 Open Journal of Medicinal Chemistry, 2013, 3, 55-59 http://dx.doi.org/10.4236/ojmc.2013.33008 Published Online September 2013 (http://www.scirp.org/journal/ojmc) -Galactosyl Phytosphingosine 2,6’-Diamide as an Inducer of Invariant Natural Killer T Cell Ying-Cheng Huang1, Wei-Ting Chen2, Shu-Fan Tien2, Ho-Lien Huang2, Chun-Nan Yeh3, Kun-I Lin2,4*, Chung-Shan Yu2,5* 1Department of Neurosurgery, Chang Gung Memorial Hospital at Linkou, Chang Gung University, Taoyuan, Taiwan 2Department of Biomedical Engineering and Environmental Sciences, National Tsing-Hua University, Hsinchu, Taiwan 3Department of Surgery, Chang Gung Memorial Hospital at Linkou, Chang Gung University, Taoyuan, Taiwan 4Department of Obsterics and Gynecology, Chang Bing Show Chwan Memorial Hospital, Changhua, Taiwan 5Institute of Nuclear Engineering and Science, National Tsing-Hua University, Hsinchu, Taiwan Email: *csyu@mx.nthu.edu.tw Received April 15, 2013; revised May 14, 2013; accepted May 28, 2013 Copyright © 2013 Ying-Cheng Huang et al. This is an open access article distributed under the Creative Commons Attribution Li- cense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Four -galactosyl phytosphingosine 2,6’-diamide analogs were prepared from 2,6’-diamino -galactosylphytosphingo- sine and the aromatic-bearing carboxylic acids. After purification with High Performance Liquid Chromatography, a flowcytometry for the four compounds for stimulation of human V 24+/V 11+ NKT cell populations was carried out. Additional keto groups on the acyl chains of the 2,6’-diamide compound were associated with the enhanced stimulating effect. Keywords: Phytosphingosine; HPLC; Keto; Flow Cytometry; Simulation 1. Introduction -galactosyl ceramide ( -GalCer) [1,2] has been associ- ated with the treatment of immunological disorders such as certain tumors, infections and autoimmune diseases (Figure 1) [3-6]. Its immune-stimulation potency is im- plicated in the activation of the invariant natural killer T cells (iNKT cells) to trigger the release of cytokines. The semi-invariant T cell receptor (TCR) which is encoded by an invariant V 24-J 18 chain in humans recognizes the complex formed from glycolipid antigen and MHC class I-like protein CD1d. After binding, iNKT cells could be induced, thereby leading to a rapid release of the relevant proinflammatory cytokines such as IFN- and IL-4 which are correlated with Th1 and Th2 pathway, respectively. Because of the opposing activities affected by the simultaneously secreted Th1 and Th2 cytokines that may counteract the therapeutic applications of - GalCer, [7] endeavor has been continuously devoted to the development of potent -GalCer analogues with a biased Th1 and Th2 profile [8-12]. We have recently reported a chemical preparation of 2,6’-diamino -galactosyl phytosphingosine ( -GalSph) analogue that could be coupled with carboxylic acids to generate a library consisting of 40 members of 2,6’-dia- mide -GalSph analogues. [13] Among them, the com- pound that exerted minimal cytotoxicity could induce a moderate proliferation of iNKT cells (Figure 2). Because the potential compound bears two aromatic groups, a further study based on this structural feature is therefore pursued. The present work is aimed to prepare the four diamide compounds 1-4 with structural variation on the linker or the aromatic ring (Figure 3). The bioactivities of the four compounds were also addressed to correlate with their structures (SAR). 2. Results and Discussion Preparation of 6-amino -galactosyl phytosphingosine 5 could be found in the relevant work as described before. [13] Conjugation of 6-amino -galactosyl phytosphin- gosine 5 with four carboxylic acids furnished the diamide products 1-4 with a fair yield (27% - 46%). Whereas a number of syntheses of ceramide analogs have been re- ported, the concern about purity has been rarely addres- sed. [14] In our case, the four products were still insuffi- ciently pure after flash chromatography. Further purifica- tion with High Performance Liquid Chromatography *Corresponding authors. C opyright © 2013 SciRes. OJMC 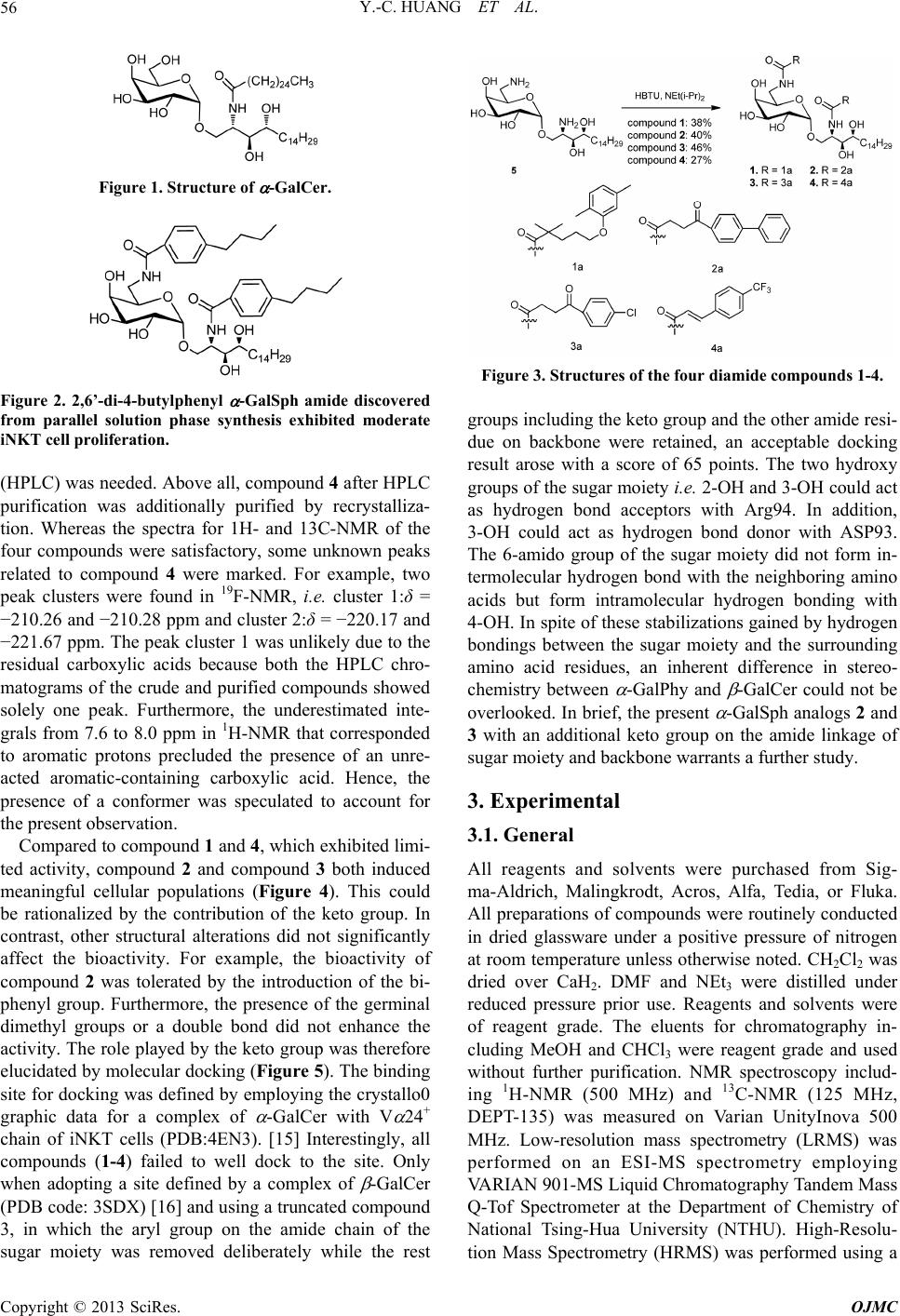 Y.-C. HUANG ET AL. 56 Figure 1. Structure of -GalCer. Figure 2. 2,6’-di-4-butylphenyl -GalSph amide discovered from parallel solution phase synthesis exhibited moderate iNKT cell proliferation. (HPLC) was needed. Above all, compound 4 after HPLC purification was additionally purified by recrystalliza- tion. Whereas the spectra for 1H- and 13C-NMR of the four compounds were satisfactory, some unknown peaks related to compound 4 were marked. For example, two peak clusters were found in 19F-NMR, i.e. cluster 1:δ = −210.26 and −210.28 ppm and cluster 2:δ = −220.17 and −221.67 ppm. The peak cluster 1 was unlikely due to the residual carboxylic acids because both the HPLC chro- matograms of the crude and purified compounds showed solely one peak. Furthermore, the underestimated inte- grals from 7.6 to 8.0 ppm in 1H-NMR that corresponded to aromatic protons precluded the presence of an unre- acted aromatic-containing carboxylic acid. Hence, the presence of a conformer was speculated to account for the present observation. Compared to compound 1 and 4, which exhibited limi- ted activity, compound 2 and compound 3 both induced meaningful cellular populations (Figure 4). This could be rationalized by the contribution of the keto group. In contrast, other structural alterations did not significantly affect the bioactivity. For example, the bioactivity of compound 2 was tolerated by the introduction of the bi- phenyl group. Furthermore, the presence of the germinal dimethyl groups or a double bond did not