S. YANG ET AL.
Copyright © 2013 SciRes. ENG
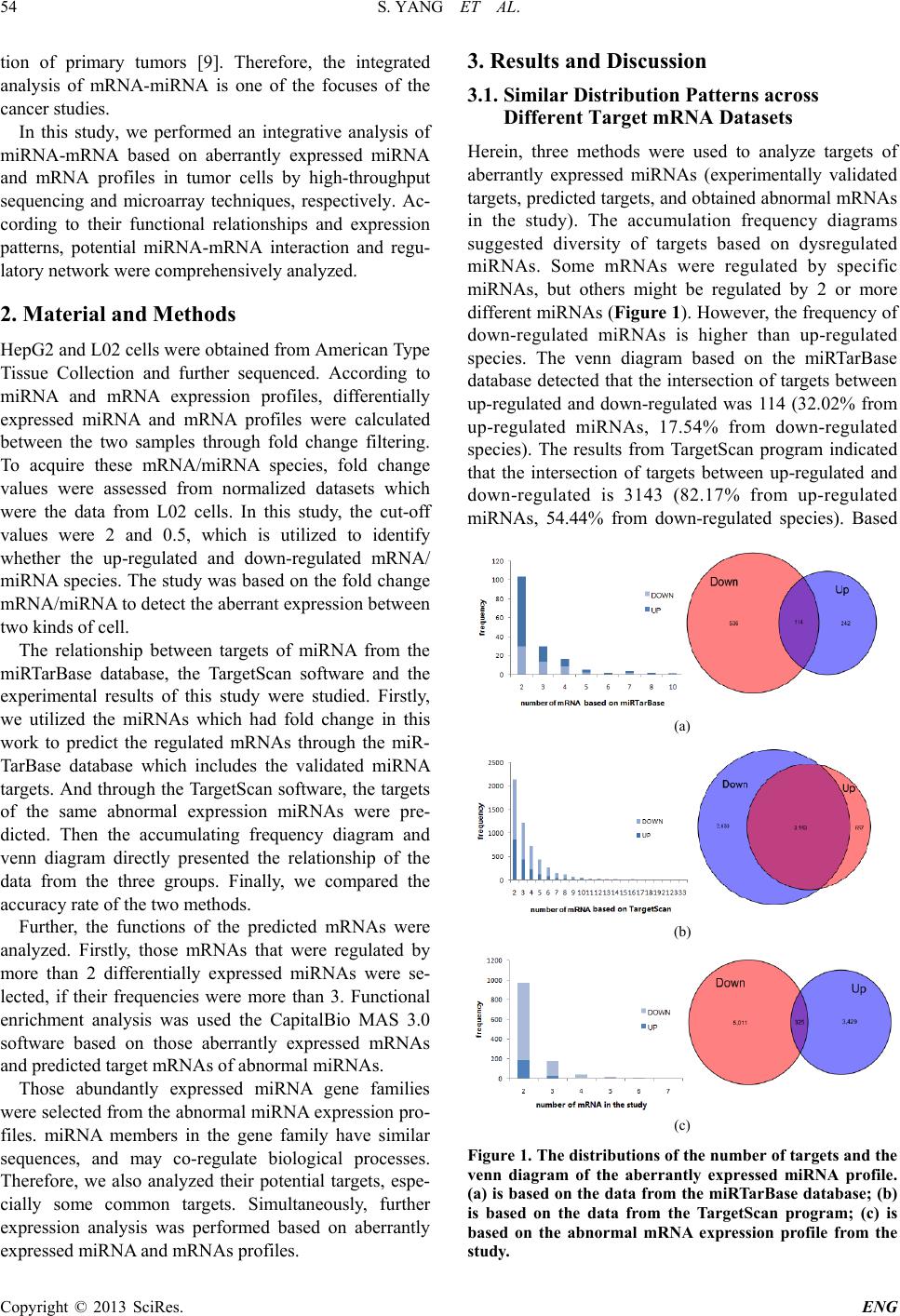
Figure 2. The experimentally validated targte mRNAs of
hsa-miR-200a-3p and hsa-miR -200b-3p (members in mir-8
gene family). mRNAs or mi RNAs in red are up-regulated in
tumor cells; mRNAs in blue are down-regulated; and
mRNAs in black stably express between normal cells. Fur-
thermore, deregulated miRNAs or mRNAs are also high-
lighted fold change values.
terns in special time and space in vivo. The co-regulation
processes will contribute to the robust network with mul-
tiple different molecules. Systematic analysis across dif-
ferent regulatory molecules should be crucial to study
tumorigenesis at the multiple molecular levels, including
mRNAs, miRNAs, lncRNA s, and etc.
REFERENCES
[1] E. Enerly, I. Steinfeld, K. Kleivi, S. K. Leivonen, M. R.
Aure, H. G. Russnes, J. A. Ronneberg, H. Johnsen, R.
Navon, E. Rodland, R. Makela, B. Naume, M. Perala, O.
Kallioniemi, V. N. Kristensen, Z. Yakhini and A. L. Bor-
resen-Dale, “miRNA-mRNA Inte grated Analysis Reveals
Roles for miRNAs in Primary Breast Tumor s,” PLoS One,
Vol. 6, 2011, Article ID: e16915.
http://dx.doi.org/10.1371/journal.pone.0016915
[2] F. Xie, P. Xiao, D. Chen, L. Xu and B. Zhang, “miRDeep
Finder: A miRNA Analysi s Tool for Deep Sequencing of
Plant Small RNAs,” Plant Molecular Biology, 2012.
