Paper Menu >>
Journal Menu >>
 Journal of Biomaterials and Nanobiotechnology, 2011, 2, 18-27 doi:10.4236/jbnb.2011.21003 Published Online January 2011 (http://www.SciRP.org/journal/jbnb) Copyright © 2011 SciRes. JBNB Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System J. A. Hickey¹, I. Lynch², K. A. Dawson², D. Cox³, A. K. Keenan¹ ¹UCD School of Biomolecular and Biomedical Science, Conway Institute, University College Dublin, Ireland; ²Irish Centre for Col- loid Science and Biomaterials, UCD School of Chemistry and Chemical Biology, University College Dublin, Ireland; ³Molecular and Cellular Therapeutics, R.C.S.I., Dublin, Ireland Email: jennifer.hickey82@gmail.com Received September 13th, 2010; revised September 25th, 2010; accepted September 30th, 2010 ABSTRACT While deployment of intracoronary stents has been shown to reduce restenosis, stenting can also damage the endothe- lial monolayer lining the vessel wall, leading to possible in-stent thrombosis. Local drug delivery from stent surfaces represents a means of delivering therapeutic doses of drug directly to the target site. The aim of this study was to elute fluvastatin, which can inh ibit vascular smooth muscle cell proliferation, and xemilofib an, which p revents p latelet adhe- sion and aggregation, together in bioactive concentrations from the same copolymer system. Combined elution from thermoresponsive N-isopropylacrylamide (NiPAAm)/N-tert-butylacrylami de (NtBAAm)-derived copolymer systems was achieved using microgels (NiPAAm/NtBAAm 65/35 wt/wt) randomly dispersed in 85/15 matrices. Fluvastatin elution from 5 m films over a 14-day period showed initial burst release, which leveled off. Of the total incorporated (8.33 ± 0.21 nmol, n = 4), 68.5% was eluted during this period. Xemilofiban relea se was measured in terms of its ability to in- hibit platelet adhesion, using a microfluidic system. To investigate the influence of location and hydrophobicity on elu- tion of bioactivity, three separate systems were employed. While elution of anti-adhesive activity from the system con- taining xemilofiban-loaded matrices was more dramatic in the short term, a more sustained level of inhibition was achieved when xemilofiban had been incorporated into microgels. All samples investigated for anti-adhesive activity also decreased human coronary artery smooth muscle cell proliferation. Therefore xemilofiban has potential as an agent for preventing in-stent thrombos is. Our study has demonstrated the feasibility o f using this novel matrix/microg el system to regulate simultaneous release of both agents in bioactive concentrations. Keywords: Fluvastatin, Xemilofiban, Nipaam/Ntbaam, Microgels, Local Drug Delivery 1. Introduction The use of intravascular coronary stents during angio- plasty procedures was first reported by Ulrich Sigwart in 1986 [1]. With the utilization of a stent there is the possi- bility of increased neointimal hyperplasia, as the metallic stent can cause physical injury to the endothelium, which can result in in-stent restenosis (ISR) and in-stent thrombosis (IST). Those higher risk patients with com- plex lesions (small vessel, bifurcation lesions) have an in-stent restenosis (ISR) occurrence rate of 30-60%, while in those with less