enhance the activity. The role played by the keto group was therefore elucidated by molecular docking (Figure 5). The binding site for docking was defined by employing the crystallo0 graphic data for a complex of -GalCer with V 24+ chain of iNKT cells (PDB:4EN3). [15] Interestingly, all compounds (1-4) failed to well dock to the site. Only when adopting a site defined by a complex of -GalCer (PDB code: 3SDX) [16] and using a truncated compound 3, in which the aryl group on the amide chain of the sugar moiety was removed deliberately while the rest Figure 3. Structures of the four diamide compounds 1-4. groups including the keto group and the other amide resi- due on backbone were retained, an acceptable docking result arose with a score of 65 points. The two hydroxy groups of the sugar moiety i.e. 2-OH and 3-OH could act as hydrogen bond acceptors with Arg94. In addition, 3-OH could act as hydrogen bond donor with ASP93. The 6-amido group of the sugar moiety did not form in- termolecular hydrogen bond with the neighboring amino acids but form intramolecular hydrogen bonding with 4-OH. In spite of these stabilizations gained by hydrogen bondings between the sugar moiety and the surrounding amino acid residues, an inherent difference in stereo- chemistry between -GalPhy and -GalCer could not be overlooked. In brief, the present -GalSph analogs 2 and 3 with an additional keto group on the amide linkage of sugar moiety and backbone warrants a further study. 3. Experimental 3.1. General All reagents and solvents were purchased from Sig- ma-Aldrich, Malingkrodt, Acros, Alfa, Tedia, or Fluka. All preparations of compounds were routinely conducted in dried glassware under a positive pressure of nitrogen at room temperature unless otherwise noted. CH2Cl2 was dried over CaH2. DMF and NEt3 were distilled under reduced pressure prior use. Reagents and solvents were of reagent grade. The eluents for chromatography in- cluding MeOH and CHCl3 were reagent grade and used without further purification. NMR spectroscopy includ- ing 1H-NMR (500 MHz) and 13C-NMR (125 MHz, DEPT-135) was measured on Varian UnityInova 500 MHz. Low-resolution mass spectrometry (LRMS) was performed on an ESI-MS spectrometry employing VARIAN 901-MS Liquid Chromatography Tandem Mass Q-Tof Spectrometer at the Department of Chemistry of National Tsing-Hua University (NTHU). High-Resolu- tion Mass Spectrometry (HRMS) was performed using a Copyright © 2013 SciRes. OJMC 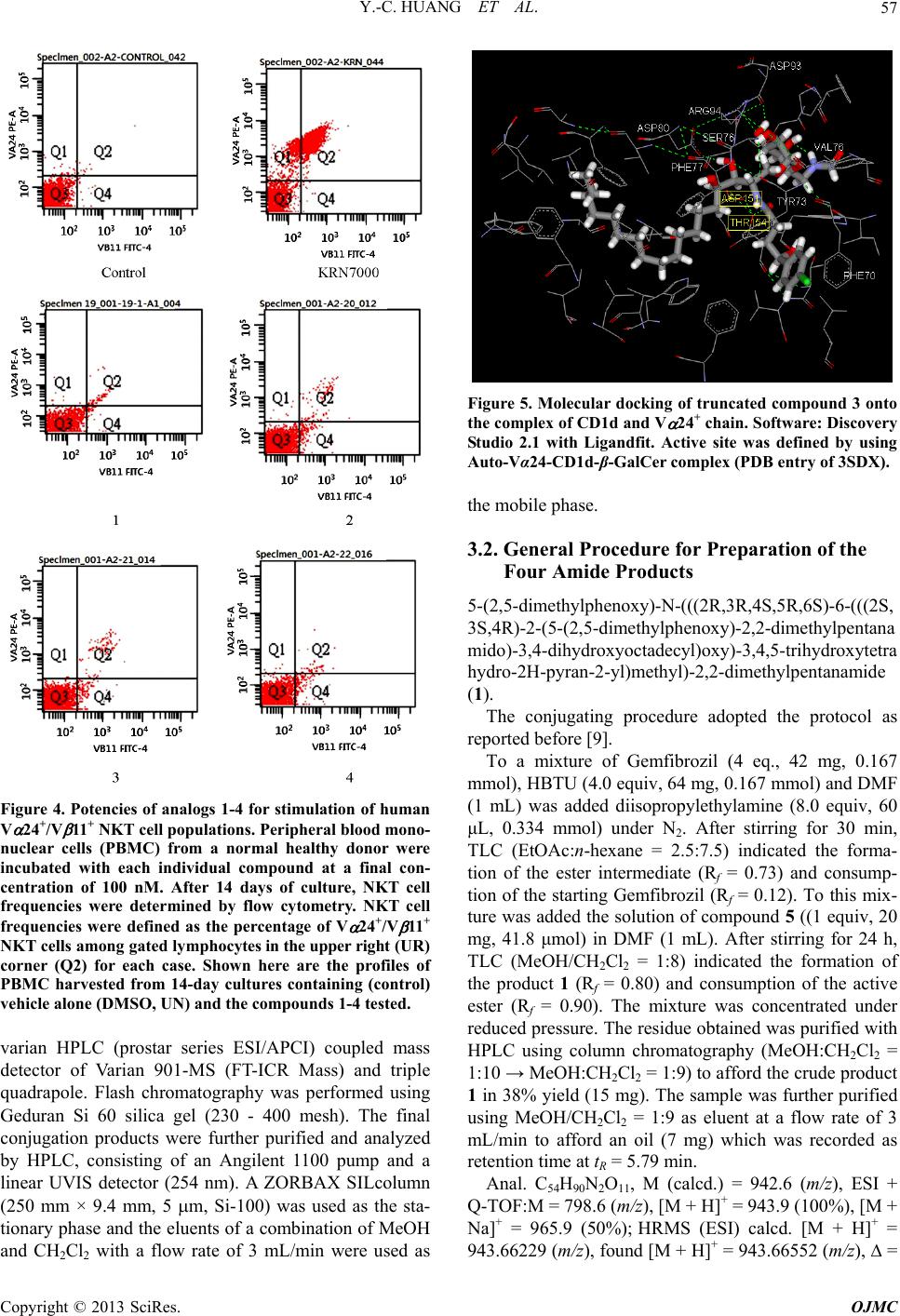 Y.