABAA BB ABIABI I qxTxqTxqTxxqTqTqTqT 2 ⅠⅡ 2 000 1 ,A BAAABABBB ABIABII Ixxxx qxTxqTxqTxxqTqTqTqT I 3 ⅠⅢ 2 000 1 ,ABAAABAB BB ABIABII IxxIxx qxTxqTxqTxxqTqTqTqT I 4 ⅡⅠ 2 000 1 ,AB ABABAA BB ABIIAB I Ixxxx qxTxqTxqTqTqTxxqTqT I 5 Ⅱ Ⅱ 22 000 11 ,ABAB ABAAAB ABBB ABI II Ixxxx Ixxxx qxTxqTxqTqTqTqTqT II 6 Ⅱ Ⅲ 22 000 11 ,ABABAB AAABABBB ABI II Ixxxx IxxIxx qxTxq Txq Tq Tq Tq Tq T II 7 Ⅲ Ⅰ 2 000 1 ,ABABAB AABB ABIIABI IxxIxx qxTxqTxqTqTqTxxqTqT I 8 Ⅲ Ⅱ 22 000 11 ,ABABAB AAABAB BB ABI II IxxIxx Ixxxx qxTxq Txq Tq Tq Tq Tq T II 9 Ⅲ Ⅲ 2 2 000 11 ,ABABABAAAB ABBB ABI II I xxIxxIxxIxx qxTxqTxqTqTqTqTqT II mation of it’s a few parts. Due to the discovery of alloy genes and the establish- ments of holographic alloy positioning (HAP) system and a big database consisting of the AG-database and the holographic network phase diagrams of alloy systems, a standard method for researchers to share predictive algo- rithms and computational methods may be produced. The whole information of an alloy system and a designed alloy may be obtained. This study will create a new way of intelligent design advanced alloys and lead to funda- mental variations in the metallic materials science. 1.6. The Choices of Potential Energy E-function and Volume V-function in the Au-Cu System The 5th E-function in Supplementary Table S1 has been chosen for describing Au-Cu system, according to the calculated values of the mixed enthalpies max ,0, m Hx , excess potential energies max ,0, ex Ex and the lowest critical temperatures c Tx of AuCu- and AuCu3-type sublattice ordered Au(1-x)Cux alloys with maximum order degrees, based on potential energies of the Au i - and Cu i -sequences separated from mixed enthalpies of the AuCu and AuCu3 compounds respectively by nine E-functions at room temperature ( Supplementary Figure S3). The 6th V-function in Supplementary Table S1 has been chosen for describing Au-Cu system, according to the calculated values of the mixed volumes max ,, m r vxT and excess volumes max ,, ex r vxT of the AuCu- and AuCu3-type sublattice ordered Au(1-x)Cux alloys with maximum order degrees based on volumes of the Au i - and Cu i -sequences separated from experimental vol- umes of AuCu and AuCu3 compounds respectively by the 2nd-9th v-functions at room temperature (Supple- mentary Figure S4). In the Au-Cu system, the correlative function between potential energies of alloy genes is, 2 AuAuAu Au 00 2 CuCuCu Cu 0. iI iI I iI IiI (5) The potential energy function of alloys is, Au Cu Au 0Cu 2 Au AuAu 0 0 2 Cu CuCu 0 0 ,I I iI i I iI i xTxTxT ixTT I Ii TT I (6) The correlative function between volumes of alloy genes is: 2 AuAuAuAu 00 CuCuCu Cu 0 2Cu Cu 0 2 iI iI I I vv iIvv vv IiIvv IiI vv (7) 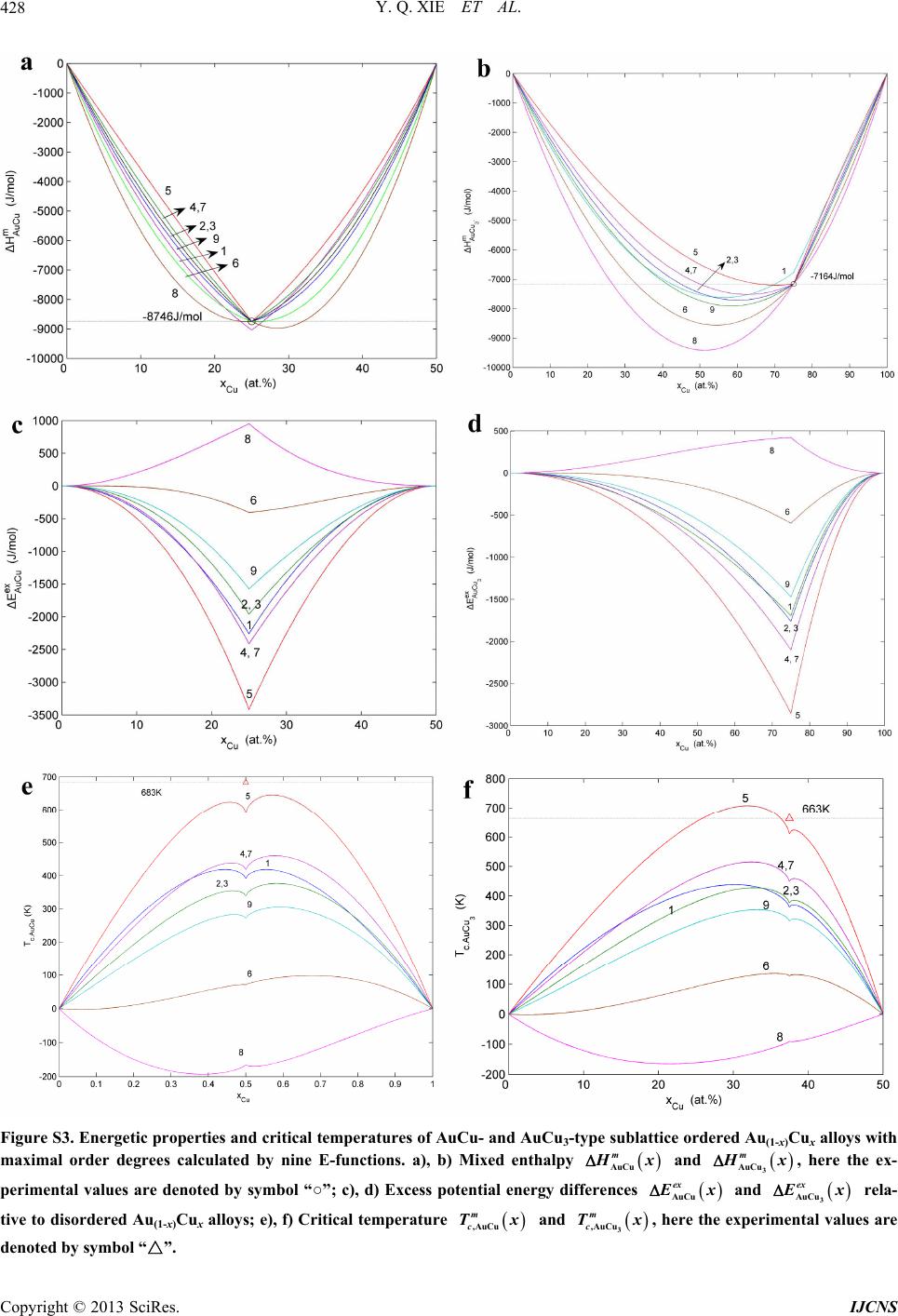 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 428 Figure S3. Energetic properties and critical temperatures of AuCu- and AuCu3-type sublattice ordered Au(1-x)Cux alloys with maximal order degrees calculated by nine E-functions. a), b) Mixed enthalpy m HxAuCu and m Hx3 AuCu , here the ex- perimental values are denoted by symbol “○”; c), d) Excess potential energy differences ex xAuCu and ex x3 AuCu rela- tive to disordered Au(1-x)Cux alloys; e), f) Critical temperature m c Tx ,AuCu and m c Tx 3 ,AuCu, here the experimental values are denoted by symbol “△”. 