complex lesions the rate is reduced to 15-20% of patients [2]. IST has an occurrence rate of ≈ 1% [3]. Drug-eluting stents (DES) have had a major influence on the reduction of in-stent restenosis. To prevent restenosis, a clear understanding of events responsible for growth factor- or cytokine-mediated vas- cular smooth muscle cell (VSMC) proliferation and mi- gration (intimal hyperplasia) has led to a precise selec- tion of potential therapeutic candidates [4]. However, the current leading agents, sirolimus and paclitaxel, can in- hibit endothelial cell as well as VSMC proliferation,  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 19 thereby affecting the re-endothelialisation process which is central to vessel recovery. HMG CoA reductase in- hibitors (statins) could be an option for the inhibition of neointimal hyperplasia, since these agents inhibit VSMC proliferation by a mechanism independent of cholesterol synthesis [5]. In an attempt to prevent the problem of IST, patients are placed on a strict regime of anti-platelet agents. However despite these adjunct therapies, stent thrombosis still occurs in 1% of patients [6]. The residual occurrence of IST may be caused by lack of penetration of such agents in adequate concentrations to the desired site of action following oral or systemic administration. Local delivery of agents designed to inhibit VSMC pro- liferation and platelet aggregation via a copolymer- coated stent platform is an attractive strategy for offset- ting the incidence of ISR and IST. Poly (n-isopropylacrylamide) (NiPAAm) is a polymer that can become inversely soluble upon heating. This change in state occurs at what is known as the lower critical solution temperature (LCST) [7]. Below the LCST the polymer chains are extended, separated and surrounded by water, but above the LCST the polymer becomes insoluble and precipitates out of solution [8]. The temperature range at which the soluble-insoluble shift occurs in relation to NiPAAm is between 31-34˚C. The addition of N-tert-butyl-acrylamide (NtBAAm) al- ters the relative hydrophobicity/hydrophilicity of NiPA- Am thereby giving a means for regulating the elution of drugs for local delivery on the basis of their polarity. In this study, a range of copolymers exhibiting increasing hydrophobicities were synthesised in varying (w/w) ra- tios of NiPAAm/NtBAAm (85/15, 65/35 and 50/50). The potential of NiPAAm/NtBAAm-derived copoly- mers to act as drug delivery vehicles for small molecules like colchicine [9-10] and therapeutic proteins like vas- cular endothelial growth factor (VEGF) [11] has been previously investigated. Another approach is to cross- link NiPAAm/NtBAAm copolymer microgel particles and disperse these through a bulk copolymer matrix. At their LCST the network volume changes from an ex- panded water-containing network, to a collapsed network where the water has been expelled. As the temperature increases the particles shrink and become hard and sphere shaped. [12] These microgels are then dispersed through the non cross-linked NiPAAm/NtBAAm matri- ces and can be used for drug loading and elution. Our group has previously shown that fluvastatin could be eluted from these matrix/microgel copolymer systems for up to 60 days