-C. HUANG ET AL. 57 Figure 4. Potencies of analogs 1-4 for stimulation of human V 24+/V 11+ NKT cell populations. Peripheral blood mono- nuclear cells (PBMC) from a normal healthy donor were incubated with each individual compound at a final con- centration of 100 nM. After 14 days of culture, NKT cell frequencies were determined by flow cytometry. NKT cell frequencies were defined as the percentage of V 24+/V 11+ NKT cells among gated lymphocytes in the upper right (UR) corner (Q2) for each case. Shown here are the profiles of PBMC harvested from 14-day cultures containing (control) vehicle alone (DMSO, UN) and the compounds 1-4 tested. varian HPLC (prostar series ESI/APCI) coupled mass detector of Varian 901-MS (FT-ICR Mass) and triple quadrapole. Flash chromatography was performed using Geduran Si 60 silica gel (230 - 400 mesh). The final conjugation products were further purified and analyzed by HPLC, consisting of an Angilent 1100 pump and a linear UVIS detector (254 nm). A ZORBAX SILcolumn (250 mm × 9.4 mm, 5 m, Si-100) was used as the sta- tionary phase and the eluents of a combination of MeOH and CH2Cl2 with a flow rate of 3 mL/min were used as Figure 5. Molecular docking of truncated compound 3 onto the complex of CD1d and V 24+ chain. Software: Discovery Studio 2.1 with Ligandfit. Active site was defined by using Auto-Vα24-CD1d-β-GalCer complex (PDB entry of 3SDX). the mobile phase. 3.2. General Procedure for Preparation of the Four Amide Products 5-(2,5-dimethylphenoxy)-N-(((2R,3R,4S,5R,6S)-6-(((2S, 3S,4R)-2-(5-(2,5-dimethylphenoxy)-2,2-dimethylpentana mido)-3,4- dihydrox yoctadecyl)ox y)-3,4,5 -trihydr oxytetra hydro-2H-pyran-2-yl)methyl)-2,2-dimethylpentanamide (1). The conjugating procedure adopted the protocol as reported before [9]. To a mixture of Gemfibrozil (4 eq., 42 mg, 0.167 mmol), HBTU (4.0 equiv, 64 mg, 0.167 mmol) and DMF (1 mL) was added diisopropylethylamine (8.0 equiv, 60 μL, 0.334 mmol) under N2. After stirring for 30 min, TLC (EtOAc:n-hexane = 2.5:7.5) indicated the forma- tion of the ester intermediate (Rf = 0.73) and consump- tion of the starting Gemfibrozil (Rf = 0.12). To this mix- ture was added the solution of compound 5 ((1 equiv, 20 mg, 41.8 μmol) in DMF (1 mL). After stirring for 24 h, TLC (MeOH/CH2Cl2 = 1:8) indicated the formation of the product 1 (Rf = 0.80) and consumption of the active ester (Rf = 0.90). The mixture was concentrated under reduced pressure. The residue obtained was purified with HPLC using column chromatography (MeOH:CH2Cl2 = 1:10 → MeOH:CH2Cl2 = 1:9) to afford the crude product 1 in 38% yield (15 mg). The sample was further purified using MeOH/CH2Cl2 = 1:9 as eluent at a flow rate of 3 mL/min to afford an oil (7 mg) which was recorded as retention time at tR = 5.79 min. Anal. C54H90N2O11, M (calcd.) = 942.6 (m/z), ESI + Q-TOF:M = 798.6 (m/z), [M + H]+ = 943.9 (100%), [M + Na]+ = 965.9 (50%); HRMS (ESI) calcd. [M + H]+ = 943.66229 (m/z), found [M + H]+ = 943.66552 (m/z), = Copyright © 2013 SciRes. OJMC  Y.