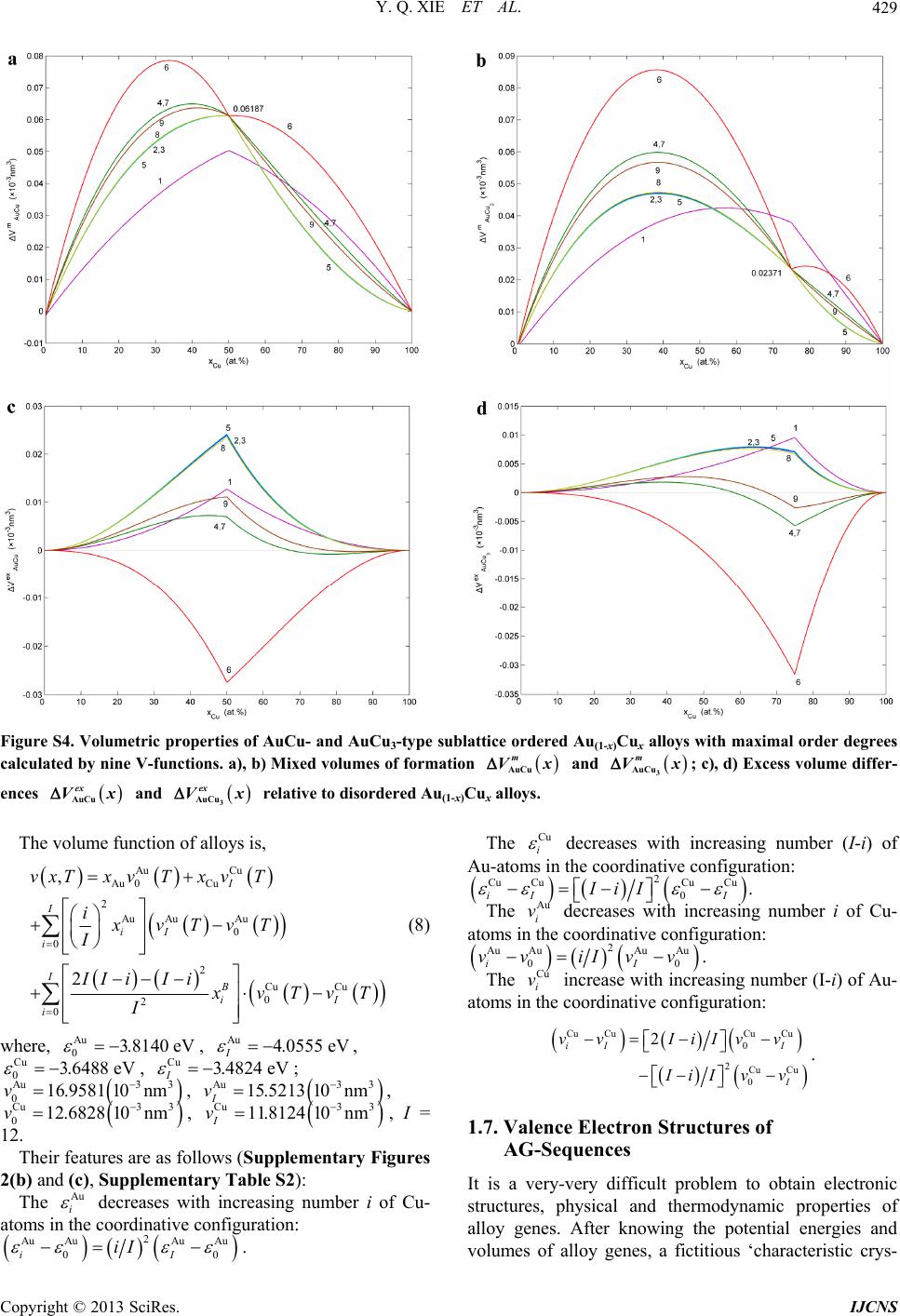 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 429 Figure S4. Volumetric properties of AuCu- and AuCu3-type sublattice ordered Au(1-x)Cux alloys with maximal order degrees calculated by nine V-functions. a), b) Mixed volumes of formation m VxAuCu and m Vx3 AuCu ; c), d) Excess volume differ- ences ex VxAuCu and ex Vx3 AuCu relative to disordered Au(1-x)Cux alloys. The volume function of alloys is, Au Cu Au 0Cu 2 Au AuAu 0 0 2 Cu Cu 0 2 0 , 2 I I iI i IB iI i vxTx vTx vT ixvTvT I II iIi vTvT I (8) where, Au 03.8140 eV , Au4.0555 eV I , Cu 03.6488 eV , Cu 3.4824 eV I ; Au3 3 016.958110nmv , Au3 3 15.5213 10nm I v , Cu3 3 012.6828 10nmv , Cu33 11.8124 10nm I v , I = 12. Their features are as follows (Supplementary Figures 2(b) and (c), Supplementary Table S2): The Au i decreases with increasing number i of Cu- atoms in the coordinative configuration: 2 Au AuAu Au 00iI iI . The Cu i decreases with increasing number (I-i) of Au-atoms in the coordinative configuration: 2 Cu CuCu Cu 0. iI I IiI The Au i v decreases with increasing number i of Cu- atoms in the coordinative configuration: 2 Au AuAu Au 00iI vv iIvv . The Cu i v increase with increasing number (I-i) of Au- atoms in the coordinative configuration: Cu CuCu Cu 0 2Cu Cu 0 2 iI I I vv IiIvv IiI vv . 1.7. Valence Electron Structures of AG-Sequences It is a very-very difficult problem to obtain electronic structures, physical and thermodynamic properties of alloy genes. After knowing the potential energies and volumes of alloy genes, a fictitious ‘characteristic crys- 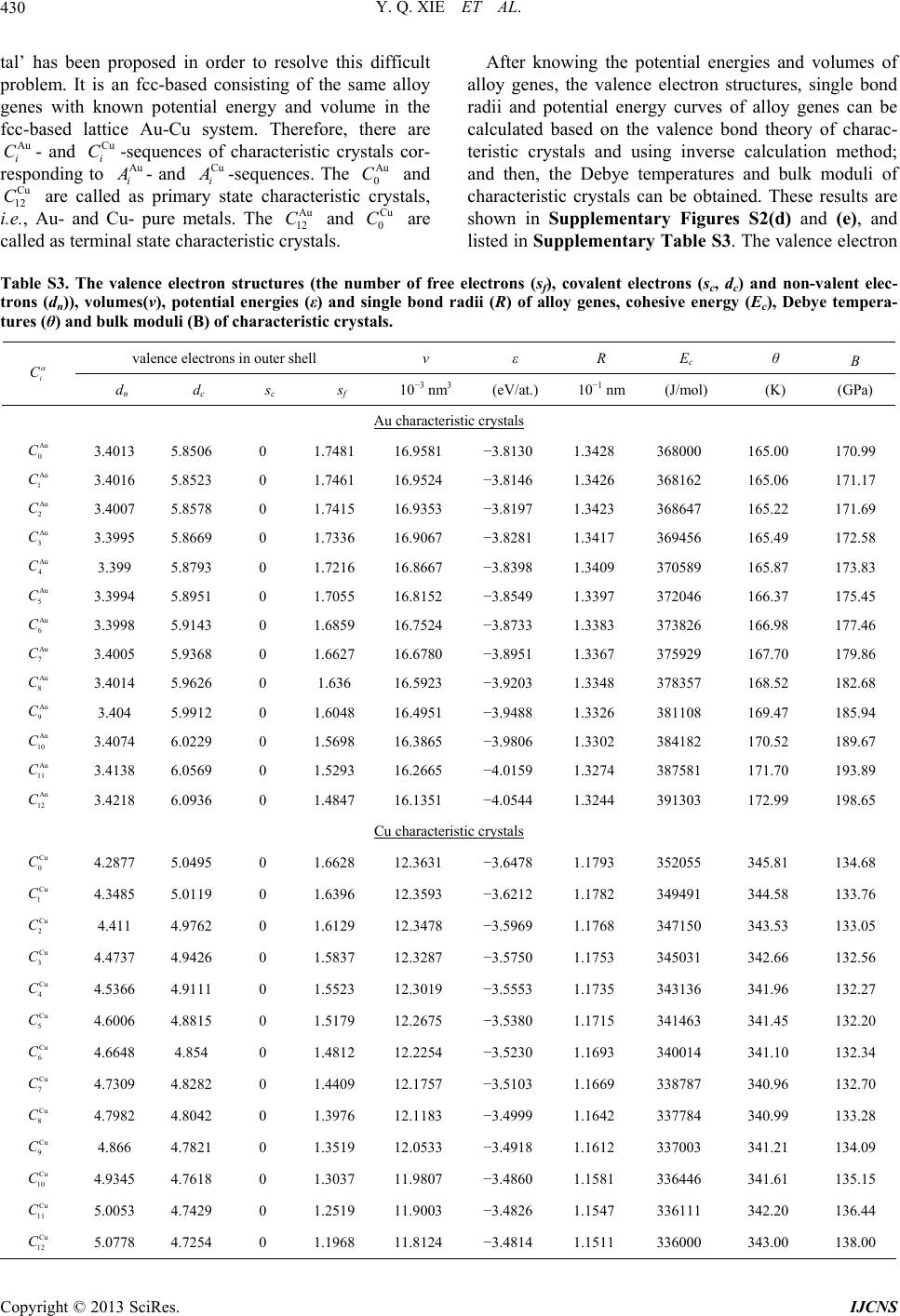 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 430 tal’ has been proposed in order to resolve this difficult problem. It is an fcc-based consisting of the same alloy genes with known potential energy and volume in the fcc-based lattice Au-Cu system. Therefore, there are Au i C- and Cu i C-sequences of characteristic crystals cor- responding to Au i - and Cu i -sequences. The Au 0 C and Cu 12 C are called as primary state characteristic crystals, i.e., Au- and Cu- pure metals. The Au 12 C and Cu 0 C are called as terminal state characteristic crystals. After knowing the potential energies and volumes of alloy genes, the valence electron structures, single bond radii and potential energy curves of alloy genes can be calculated based on the valence bond theory of charac- teristic crystals and using inverse calculation method; and then, the Debye temperatures and bulk moduli of characteristic crystals can be obtained. These results are shown in Supplementary Figures S2(d) and (e), and listed in Supplementary Table S3. The valence electron Table S3. The valence electron structures (the number of free electrons (sf), covalent electrons (sc, dc) and non-valent elec- trons (dn)), volumes(v), potential energies (ε) and single bond radii (R) of alloy genes, cohesive energy (Ec), Debye tempera- tures (θ) and bulk moduli (B) of characteristic crystals. valence electrons in outer shell v ε R Ec θ B i C dn d c s c s f 10−3 nm3 (eV/at.) 10−1 nm(J/mol) (K) (GPa) Au characteristic crystals Au 0 C 3.4013 5.8506 0 1.7481 16.9581 −3.8130 1.3428 368000 165.00 170.99 Au 1 C 3.4016 5.8523 0 1.7461 16.9524 −3.8146 1.3426 368162 165.06 171.17 Au 2 C 3.4007 5.8578 0 1.7415 16.9353 −3.8197 1.3423 368647 165.22 171.69 Au 3 C 3.3995 5.8669 0 1.7336 16.9067 −3.8281 1.3417 369456 165.49 172.58 Au 4 C 3.399 5.8793 0 1.7216 16.8667 −3.8398 1.3409 370589 165.87 173.83 Au 5 C 3.3994 5.8951 0 1.7055 16.8152 −3.8549 1.3397 372046 166.37 175.45 Au 6 C 3.3998 5.9143 0 1.6859 16.7524 −3.8733 1.3383 373826 166.98 177.46 Au 7 C 