with retention of bioactivity. In this study, dual drug release from 3 different sys- tems was investigated. System A was comprised 65/35 microgels containing fluvastatin embedded in a 50/50 matrix containing xemilofiban, while System B com- prised 65/35 microgels containing fluvastatin embedded in an 85/15 matrix containing xemilofiban. Finally, Sys- tem C contained 65/35 microgels incorporating xemilofiban, embedded in an 85/15 copolymer containing fluvastatin. Bioactivity of fluvastatin was assessed in terms of effects on human coronary artery smooth muscle cell (HCA- SMC) and human coronary artery endothelial cell (HCA- EC) proliferation while the bioactivity of xemilofiban was evaluated in terms of effects on platelet adhesion under conditions of flow. 2. Materials and Methods 2.1. Materials N-isopropylacrylamide (NiPAAm; Acros Organics, USA) and N-tert-butylacrylamide (NtBAAm; Fluka, Switzer- land) were recrystallized twice from n-hexane and dried at room temperature under vacuum. N, N-Azobisisobu- tyronitrile (AIBN; Phase Separation, UK) was recrystal- lized from methanol. Solvents were reagent grade and were purified by conventional methods. 14C-fluvastatin (specific activity 56.9 Ci/mmol) was generously donated by Novartis, Hanover. Xemilofiban (SC-54701) was kindly donated by Dr. Dermot Cox, RCSI. HCASMC with the corresponding growth medium (Medium 231) were purchased from Cascade Biologics (Belgium). HC- AEC with the corresponding medium (Endothelial Cell Growth Medium) were purchased from Promocell. 5- Bromo-2’-deoxyuridine (BrdU) was obtained from Roche Diagnostics (Germany). All other chemicals and reagents were of the highest grade commercially available. 2.2. Preparation of NiPAAm/NtBAAm Copolymers The copolymers were synthesized through free radical polymerisation of the corresponding monomers with AIBN used as an initiator. The methodology was as out- lined by McGillicuddy et al. [13] Briefly, mixtures of NiPAAm and NtBAAm monomers in different ratios were dissolved in benzene with AIBN then added. The removal of oxygen was achieved by bubbling N2 gas through the solution for 30 min. Polymerisation then proceeded at 60˚C for 24 h. Benzene was evaporated off and the copolymer was dissolved in acetone and precipi- tated in n-hexane. This whole procedure was repeated three times. The purified copolymer was dried at room temperature under vacuum. 2.3. Preparation of Microgels Microgels were synthesized using the method of disper- sion polymerisation. [14] In their desired w/w ratios  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 20 (85/15, 65/35, 50/50, total 0.2 g) NiPAAm/NtBAAm and bisacrylamide (0.02 g) were dissolved in 36 ml of water. One ml of 0.1 wt% Triton solution was added and heated to 70˚C. The solution was degassed by bubbling with N2. Ammonium persulphate (0.02 g) was dissolved in 4 ml of water, degassed, and added slowly to the stirring monomer solution, under an atmosphere of N2. The reac- tion was left for 12 h at 70˚C and the resulting microgel dispersion (1 wt% in water) cleaned by dialysis, and freeze-dried before use. 2.4. Preparation of, and Drug Incorporation into Matrix/Microgel Copolymer Systems Fluvastatin was incorporated into 65/35 microgel parti- cles (5 