-C. HUANG ET AL. 58 3.42 ppm; 1H-NMR (500 MHz, CD3OD): 0.88 (t, 3H, Haliphatic), 1.18 - 1.20 (m, 12H, Haliphatic), 1.25 - 1.39 (m, 24H, Haliphatic), 1.54 - 1.72 (m, 10H, Haliphatic), 2.12 (s, 6H, 2 × OPhMe), 2.26 (s, 6H, 2 × OPhMe), 3.24 - 3.29 (m, 1H), 3.50 - 3.57 (m, 2H), 3.60 - 3.71 (m, 3H), 3.74 - 3.84 (m, 4H), 3.88 - 3.89 (m, 4H, PhO-CH2), 4.18 - 4.22 (m, 1H), 4.88 (d, 1H, J1,2 = 3.5 Hz, H-1), 6.59 (d, 2H, J = 7.5 Hz, ArH), 6.64 (s, 2H, ArH), 6.93 (d, 2H, J = 7.5 Hz, ArH); 13C-NMR (125 MHz, CD3OD):δ 14.44 (CH3, Caliphatic), 16.08 (CH3), 16.12 (CH3), 21.50 (CH3), 23.74 (CH2), 25.68 (CH3), 25.86 (CH3), 25.95 (CH3), 26.02 (CH3), 26.28 (CH2), 26.34 (CH2), 27.04 (CH2), 30.48 (CH2), 30.77 (CH2), 30.81 (CH2), 30.85 (CH2), 30.93 (CH2), 33.08 (CH2), 38.64 (CH2), 41.39 (CH2), 43.02 (C), 43.10 (C), 51.97 (CH), 68.09 (CH2), 69.13 (CH2), 69.23 (CH2), 70.03 (CH), 70.43 (CH), 71.25 (CH), 71.37 (CH), 72.93 (CH), 75.55 (CH), 101.29 (CH, C-1), 113.09 (CH, aromatic), 121.78 (CH, aromatic), 121.83 (CH, aromatic), 124.42 (C, aromatic), 131.20 (CH, aromatic), 131.23 (CH, aromatic), 137.61 (C, aromatic), 158.33 (C, aro- matic), 179.65 (C, amide), 180.91 (C, amide). 4-([1,1’-b iphenyl]-4- yl)-N-(((2 R,3R,4S,5 R,6S)-6-(((2 S ,3S,4R)-2-(4-([1,1’-biphenyl]-4-yl)-4-oxobutanamido)-3, 4-dihydroxyoctadecyl)oxy)-3,4,5-trihydroxytetrahydro-2 H-pyran-2-yl)methyl)-4-oxobutanamide (2). The same procedure as that for amide coupling of compound 1 was used. Fenbufen (42 mg, 0.167 mmol) was used as the carboxylic acid for the conjugation. The required reagents were the same as that described above. Column chromatography was performed by using eluents of MeOH:CH2Cl2 = 1:11 to provide the white crude product 2 in 40% yield (16 mg). After further purifica- tion with HPLC using eluents of MeOH:CH2Cl2 = 1:11, a white solid was obtained in an overall yield of 15% (6 mg). tR = 9.04 min. Anal. C56H74N2O11, HRMS (ESI) M (calcd.) = 950.5293 (m/z), [M + H]+ = 951.5371 m/z, [M − H]− = 949.5214 m/z; found [M + H]+ = 951.5289 (m/z), = 8.62 ppm; [M − H]− = 949.5216 (m/z), = 0.21 ppm; 1H-NMR (600 MHz, CD3OD):δ 0.79 (t, 3H, J = 7.2 Hz, Haliphatic), 1.15 - 1.34 (m, 24 H, Haliphatic), 1.44 - 1.46 (m, 1H, Haliphatic), 1.57 - 1.60 (m, 1H, Haliphatic), 2.54 - 2.62 (m, 4H aliphatic), 3.18 - 3.36 (m, 6H), 3.50 - 3.65 (m, 4H), 3.72-3.78 (m, 3H), 3.85 (dd, 1H, J2,3 = 10.5, J2,1 = 3.5 Hz, H-2), 4.15 - 4.16 (m, 1H), 4.82 (d, 1H, J1,2 = 3.5 Hz, H-1), 7.38 - 7.97 (m, 18H, Phamide); 13C-NMR (150 MHz, CD3OD):δ 20.50 (C), 29.17 (CH2), 32.35 (CH2), 35.86 (CH2), 36.16 (CH2), 36.22 (CH2), 36.26 (CH2), 36.31 (CH2), 38.42 (CH2), 39.27 (CH2), 40.26 (CH2), 40.32 (CH2), 46.15 (CH2), 55.81 (CH2), 73.55 (CH2), 75.55 (CH), 76.44 (CH), 78.64 (CH), 81.04 (CH), 105.94 (CH, C-1), 133.72 (CH, aromatic), 134.82 (CH, aromatic), 135.25 (CH, aromatic), 135.48 (CH, aromatic), 141.61 (C, aromatic), 146.17 (C, aromatic), 146.22 (C, aromatic), 152.59 (C, aromatic), 152.67 (C, aromatic), 179.20 (C, CO), 180.63 (C, CO), 205.60 (C, CO), 206.17 (C, CO) 4-(4-chlorophenyl)-N-(((2R,3R,4S,5R,6S)-6-(((2S,3S,4R )-2-(4-(4-ch lorophen yl)-4-oxobu tanamido)-3 ,4-dih ydrox yoctadecyl)oxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2- yl)methyl)-4-oxobutanamide (3). 