3.4005 5.9368 0 1.6627 16.6780 −3.8951 1.3367 375929 167.70 179.86 Au 8 C 3.4014 5.9626 0 1.636 16.5923 −3.9203 1.3348 378357 168.52 182.68 Au 9 C 3.404 5.9912 0 1.6048 16.4951 −3.9488 1.3326 381108 169.47 185.94 Au 10 C 3.4074 6.0229 0 1.5698 16.3865 −3.9806 1.3302 384182 170.52 189.67 Au 11 C 3.4138 6.0569 0 1.5293 16.2665 −4.0159 1.3274 387581 171.70 193.89 Au 12 C 3.4218 6.0936 0 1.4847 16.1351 −4.0544 1.3244 391303 172.99 198.65 Cu characteristic crystals Cu 0 C 4.2877 5.0495 0 1.6628 12.3631 −3.6478 1.1793 352055 345.81 134.68 Cu 1 C 4.3485 5.0119 0 1.6396 12.3593 −3.6212 1.1782 349491 344.58 133.76 Cu 2 C 4.411 4.9762 0 1.6129 12.3478 −3.5969 1.1768 347150 343.53 133.05 Cu 3 C 4.4737 4.9426 0 1.5837 12.3287 −3.5750 1.1753 345031 342.66 132.56 Cu 4 C 4.5366 4.9111 0 1.5523 12.3019 −3.5553 1.1735 343136 341.96 132.27 Cu 5 C 4.6006 4.8815 0 1.5179 12.2675 −3.5380 1.1715 341463 341.45 132.20 Cu 6 C 4.6648 4.854 0 1.4812 12.2254 −3.5230 1.1693 340014 341.10 132.34 Cu 7 C 4.7309 4.8282 0 1.4409 12.1757 −3.5103 1.1669 338787 340.96 132.70 Cu 8 C 4.7982 4.8042 0 1.3976 12.1183 −3.4999 1.1642 337784 340.99 133.28 Cu 9 C 4.866 4.7821 0 1.3519 12.0533 −3.4918 1.1612 337003 341.21 134.09 Cu 10 C 4.9345 4.7618 0 1.3037 11.9807 −3.4860 1.1581 336446 341.61 135.15 Cu 11 C 5.0053 4.7429 0 1.2519 11.9003 −3.4826 1.1547 336111 342.20 136.44 Cu 12 C 5.0778 4.7254 0 1.1968 11.8124 −3.4814 1.1511 336000 343.00 138.00  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 431 structures of characteristic crystals may represent ones of alloy genes and their physical properties may represent the contributions of alloy genes to properties of alloys. In the free atom system, the electron structures of at- oms are described by four quantum numbers. The out- shell electron structures of the Au- and Cu- free atoms at the basic states are respectively 10 1 56ds and 10 1 34ds. The electronic structure units of the alloy genes are valence electrons described by four quantum numbers: free electron , covalent electron ,,, cccc pd f, magnetic electron , mm df and non-valence electron ,, nnn pd f. In the Au-Cu system, the valence electron structures of AG-sequences are listed in Supplementary Table S3. The valence electron structures of the primary states Au Cu 012 , and terminal states Au Cu 12 0 , of alloy genes are as follows: Au3.4013 5.85061.7481 0556 ncf dds ; Au3.4218 6.0936 1.4847 12 556 ncf dd s . Cu5.0778 4.7254 1.1968 12 334 nc f dds ; Cu4.2877 5.04954 1.6628 033 4 nc f dd s . The essence of properties of alloy genes can be ex- plained by their valence electron structures. The essence for forming fcc-based lattice: As changing from Au 0 to Au 12 ,the n d-electrons slightly increase (3.4012/atom → 3.4218/atom)and occupy at the d-energy band e state; and the dc-electrons obviously increase (5.8506/atom → 6.0936/atom)and occupy at the d-energy band g t2 state. Therefore, each Au i -atom possesses 12 covalent bonds to form fcc-based lattice. As changing from Cu 12 to Cu 0 ,the dn-electrons obviously decrease (5.0778/atom → 4.2877/atom), few of them occupy at the d-energy band 2 t state, besides filling the e state fully; the dc-electrons obviously in- crease (4.7254/atom → 5.0495/atom)and occupy at the d-energy band 2 t state. Each Cu i -atom possesses 12 covalent bonds to form fcc-based lattice. For these reasons, the intermetallics, ordered and dis- ordered alloys formed by Au i - and Cu i -sequences possess fcc-based lattice. The essence for variations