mg) by incubation with 1 mL of an ethanolic so- lution of 5mM drug containing tracer levels of [14C]-fluvastatin for 24 h at 4˚C. The microgel/drug sus- pension was then centrifuged at 5000 rpm for 10 min, the supernatant containing unincorporated drug was removed, and the microgels resuspended in 0.5 ml ethanol. Drug- loaded microgel suspensions were subsequently added to 0.5 mL of a 10% 50/50 or 85/15 matrix copolymer solu- tion containing 2.5 mM xemilofiban. Alternatively xe- milofiban was incorporated into 65/35 microgel particles which were subsequently incorporated into a 50/50 ma- trix containing fluvastatin. Aliquots (27 µl) of the result- ing suspensions were applied evenly to the wells of 24- well tissue culture plates and were allowed to dry over- night in an ethanolic atmosphere. All manipulations in aqueous medium were done at 37˚C which is above the LCST and therefore all copolymer systems were in the collapsed/insoluble state. 2.5. Drug Release from Copolymer Films Dried films were washed with prewarmed PBS (37˚C) to retain the collapsed state of the copolymer, prevent its dissolution, and remove any surface-bound drug. Drug elution from the resulting films at 37˚C was monitored (as [14C]-fluvastatin) every 24 h by removing, storing and replacing the PBS solution. The amount of [14C]- fluvastatin eluted into the overlying solution was deter- mined by scintillation counting against a suitable stan- dard curve, from which total eluted fluvastatin was de- termined. 2.6. Cell Culture HCASMC that had been isolated from a 21-year old Caucasian male by Cascade Biologics were received as cryopreserved cultures (passage 2) from Cytotech Ltd. (Denmark). The cells were maintained in their appropri- ate medium, Medium 231, supplemented with foetal bo- vine serum (4.9%), basic fibroblast growth factor (2ng/ ml), epidermal growth factor (0.5g/ml), heparin (5ng/ml), insulin (5µg/ml), BSA (0.2µg/ml), gentamicin (10µg/ml) and amphotericin B (0.25µg/ml). The cells were grown to confluency in 75cm2 filter top tissue culture flasks and were maintained at 37˚C in a humidified atmosphere containing 95% O2 and 5% CO2. Subcultures were cre- ated by passaging using a trypsin/EDTA (T/E) (0.025 %/0.01%) mixture in phosphate buffer saline (PBS), harvested by centrifugation (3 min at 433 × g) and seeded at stated densities. Cells of passages 4-10 were used for experiments. HCAEC were purchased from Promocell (Germany). The cryopreserved cells (passage 2) received had been isolated from a 63-year old Caucasian male. The appro- priate endothelial cell growth medium was used to main- tain the HCAEC. The medium was supplemented with 20 % heat-inactivated foetal calf serum (FCS), endothelial cell growth supplement/heparin (2 ml), human recombi- nant epidermal growth factor (5 µg/500 µl), hydrocorti- sone (500 µg/500 µl), gentamicin (10 µg/ml) and am- photericin B (0.25 µg/ml). The cells were grown to con- fluency in 75 cm2 filter top tissue culture flasks and were maintained at 37˚C in a humidified atmosphere contain- ing 95% O2 and 5% CO2. Subcultures were created by passaging using a T/E (0.025%/0.01%) mixture in PBS, harvested by centrifugation (3 min at 433 × g) and seeded at stated densities. Cells of passages 4-10 were used for experiments. Cells were also routinely tested for the presence of mycoplasm. 