3-(4-Chlorobenzoyl)propionic acid (53 mg, 0.251 mmol) was used as the carboxylic acid for the conjuga- tion. Column chromatography was performed by using eluents of MeOH:CH2Cl2 = 1:9 to provide the white crude product 3 in 46% yield (25 mg). After further puri- fication with HPLC using eluents of MeOH:CH2Cl2 = 1:11, a white solid was obtained in an overall yield of 28% (15 mg). tR = 10.94 min. Anal. C44H64Cl2N2O11, M (calcd.) = 866.4 (m/z); found: ESI + Q-TOF:[M + Na]+ = 889.5 (47.1%); HRMS (ESI) M (calcd.) = 866.38872 (m/z), [M + H]+ = 867.39654 amu; found [M + H]+ = 867.39501, = 1.76 ppm; 1H-NMR (600 MHz, CD3OD): δ 0.89 (t, 3H, J = 7.2 Hz, Haliphatic), 1.23-1.38 (m, 24H, Haliphatic), 1.51 (m, 1H, Haliphatic), 1.60 - 1.62 (m, 1H, Haliphatic), 2.58 - 2.64 (m, 4H aliphatic), 3.26 - 3.29 (m, 2H), 3.30 - 3.34 (m,2H), 3.47 - 3.50 (m, 1H), 3.58 - 3.63 (m, 2H), 3.68 - 3.70 (m, 1H), 3.77 - 3.80 (m, 3H), 3.84 - 3.90 (m, 2H), 4.21 - 4.22 (m, 1H), 7.46 - 7.48 (m, 4H, arom., 7.96 - 7.98 (m, 4H, arom.); 13C-NMR (150 MHz, CD3OD):δ14.45 (CH3), 23.75 (CH2), 27.00 (CH2), 30.49 (CH2), 30.72 (CH2), 30.78 (CH2), 30.83 (CH2), 30.87 (CH2), 30.90 (CH2), 33.08 (CH2), 34.82 (CH2), 41.33 (CH2), 51.94 (CH), 68.03 (CH2), 70.21 (CH), 70.55 (CH), 71.28 (CH), 71.30 (CH), 72.98 (CH), 75.49 (CH), 101.01 (CH, C-1), 129.95 (CH, arom.), 129.96 (CH, arom.), 130.84 (CH, arom.), 136.56 (C, arom.), 136.58 (C, arom.), 140.49 (C, arom.), 140.53 (C, arom.), 174.42 (C, CO), 175.32 (C, CO), 199.39 (C, CO), 199.52 (C, CO). (E)-N-((2S,3S,4R)-3,4-dihydroxy-1-(((2S,3R,4S,5R,6 R)-3,4,5- trihydrox y-6-((( E)-3-(4-(trifluoromethyl)phenyl )acrylamido)methyl)tetrahydro-2H-pyran-2-yl)oxy)oc- tadecan-2-yl)-3-(4-(trifluoromethyl)phenyl)acrylamide (4). trans-4-(Trifluoromethyl) cinnamic acid (54 mg, 0.251 mmol) was used as the carboxylic acid for the conjuga- tion. Column chromatography was performed using elu- ents of MeOH:CH2Cl2 = 1:10 → MeOH:CHCl2 = 1:8 to provide the white crude product 4 in 27% yield (15 mg). After additional purification with HPLC using eluents of MeOH:CH2Cl2 = 1:7, a white solid was obtained in 22% yield (12 mg). The sample was further purified by using recrystallization from MeOH to provide the white solid in an overall yield of 13% (7 mg). tR = 8.49 min. Anal. C44H60F6N2O9, M (calcd.) = 874.4 (m/z); found:ESI + Q-TOF:[M + H]+ = 875.4, [M + Na]+ = 897.4; 1H-NMR (500 MHz, CD3OD) :δ 0.88 (t, 3H, J = 7.0Hz, Haliphatic), 1.20 - 1.42 (m, 24H, Haliphatic), 1.45 - 1.65 (m, 2H, Haliphatic), 3.35 - 3.40 (m, 1H), 3.44 - 3.49 (m, 1H), 3.52 - 3.56 (m, 1H), 3.61 - 3.77 (m, 3H), 3.79 - 3.82 (m, 1H), Copyright © 2013 SciRes. OJMC  Y.