in potential energies and volumes of alloy genes: As changing from Au 0 to Au 12 , the -electrons decrease: 1.7481/atom → 1.4847/atom, and the de- creased -electrons are translated into dc-electrons. It results in falling Au 12 potential energy: −3.813 eV/atom → −4.0544eV/atom, shorting single bond radus: 1.3428 × 10−1 nm → 1.3244 × 10−1 nm, and reducing Au 12 v volume: 16.9581 × 10−3 nm3 → 16.1351 × 10−3 nm3. Therefore, The Au i -potential energies decrease and the Au i v-volumes reduce with increasing number i of Cu- atoms in the coordinative configurations. As changing from Cu 12 to Cu 0 , the -electrons increase: 1.1968/atom → 1.6628/atom,and the dc- elec- trons increase: 4.7254/atom → 5.0495/atom. It results in falling Cu 0 potential energy: −3.4814 eV/atom → −3.6478 eV/atom, lengthening single bond radius: 1.1511 × 10−1 nm → 1.1793 × 10−1 nm, and expanding Cu 0 v volume: 11.8124 × 10−3 nm3 → 12.3631 × 10−3 nm3. Therefore, the Cu i -potential energies decrease and Cu i v-volumes increases with increasing number (I-i) of Au-atoms in the coordinative configurations, that is con- trary to the expected behavior. 1.8. The Thermodynamics Properties of AG-Sequences In the SMMS framework, the Gibbs energy i GT of each characteristic crystal (or ally gene) may be split into two parts: a temperature-independent contribution of po- tential energy 0E, of which the variation with tem- peratures has been accounted in the attaching vibration energy, and a temperature-dependent contribution of generalized vibration free energy v T, but both are configuration(i)-dependent (Equation (9)). The enthalpy i T of each characteristic crystal may be also split into two parts: a temperature-independent contribution of potential energy 0E and a temperature-dependent contribution of generalized vibration energy v UT (Equation (10)). The generalized vibration free energy includes the generalized vibration energies v UT , which include Debye vibration energies D UT and attaching vibration energies E UT, and the contribu- tion of the generalized vibration entropies v ST , which include Debye vibration entropies D ST and attaching vibration entropies E ST,the generalized vibration heat capacity v p CT, which include Debye vibration heat capacity D p CT, attaching vibration heat capacity E p CT. The attaching vibration energy includes contributions of electron excitation, energy of formation of holes, variation of potential energy with temperature, and so on. Therefore, the multi-level ener- getic functions of characteristic crystals at T K as fol- lows: AuAuAu.AuAu. CuCu Cu.CuCu. 0 0 vv iiii i vv iiii i GT EXTHTTST GT EXTHTTST (9) AuAu Au. CuCuCu. 0 0 v iii v iii TEU T TEUT (10) Au. Au.Au. Cu. Cu.Cu. vv v ii i vv v ii i TUTTST TUTTS T (11)  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 432 Au. Au.Au. Cu. Cu.Cu. DD D ii i DD D ii i TU TTS T TUTTS T (12) Au. Au.Au. Cu. Cu.Cu. EE E ii i EE E ii i TUTTS T TU TTST (13) Au. Au. Au. Cu. Cu.Cu. vDE iii vDE iii UTU TUT UTUTU T (14) Au. Au. . 0 Cu. Cu. . 0 d d T DD ipi T DD ipi UTCTT UTCTT (15) Au. Au. . 0 Cu. Cu. . 