[3] D. P. Bartel, “MicroRNAs: Target Recognition and Reg-
ulatory Functions,” Cell, Vol. 136, 2009, pp. 215-233.
http://dx.doi.org/10.1016/j.cell.2009.01.002
[4] C. Vaz, H. M. Ahmad, P. Shar ma, R. Gupta , L. Kumar, R.
Kulshreshtha and A. Bhattacharya, “Analysis of micro-
RNA Transcriptome by Deep Sequencing of Small RNA
Libraries of Peripheral Blood,” BMC Genomics, Vol. 11,
2010, p. 288 . http://dx.doi.org/10.1186/1471-2164-11-288
[5] J. A. Broderick, W. E. Salomon, S. P. Ryder, N. Aronin
and P. D. Zamore, “Argonaute Protein Identity and Pair-
ing Geometry Determine Cooperativity in Mammalian
RNA Silencing,” RNA —A Public ation of the RNA Soci ety,
Vol. 17, 2011, pp. 1858-1869.
http://dx.doi.org/10.1261/rna.2778911
[6] F. M. Buffa, C. Camps, L. Winchester, C. E. Snell, H. E.
Gee, H. Sheldon, M. Taylor, A. L. Harris and J. Ragous-
sis, “microRNA-Associated Progression Pathways and
Potential Therapeutic Targets Identified by Integrated
mRNA and microRNA Expression Profiling in Breast
Cancer,” Cancer Research, Vol. 71, 2011, pp. 5635-5645.
http://dx.doi.org/10.1158/0008-5472.CAN-11-0489
[7] R. Rosell, M. A. Molina, C. Costa, S. Simonetti, A. Gi-
menez-Capitan, J. Bertran-Alamillo, C. Mayo, T. Moran,
P. Mendez, F. Cardenal, D. Isla, M. Provencio, M. Cobo,
A. Insa, R. Garcia-Campelo, N. Reguart, M. Majem, S.
Viteri, E. Carcereny, R. Porta, B. Massuti, C. Queralt, I.
de Aguirre, J. M. Sanchez, M. Sanchez-Ronco, J. L. Mate,
A. Ariza, S. Benlloch, J. J. Sanchez, T. G. Bivona, C. L.
Sawyers and M. Taron, “Pretreatment EGFR T790M
Mutation and BRCA1 mRNA Expression in Erlotinib-
Treated Advanced Non-Small-Cell Lung Cancer Patients
with EGFR Mutations,” Clinical Cancer Research, Vol.
17, 2011, pp. 1160-1168.
http://dx.doi.org/10.1158/1078-0432.CCR-10-2158
[8] A. Font, M. Taron, J. L. Gago, C. Costa, J. J. Sanchez, C.
Carrato, M. Mora, P. Celiz, L. Perez, D. Rodriguez, A.
Gimenez-Capitan, V. Quiroga, S. Benlloch, L. Ibarz and
R. Rosell, “BRCA1 mRNA Expression and Outcome to
Neoadjuvant Cisplatin-Based Chemotherapy in Bladder
Cancer,” Annals of Oncology, Vol. 22, 2011, pp. 139-144.
http://dx.doi.org/10.1093/annonc/mdq333
[9] A. M. Sieuwerts, B. Mostert, J. Bolt-de Vries, D. Peeters,
F. E. de Jongh, J. M. Stouthard, L. Y. Dirix, P. A. van
Dam, A. Van Galen, V. de Weerd, J. Kraan, P. van der
Spoel, R. Ramirez-Moreno, C. H. van Deurzen, M. S mid,
J. X. Yu, J. Jia ng, Y . Wang, J. W. Gratama, S. Sl eijfer, J.
A. Foekens and J. W. Martens, “mRNA and microRNA
Expression Profiles in Circulating Tumor Cells and Pri-
mary Tumors of Metastatic Breast Cancer Patients,” Cli-
nical Cancer Research, Vol. 17, 2011, pp. 3600-3618.
http://dx.doi.org/10.1158/1078-0432.CCR-11-0255
[10] H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono and M.
Kanehisa, “KEGG: Kyoto Encyclopedia of Genes and
Genomes,” Nucleic Acids Research, Vol. 27, 1999, pp.
29-34. http://dx.doi.org/10.1093/nar/27.1.29
[11] H. Yamaguchi and J. Condeelis, “Regulati on of the Actin
Cytoskeleton in Cancer Cell Migration and Invasion,”
Biochimica et Biophysica Acta, Vol. 1773, 2007, pp. 642-
652. http://dx.doi.org/10.1016/j.bbamcr.2006.07.001
[12] A. Lachenmayer, C. Alsinet, R. Savic, L. Cabellos, S.
Toffanin, Y. Hoshida, A. Villanueva, B. Minguez, P.
Newell, H. W. Tsai , J. Barretina, S. Thung, S. C. Ward, J.
Bruix, V. Mazzaferro, M. Schwartz, S. L. Friedman and J.
M. Llovet, “Wnt-Pathway Activation in Two Molecular
Classes of Hepatocellular Carcinoma and Experimental
Modulation by Sorafenib,” Clinical Cancer Research,
Vol. 18, 2012, pp. 4997-5007.
http://dx.doi.org/10.1158/1078-0432.CCR-11-2322