-C. HUANG ET AL. Copyright © 2013 SciRes. OJMC 59 3.85 (m, 1H), 3.92 - 3.98 (m, 2H), 4.34 - 4.37 (m, 1H), 4.89 (d, 1H, J1,2 = 3.5 Hz, H-1), 6.72 (d, 1H, J = 16.0 Hz), 6.76 (d, 1H, J = 16.0 Hz), 7.45 (d, 1H, J = 16.0 Hz), 7.48 (d, 1H, J = 16.0 Hz), 7.63 - 7.66 (m, 8H, Hamide-aromatic); 13C-NMR (125 MHz, CD3OD):δ 14.44 (CH3, Caliphatic), 23.74 (CH2), 26.79 (CH2), 30.47 (CH2), 30.71 (CH2), 30.75 (CH2), 30.78 (CH2), 30.85 (CH2), 33.07(CH2), 33.20 (CH2), 41.75 (CH2), 52.56 (CH), 68.13 (CH2), 70.50 (CH), 71.02 (CH), 71.26 (CH), 71.57 (CH), 72.83 (CH), 75.40 (CH), 100.93 (CH, C-1), 124.50 (CH), 124.74 (CH), 126.77 (CH), 129.35 (CH), 140.02 (CH), 140.08 (CH), 167.41 (C, COamide), 168.26 (C, COamide). 19F-NMR (470 MHz, CD3OD) δ −210.26, −210.28, −220.17, −221.67. Conformers were speculated to assign for δ −220.17 and −221.67. 4. Acknowledgements We are grateful to the National Science Council of Tai- wan, Chang-Bing Show Chwan Memorial Hospital, CGMH_NTHU Joint Research, and Chang-Gung Medi- cal Research Project for providing financial support through grant numbers NSC-101-2113-M-007-010, NSC-97-2314- B-182A-020-MY3, NSC-97-2314-B-182A-020-MY3, CGTH96N2342E1, CMRPG3B0531, CMRPG390661, CMRPG390931, and CMRPG3B0361. REFERENCES [1] T. Natori, M. Morita, K. Akimoto and Y. Koezuka, “Age- lasphins, Novel Antitumor and Immunostimulatory Cere- brosides from the Marine Sponge Agelas-Mauritianus,” Tetrahedron, Vol. 50, No. 9, 1994, pp. 2771-2784. doi:10.1016/S0040-4020(01)86991-X [2] M. Morita, K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi and H. Fu- kushima, “Structure-Activity Relationship of Alpha-Ga- lactosylceramides Against B16-Bearing Mice,” Journal of Medicinal Chemistry, Vol. 38, No. 12, 1995, pp. 2176- 2187. doi:10.1021/jm00012a018 [3] M. Skold and S. M. Behar, “Role of CD1d-Restricted NKT Cells in Microbial Immunity,” Infection and Immu- nity, Vol. 71, No. 10, 2003, pp. 5447-5455. doi:10.1128/IAI.71.10.5447-5455.2003 [4] A. Bendelac, P. B. Savage and L. Teyton, “The Biology of NKT Cells,” Annual Review of Immunology, Vol. 25, 2007, pp. 297-336. doi:10.1146/annurev.immunol.25.022106.141711 [5] M. J. Smyth, N. Y. Crowe, Y. Hayakawa, K. Takeda, H. Yagita and D. Godfrey, “NKT Cells-Conductors of Tu- mor Immunity?” Current Opinion in Immunology, Vol. 14, No. 2, 2002, pp. 165-171. doi:10.1016/S0952-7915(02)00316-3 [6] K. J. L. Hammond and D. I. Godfrey, “NKT Cells: Poten- tial Targets for Autoimmune Disease Therapy?” Tissue Antigens, Vol. 59, No. 5, 2002, pp. 353-363. doi:10.1034/j.1399-0039.2002.590501.x [7] C. R. Berkers and H. Ovaa, “Immunotherapeutic Potential for Ceramide-Based Activators of iNKT Cells,” Trends in Pharmacological Sciences, Vol. 26, No. 5, 2005, pp. 252- 257. doi:10.1016/j.tips.2005.03.005 [8] J. Wojno, J. P. Jukes, H. Ghadbame, D. Shepherd, G. S. Bersa, V. Gerundolo and L. R. Cox, “Amide Analogs of CD1d Agonists Modulate iNKT-Cell-Mediated Cytokine Production,” ACS Chemical Biology, Vol. 7, No. 5, 2012, pp. 847-855. doi:10.1021/cb2005017 [9] J. Hunault, M. Diswall, J. C. Trison, V. Blet, J. Pocher, S. Marionneau-Lambt, T. Oullier, J.