0 () d d T EE ipi T EE ipi UTCTT UTCTT (16) Au. Au.Au. Cu. Cu.Cu. vDE iii vDE iii STSTS T STSTS T (17) Au. . Au. 0 Cu. . Cu. 0 ()d d D T pi D i D T pi D i CT ST T T CT ST T T (18) Au. . Au. 0 Cu. . Cu. 0 d d E T pi E i E T pi E i CT STT T CT ST T T (19) 23 23 Au AuAuAuAuAu 000 23 23 Cu CuCuCuCuCu 12 12 00 00 0000 iii iI ii EVEV EV EV (20) Au Cu 34 Au. .Au 2 0 34 Cu. .Cu 2 0 e 9d e1 e 9d e1 i i x T D pi x i x T D pi x i Tx CTR x Tx CTR x (21) Au.106 2 .0 Au.46 2 .12 1.858993 104.22258410 2.763134 105.481502 10 E p E p CTTT CTTT (22) Cu.6 2 .12 Cu.62 .0 0.001680352.826912 10 0.005646143.632353 10 E p E p CT TT CT TT (23) where, Au 0165 K , Au 12 175.21 K , Cu 12 343 K , Cu 0343.07 K . It can be demonstrated that the general correlative functions between other energetic properties (q) of char- acteristic crystals (ally genes) at T K are as follows 2 AuAuAuAu 00 2 CuCuCuCu 0I iI iI qT qTiI qTqT qT qTIiIqTqT (24) The AG-thermodynamic properties of characteristic crystals (ally genes) of Au-Cu system are shown in Sup- plementary Figure S5. 2. Alloy Gene Gibbs Energy Partition Function In this section we present the AG-Gibbs energy partition function of AuCu-type sublattice alloy system, which includes AG-Gibbs energy transmission law, and AG- arranging function, as well as other functions of thermo- dynamic properties. Characteristic Gibbs energy function: The characteristic Gibbs energy *,GxT function can be obtained by the AGT law (S1): 12 AuAu CuCu 0 ,, , iiii i GxTxxTGTxxT GT (25) Partition function of the alloy: Au Cu * ,,,, exp , ii TgxxTxxT GxT kT (26) 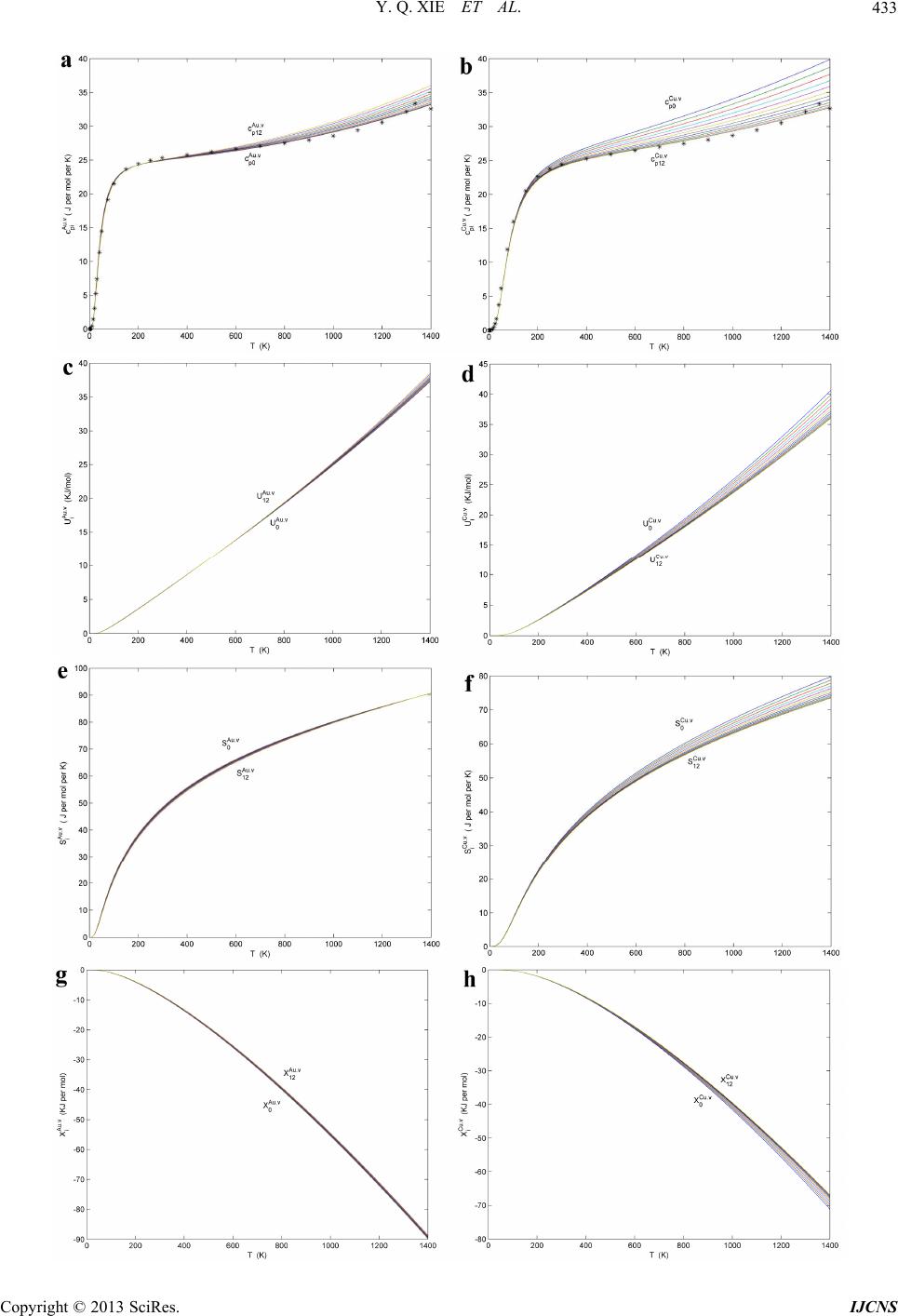 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 433 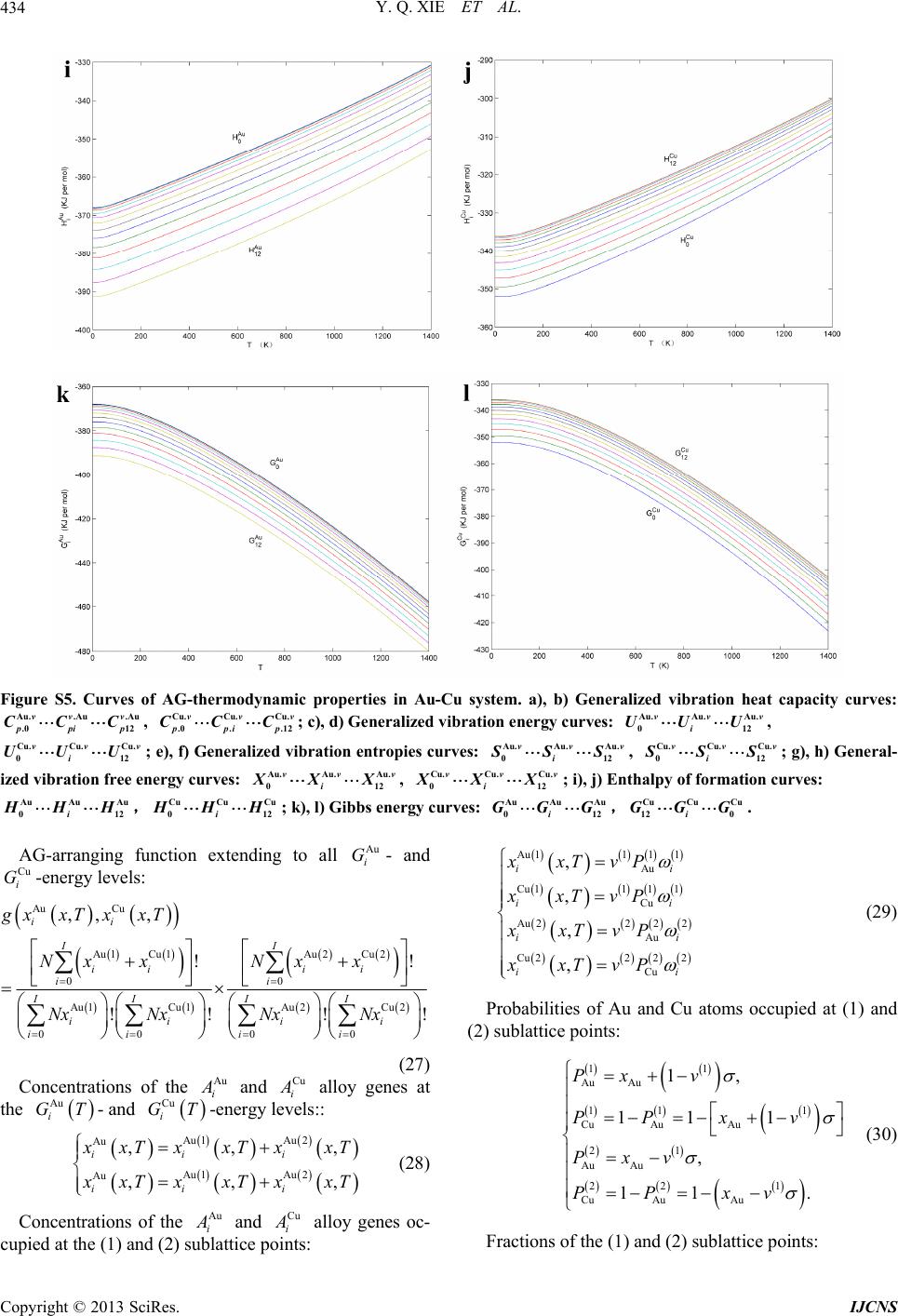 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 434 Figure S5. Curves of AG-thermodynamic properties in Au-Cu system. a), b) Generalized vibration heat capacity curves: vvv ppip CCC Au..Au .Au .0 12 , vvv ppip CCC Cu. Cu. Cu. .0 ..12 ; c), d) Generalized vibration energy curves: vvv i UUU Au. Au. Au. 012 , vvv i UUU Cu. Cu. Cu. 012 ; e), f) Generalized vibration entropies curves: vvv i SSS Au. Au. Au. 012 , vvv i SSS Cu. Cu.Cu. 012 ; g), h) General- ized vibration free energy curves: vvv i XXX Au. Au. Au. 012 , vvv i XXX Cu. Cu.Cu. 012 ; i), j) Enthalpy of formation curves: i HHH Au Au Au 012 ,i HHH Cu Cu Cu 012 ; k), l) Gibbs energy curves: i GGG Au Au Au 012 ,i GGG Cu CuCu 12 0 . AG-arranging function extending to all Au i G- and Cu i G-energy levels: Au Cu Au 1Cu 1Au 2Cu2 00 Au 1Cu 1Au2Cu 2 0000 ,, , !! !! !! ii II iii i ii III I iii i iiii gxxTxxT Nx xNxx Nx NxNxNx (27) Concentrations of the Au i and Cu i alloy genes at the Au i GT- and Cu i GT-energy levels:: Au 1Au2 Au Au 1Au2 Au ,,, ,,, ii i ii i xTxxTxxT xTxxTxxT (28) Concentrations of the Au i and Cu i alloy genes oc- cupied at the (1) and (2) sublattice points: Au 1111 Au Cu 1111 Cu Au 2222 Au Cu 2222 Cu , , , , ii ii ii ii xxTvP xxTvP xxTvP xxTvP (29) Probabilities of Au and Cu atoms occupied at (1) and (2) sublattice points: 11 Au Au 11 1 Cu AuAu 21 Au Au 22 1 Cu AuAu 1, 11 1 , 11 . Px v PPx v Pxv PP xv (30) Fractions of the (1) and (2) sublattice points:  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 435 11 22 12 12 vNN vNN (31) Definition of order degree: 12 Au1Cu2 Au AuCuCuAuCu 1212 121221 11 ii ii PxPxxxxx vvvvwvvvwv (32) The probability 1 i of the coordinative cluster Au,CuIii surrounding the (1)-sublattice point: 84 1 122 211 8AuCu4AuCu Kiiii ii ii i ii ii ik CPPC PP (33) In the 1 i equation, the probability 2 i of co- ordinative cluster 8Au,Cuii occupied at the (2)-sublattice points, and the probability 2 ii of co- ordinative cluster 4Au,Cuii ii occupied at the (1)-sublattice points: 8 222 8Au Cu 4 111 4AuCu Kii i i ik Kiiii ii ii ik CP P CP P (34) where, the i is the number of the Cu atoms occupied at the (2)-sublattice points. The probability 2 i of coordinative cluster Au,CuIii surrounding the (2)-sublattice point: 84 2 211122 8AuCu4 AuCu Kiiii ii ii i ii ii ik CPPCPP (35) where, the i is the number of the Cu atoms occupied at the (1)-sublattice points. In the 2 i equation, the probability 1 i of co- ordinative cluster 8Au,Cuii occupied at the (1)- sublattice points, and the probability 2 ii of coordina- tive cluster 4Au,Cuii ii occupied at the (2)-sublattice points: 8 111 8Au Cu ' 4 222 4Au Cu ' Kii i i ik Kiiii ii ii ik CPP CP P (36) In the Equations (32) to (35), k and K take their values, according to 0,,if 4; 4,,if 48; 4,8,if 812. kKii ki Kii ki Ki (37) The maximum order degree max as function of com- position Au : For the stoichiometric alloy, due to 1 Au 1P, max 1 ; For the alloys with 1 Au v, due to 2 Au 0P , 1 max Au xv ; For the alloys with 1 Au v, due to 1 Au 1P, 1 max Au 11 xv . The methods for calculating 1 i and 2 i are listed in Supplementary Tables S4 and S5. The configurational entropies of alloy and compo- nents: Au CuAu.Cu. Au Cu Au 111Au222 Au. Au Au 000 0 Au Cu 111Cu2 Cu. Cu 00 Cu ,ln,, , ,ln,ln ,ln, ccc Bii III I c iiii iii i II c iii iii SxTkgx xxSxTxSxT R SxxTPxxTP x R Sx xTPxxT x 22 Cu 00 ln II i i P (38) 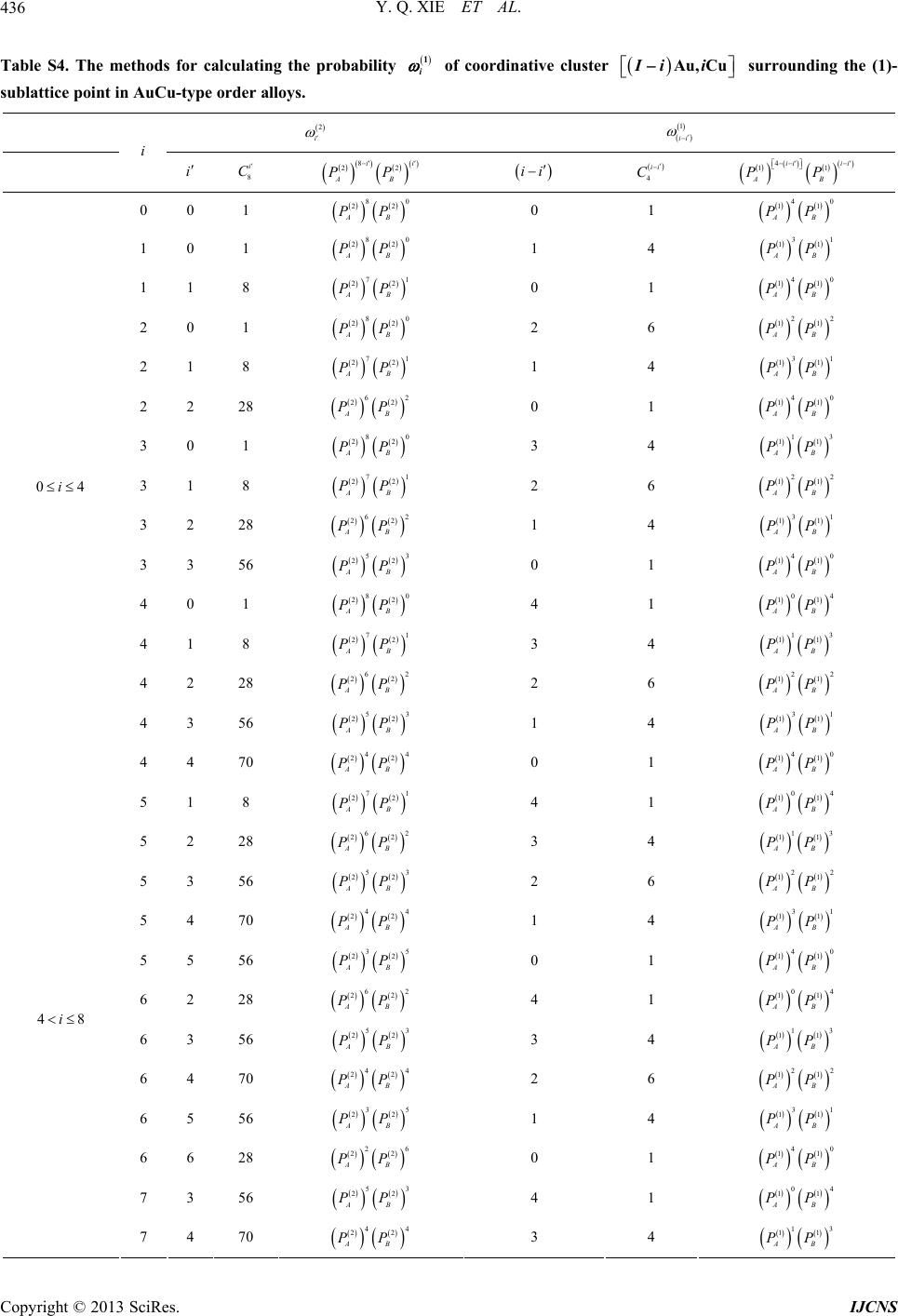 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 436 Table S4. The methods for calculating the probability i 1 of coordinative cluster Ii iAu, Cu surrounding the (1)- sublattice point in AuCu-type order alloys. 2 i 1 ii i i 8 i C 8 22 ii AB PP ii 4 ii C 4 11 ii ii AB PP 0 0 1 80 22 AB PP 0 1 40 11 AB PP 1 0 1 80 22 AB PP 1 4 31 11 AB PP 1 1 8 71 22 AB PP 0 1 40 11 AB PP 2 0 1 80 22 AB PP 2 6 22 11 AB PP 2 1 8 71 22 AB PP 1 4 31 11 AB PP 2 2 28 62 22 AB PP 0 1 40 11 AB PP 3 0 1 80 22 AB PP 3 4 13 11 AB PP 3 1 8 71 22 AB PP 2 6 22 11 AB PP 3 2 28 62 22 AB PP 1 4 31 11 AB PP 3 3 56 53 22 AB PP 0 1 40 11 AB PP 4 0 1 80 22 AB PP 4 1 04 11 AB PP 4 1 8 71 22 AB PP 3 4 13 11 AB PP 4 2 28 62 22 AB PP 2 6 22 11 AB PP 4 3 56 53 22 AB PP 1 4 31 11 AB PP 04i 4 4 70 44 22 AB PP 0 1 40 11 AB PP 5 1 8 71 22 AB PP 4 1 04 11 AB PP 5 2 28 62 22 AB PP 3 4 13 11 AB PP 5 3 56 53 22 AB PP 2 6 22 11 AB PP 5 4 70 44 22 AB PP 1 4 31 11 AB PP 5 5 56 35 22 AB PP 0 1 40 11 AB PP 6 2 28 62 22 AB PP 4 1 04 11 AB PP 6 3 56 53 22 AB PP 3 4 13 11 AB PP 6 4 70 44 22 AB PP 2 6 22 11 AB PP 6 5 56 35 22 AB PP 1 4 31 11 AB PP 6 6 28 26 22 AB PP 0 1 40 11 AB PP 7 3 56 53 22 AB PP 4 1 04 11 AB PP 48i 7 4 70 44 22 AB PP 3 4 13 11 AB PP  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 437 Continued 7 5 56 35 22 AB PP 2 6 22 11 AB PP 7 6 28 26 22 AB PP 1 4 31 11 AB PP 7 7 8 17 22 AB PP 0 1 40 11 AB PP 8 4 70 44 22 AB PP 4 1 04 11 AB PP 8 5 56 35 22 AB PP 3 4 13 11 AB PP 8 6 28 26 22 AB PP 2 6 22 11 AB PP 8 7 8 17 22 AB PP 1 4 31 11 AB PP 8 8 1 08 22 AB PP 0 1 40 11 AB PP 9 5 56 35 22 AB PP 4 1 04 11 AB PP 9 6 28 26 22 AB PP 3 4 13 11 AB PP 9 7 8 17 22 AB PP 2 6 22 11 AB PP 9 8 1 08 22 AB PP 1 4 31 11 AB PP 10 6 28 26 22 AB PP 4 1 04 11 AB PP 10 7 8 17 22 AB PP 3 4 13 11 AB PP 10 8 1 8)2(0)2( )()( BAPP 2 6 22 11 AB PP 11 7 8 17 22 AB PP 4 1 04 11 AB PP 11 8 1 08 22 AB PP 3 4 13 11 AB PP 812i 12 8 1 08 22 AB PP 4 1 04 11 AB PP The mole potential energies of the alloy and compo- nents: Au Cu Au Cu 12 AuAu Au Au 0 12 CuCuCu Cu 0 ,,, ,1, 0 ,1, 0 I ii i I ii i ExTxExTx ExT ExT xxxTE ExTxxxTE (39) The Debye vibration energies of alloy and compo- nents: Au. Cu. Au Cu 12 Au.Au Au. Au , 0 0 12 Cu.CuCu. Cu , 0 0 ,,, ,1 ,d ,1 ,d DDD IT DD