-Y. Douillard, S. Guil- larme, C. Saluzzo, G. Dujardin, D. Jacqomin, J. Graton, J.-Y. Le Ouestel, M. Evain, J. Lebreton, D. Dubreuil, J. Le Pendu and M. Pipelier, “3-Fluoro- and 3,3-Difluoro- 3,4-dideoxy-KRN7000 Analogues as New Potent Immu- nostimulator Agents: Total Synthesis and Biological Evaluation in Human Invariant Natural Killer T Cells and Mice,” Journal of Medicinal Chemistry, Vol. 55, No. 3, 2012, pp. 1227-1241. doi:10.1021/jm201368m [10] N. Veerapen, E. A. Leadbetter, M. B. Brenner, L. R. Cox and G. S. Bersa, “Synthesis of a Novel-Galactosyl Cera- midic Hepatenated-Lipid Antigen, a Useful Tool in Dem- onstrating the Involvement of iNKT Cells in the Produc- tion of Antilipid Antibodies,” Bioconjugate Chemistry, Vol. 21, No. 4, 2010, pp. 741-747. doi:10.1021/bc9005255 [11] P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Arm- strong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bun- dle, “Shiga-Like Toxins Are Neutralized by Tailored Multivalent Carbohydrate Ligands,” Nature, Vol. 403, No. 6770, 2000, pp. 669-672. doi:10.1038/35001095 [12] R. W. Franck and M. Tsuji, “ -C-Galactosyl Ceramides: Synthesis and Immunology,” Accounts of Chemical Re- search, Vol. 39, No. 10, 2006, pp. 692-701. doi:10.1021/ar050006z [13] Y.-C. Huang, L.-W. Chiang, K.-S. Chang, W.-C. Su, Y.- H. Lin, K.-C. Jeng, K.-I. Lin, K.-Y. Liao, H.-L. Huang and C.-S. Yu, “Synthesis of Amino Core Compounds of Galactosyl Phytosyl Ceramide Analogs for Developing iNKT-Cell Inducers,” Molecules, Vol. 17, No. 3, 2012, pp. 3058-3081. doi:10.3390/molecules17033058 [14] M. Michieletti, A. Bracci, F. Compostella, G. De Libero, L. Mori, S. Fallarini, G. Lombard and L. Panza, “Synthe- sis of -Galactosyl Ceramide (KRN7000) and Analogs Thereof via a Common Precursor and Their Preliminary Biological Assessment,” The Journal of Organic Chem- istry, Vol. 73, No. 22, 2008, pp. 9192-9195. doi:10.1021/jo8019994 [15] J. Lopez-Sagaseta, J. E. Kung, P. B. Savage, J. Gumperz and E. J. Adams, “The Molecular Basis for Recognition of CD1d/Alpha-Galactosylceramide by a Human Non-V alpha 24 T Cell Receptor,” PLOS Biology, Vol. 10, No. 10, 2012, p. e1001412. doi:10.1371/journal.pbio.1001412 [16] D. G. Pellicci1, A. J. Clarke, O. Patel, T. Mallevaey, T. Beddoe, J. Le Nours, A. P. Uldrich, J. McCluskey, G. S. Besra, S. A. Porcelli, L. Gapin, D. I. Godfrey and J. Ross- john, “Recognition of -Linked Self Glycolipids Medi- ated by Natural Killer T Cell Antigen Receptors,” Nature Immunology, Vol. 12, No. 9, 2011, pp. 827-834. doi:10.1038/ni.2076
|