ipi i IT DD ipi i UxTxUxTxXxT UxT xxxTCTT UxTxxxTCTT (40) The attaching vibration energies of alloy and compo- nents: Au. Cu. Au Cu 12 Au.Au Au. Au , 0 0 12 Cu.Cu Cu. Cu , 0 0 ,,, ,1 ,d ,1 ,d EEE IT EE ipi i IT EE ipi i UxTxUxT xXxT UxT xxxTCTT UxTxxxTCTT (41) The generalized vibration energies of the alloy and components: Au. Cu. Au Cu 12 Au.Au Au. Au 0 12 Cu.Cu Cu. Cu 0 ,,, ,1, ,1 , vvv I vv ii i I vv ii i UxTxUxT xUxT UxT xxxTUT UxTxxxTUT (42) 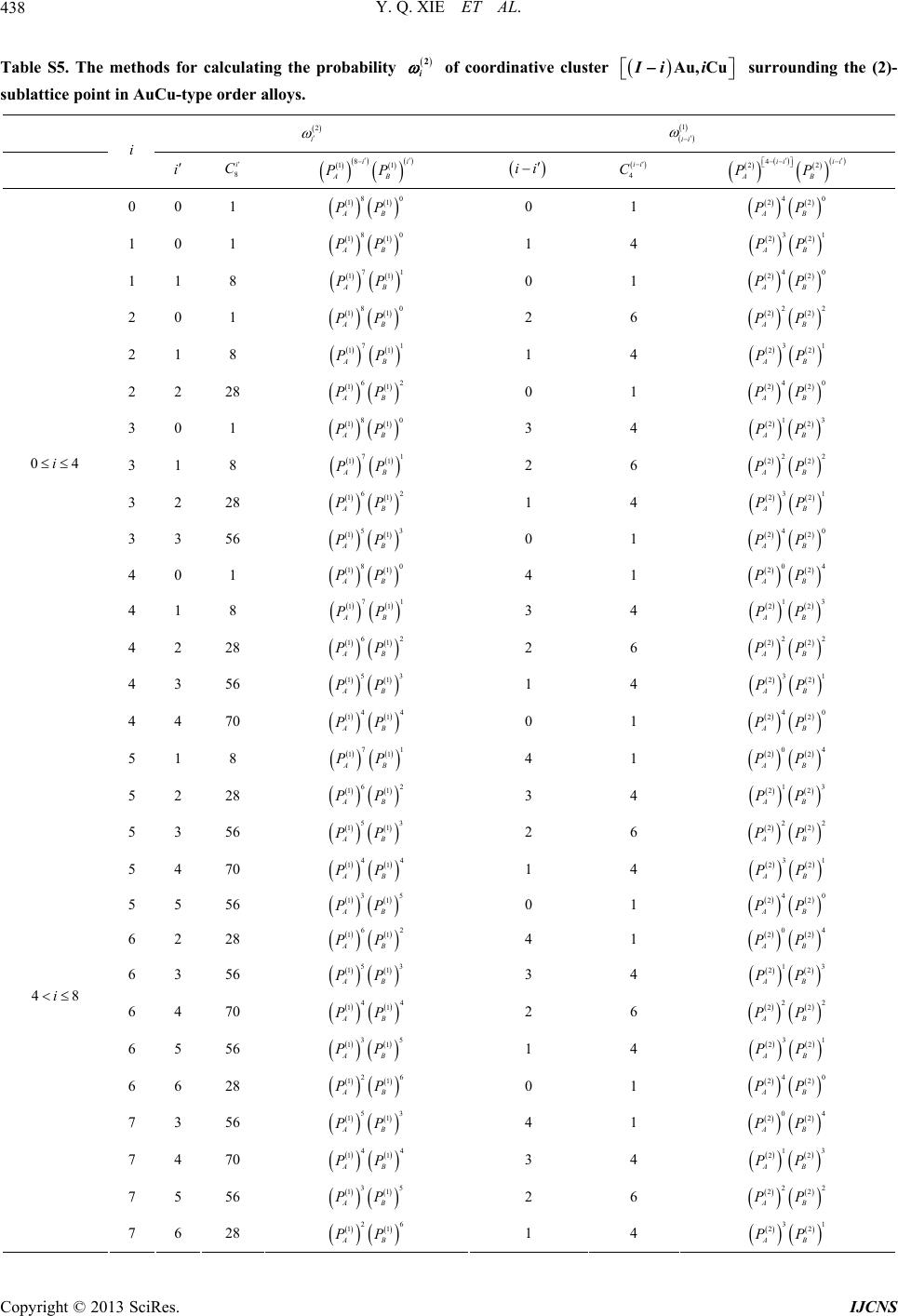 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 438 Table S5. The methods for calculating the probability i 2 of coordinative cluster Ii iAu, Cu surrounding the (2)- sublattice point in AuCu-type order alloys. 2 i 1 ii i i 8 i C 8 11 ii AB PP ii 4 ii C 4 22 ii ii AB PP 0 0 1 80 11 AB PP 0 1 40 22 AB PP 1 0 1 80 11 AB PP 1 4 31 22 AB PP 1 1 8 71 11 AB PP 0 1 40 22 AB PP 2 0 1 80 11 AB PP 2 6 22 22 AB PP 2 1 8 71 11 AB PP 1 4 31 22 AB PP 2 2 28 62 11 AB PP 0 1 40 22 AB PP 3 0 1 80 11 AB PP 3 4 13 22 AB PP 3 1 8 71 11 AB PP 2 6 22 22 AB PP 3 2 28 62 11 AB PP 1 4 31 22 AB PP 3 3 56 53 11 AB PP 0 1 40 22 AB PP 4 0 1 80 11 AB PP 4 1 04 22 AB PP 4 1 8 71 11 AB PP 3 4 13 22 AB PP 4 2 28 62 11 AB PP 2 6 22 22 AB PP 4 3 56 53 11 AB PP 1 4 31 22 AB PP 04i 4 4 70 44 11 AB PP 0 1 40 22 AB PP 5 1 8 71 11 AB PP 4 1 04 22 AB PP 5 2 28 62 11 AB PP 3 4 13 22 AB PP 5 3 56 53 11 AB PP 2 6 22 22 AB PP 5 4 70 44 11 AB PP 1 4 31 22 AB PP 5 5 56 35 11 AB PP 0 1 40 22 AB PP 6 2 28 62 11 AB PP 4 1 04 22 AB PP 6 3 56 53 11 AB PP 3 4 13 22 AB PP 6 4 70 44 11 AB PP 2 6 22 22 AB PP 6 5 56 35 11 AB PP 1 4 31 22 AB PP 6 6 28 26 11 AB PP 0 1 40 22 AB PP 7 3 56 53 11 AB PP 4 1 04 22 AB PP 7 4 70 44 11 AB PP 3 4 13 22 AB PP 7 5 56 35 11 AB PP 2 6 22 22 AB PP 48i 7 6 28 26 11 AB PP 1 4 31 22 AB PP  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 439 Continued 7 7 8 17 11 AB PP 0 1 40 22 AB PP 8 4 70 44 11 AB PP 4 1 04 22 AB PP 8 5 56 35 11 AB PP 3 4 13 22 AB PP 8 6 28 26 11 AB PP 2 6 22 22 AB PP 8 7 8 17 11 AB PP 1 4 31 22 AB PP 8 8 1 08 22 AB PP 0 1 40 22 AB PP 9 5 56 35 11 AB PP 4 1 04 22 AB PP 9 6 28 26 11 AB PP 3 4 13 22 AB PP 9 7 8 17 11 AB PP 2 6 22 22 AB PP 9 8 1 08 22 AB PP 1 4 31 22 AB PP 10 6 28 26 11 AB PP 4 1 04 22 AB PP 10 7 8 17 11 AB PP 3 4 13 22 AB PP 10 8 1 08 22 AB PP 2 6 22 22 AB PP 11 7 8 17 11 AB PP 4 1 04 22 AB PP 11 8 1 08 22 AB PP 3 4 13 22 AB PP 812i 12 8 1 08 22 AB PP 4 1 04 22 AB PP The Debye vibration entropies of the alloy and com- ponents: Au. Au Cu. Cu Au. 12 , Au. Au Au 0 0 Cu. 12 , Cu. Cu Cu 0 0 ,, , ,1 ,d ,1 ,d DD D D ITpi D i i D ITpi D i i SxT xSxT xS xT CT SxT xxxTT T CT SxTxxxTT T (43) The attaching vibration entropies of the alloy and components: Au. Au Cu. Cu Au. 12 , Au. Au Au 0 0 Cu. 12 , Cu. Cu Cu 0 0 ,, , ,1 ,d ,1 ,d EE E E ITpi E i i E ITpi E i i SxTxSxT xS xT CT SxTxxxTT T CT SxT xxxTT T (44) The generalized vibration entropies of the alloy and components: Au. Cu. Au Cu 12 Au.AuAu. Au 0 12 Cu.Cu Cu. Cu 0 ,,, ,1, ,1 , vvv I vv ii i I vv ii i SxTxSxTxSxT SxTxxxTST SxT xxxTST (45) The generalized vibration free energies of the alloy and components: Au. Cu. Au Cu 12 Au.AuAu. Au 0 12 Cu.Cu Cu. Cu 0 ,,, ,1 , ,1 , vvv I vv ii i I vv ii i xTxXxTx XxT xTxxxTXT xTxxxT XT (46) The enthalpies of the alloy and components: Au Cu Au Cu 12 AuAuAu Au. Au 0 12 CuCuCuCu. Cu 0 ,,, ,1, 0 ,1, 0 Iv iii i Iv iii i HxTx HxTx HxT xTxxxT EUT xTxxxTEUT (47)  Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 440 The characteristic Gibbs energies of the alloy and components: Au Cu Au Cu 12 AuAu Au Au 0 12 CuCuCu Cu 0 ,,, ,1, ,1 , I ii i I ii i GxTxGxT xGxT GxT xxxTGT GxT xxxTGT (48) The Gibbs energies of the alloy and components: Au Cu Au Cu Au*Au Au. Au Cu*CuCu. Cu ,,, ,1 , , ,1 ,, c c GxTxGxTx GxT GxTxGxTTSxT GxTx G xTTSxT (49) The mixed heat capacity of the alloy: .. .AuAu.Au. ,,0 0 CuCu. Cu. ,,12 0 Au .AuAu 0 0 Cu Cu Cu 12 0 ,,, ,, , ,d ,d ,d d mmvms pp p I mvv v pipip i Ivv ipip i Ii ms pi i Ii i i CxTC xTC xT CxTxxTC TCT xxTCTC T xxT CxT HTHT T xxT THT T (50) The activities of the components: * AuAu Au * CuCu Cu 12 *AuAuAu Au Au 0 0 12 *CuCuCu Cu Cu 0 ,exp, ,exp, ,1 , ,1 , m m I ii i I iiI i axT xGxTRT axT xGxTRT GxT xxxTGTGT GxT xxxTGTGT (51) The volumes of the alloy and components: Au Cu Au Cu 12 AuAu Au Au 0 12 CuCu Cu Cu 0 ,,, ,1, ,1 , I ii i I ii i vxTx vxTxvxT vxT xxxTvT ExTxxxTvT (52) The mixed volume thermal expansion coefficient of the alloy: .. .AuAu. Au.CuCu. Cu. 012 00 Au Cu .AuAuCuCu 012 Au Cu 0 0 ,,, ,,, ,, 1d 1d ,dd mmvms II mvv vv v ii ii ii I I i i ms i i i i i i xTxT xT xTxxTTTx xTTT xxT xxT T vTvTvTvT TT vT vT (53) It should be emphasized that the new , T-func- tion can be used to describe both long-distance order and short-distance order states of alloy phases, because our model includes the sublattice model and coordinative model (Equation (32)), and that, the thermodynamic properties of alloy components need no representatives by the partial molar properties. We have proved that the partial molar properties cannot represent the average properties of the corresponding components [20]. 3. EHNP Diagrams of Thermodynamic Properties of AuCu-type Sublattice Alloy System According to three-dimension m GxT and T EHNP diagrams, we have obtained other qxT EHNP diagrams of the AuCu-type sublattice system in Supplementary Figure S6. It should be emphasized that from each three-dimen- sion qxT EHNP diagram we can obtain isocompo- 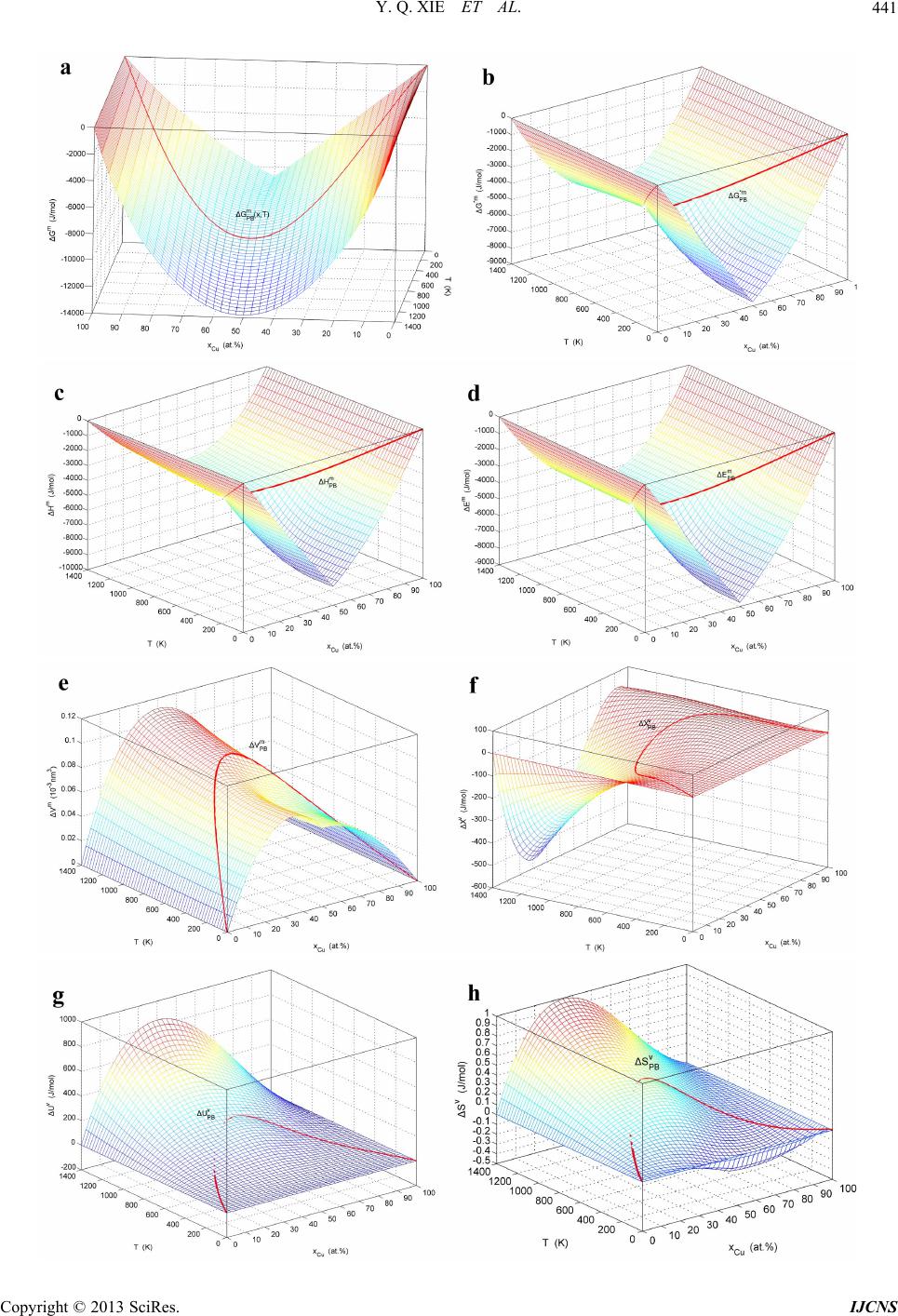 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 441 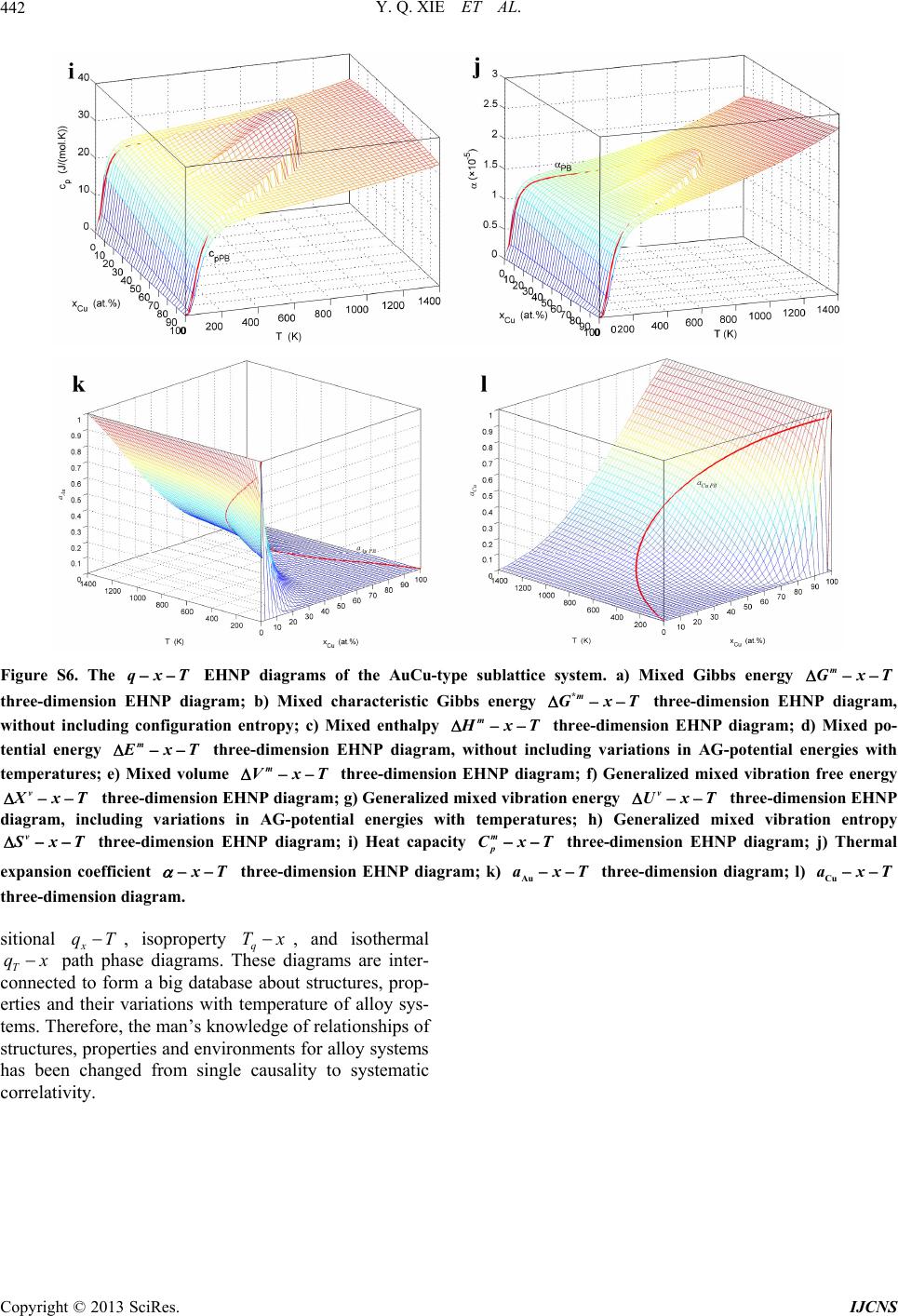 Y. Q. XIE ET AL. Copyright © 2013 SciRes. IJCNS 442 Figure S6. The xT EHNP diagrams of the AuCu-type sublattice system. a) Mixed Gibbs energy m GxT three-dimension EHNP diagram; b) Mixed characteristic Gibbs energy m GxT * three-dimension EHNP diagram, without including configuration entropy; c) Mixed enthalpy m HxT three-dimension EHNP diagram; d) Mixed po- tential energy m xT three-dimension EHNP diagram, without including variations in AG-potential energies with temperatures; e) Mixed volume m VxT three-dimension EHNP diagram; f) Generalized mixed vibration free energy v xT three-dimension EHNP diagram; g) Generalized mixed vibration energy v UxT three-dimension EHNP diagram, including variations in AG-potential energies with temperatures; h) Generalized mixed vibration entropy v xT three-dimension EHNP diagram; i) Heat capacity m p CxT three-dimension EHNP diagram; j) Thermal expansion coefficient xT three-dimension EHNP diagram; k) axT Au three-dimension diagram; l) axT Cu three-dimension diagram. sitional x qT, isoproperty q Tx, and isothermal T qx path phase diagrams. These diagrams are inter- connected to form a big database about structures, prop- erties and their variations with temperature of alloy sys- tems. Therefore, the man’s knowledge of relationships of structures, properties and environments for alloy systems has been changed from single causality to systematic correlativity.
|