2.7. Measurement of Cellular Proliferation (BrdU ELISA) HCASMC or HCAEC (3 × 104 cells/ml/well) were seed- ed into 96-well plates and left to adhere overnight. Cells were then incubated with samples of fluvastatin eluted from the various copolymer systems and left for 48h. Proliferation was subsequently assessed by measurement of BrdU incorporation into the DNA of proliferating cells using a colorimetric ELISA. 2.8. Measurement of Platelet Adhesion under Conditions of Flow A novel microflow system consisting of a novel syringe pump with microfluidic biochip and flow sensor con- trolled by a PC using dedicated software was used to assess the effects of eluted samples of xemilofiban. Blood was collected from healthy volunteers who were not taking any medication and were free from aspirin and other anti-platelet agents within the previous 2 weeks. The blood was drawn by venupuncture into tubes con- taining a 1:10 volume of 3.8% (wt/vol) trisodium citrate and gently mixed. Each microgel channel of the novel 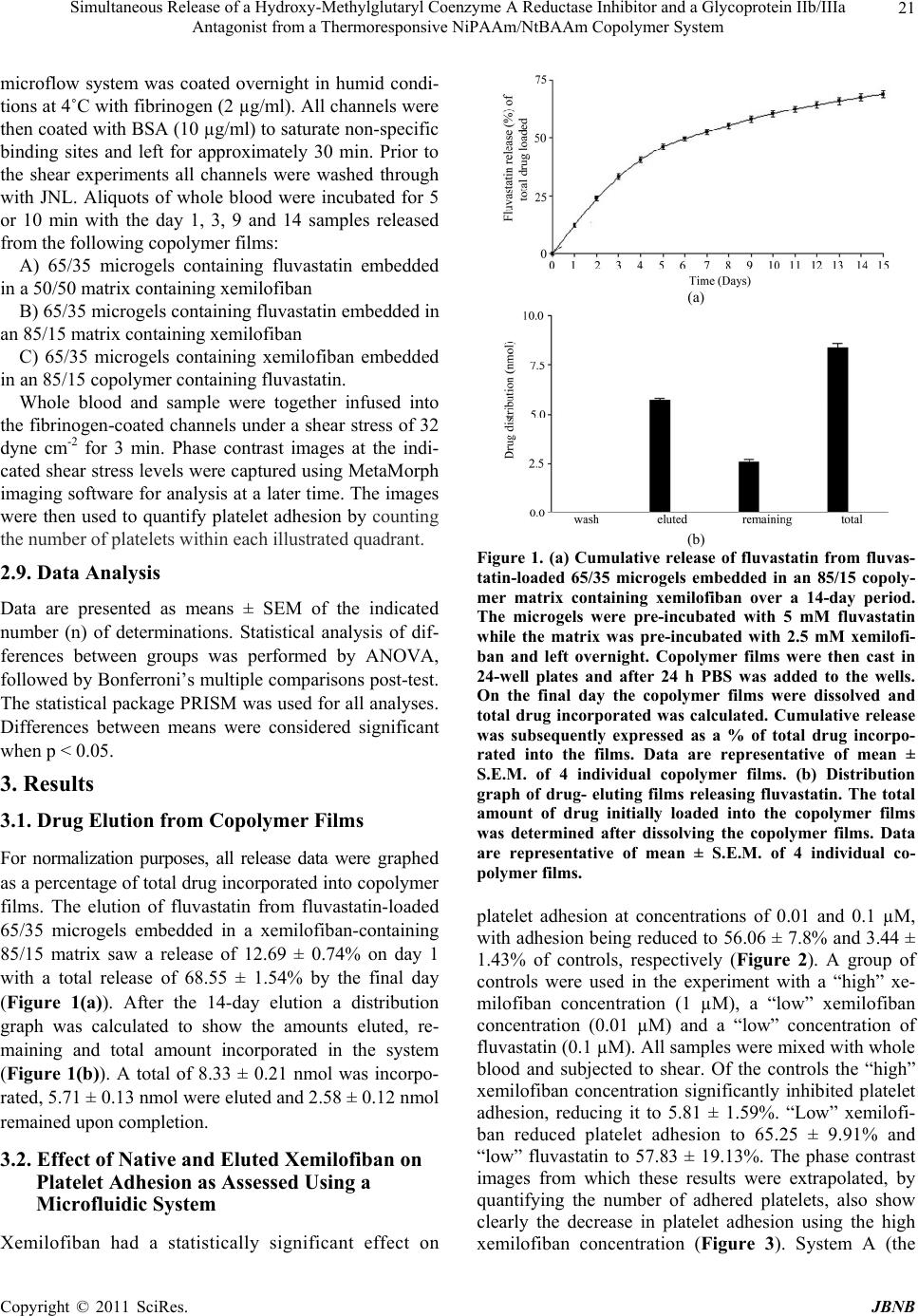 Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 21 microflow system was coated overnight in humid condi- tions at 4˚C with fibrinogen (2 µg/ml). All channels were then coated with BSA (10 µg/ml) to saturate non-specific binding sites and left for approximately 30 min. Prior to the shear experiments all channels were washed through with JNL. Aliquots of whole blood were incubated for 5 or 10 min with the day 1, 3, 9 and 14 samples released from the following copolymer films: A) 65/35 microgels containing fluvastatin embedded in a 50/50 matrix containing xemilofiban B) 65/35 microgels containing fluvastatin embedded in an 85/15 matrix containing xemilofiban C) 65/35 microgels containing xemilofiban embedded in an 85/15 copolymer containing fluvastatin. Whole blood and sample were together infused into the fibrinogen-coated channels under a shear stress of 32 dyne cm-2 for 3 min. Phase contrast images at the indi- cated shear stress levels were captured using MetaMorph imaging software for analysis at a later time. The images were then used to quantify platelet adhesion by counting the number of platelets within each illustrated quadrant. 2.9. Data Analysis Data are presented as means ± SEM of the indicated number (n) of determinations. Statistical analysis of dif- ferences between groups was performed by ANOVA, followed by Bonferroni’s multiple comparisons post-test. The statistical package PRISM was used for all analyses. Differences between means were considered significant when p < 0.05. 3. Results 3.1. Drug Elution from Copolymer Films For normalization purposes, all release data were graphed as a percentage of total drug incorporated into copolymer films. The elution of fluvastatin from fluvastatin-loaded 65/35 microgels embedded in a xemilofiban-containing 85/15 matrix saw a release of 12.69 ± 0.74% on day 1 with a total release of 68.55 ± 1.54% by the final day (Figure 1(a)). After the 14-day elution a distribution graph was calculated to show the amounts eluted, re- maining and total amount incorporated in the system (Figure 1(b)). A total of 8.33 ± 0.21 nmol was incorpo- rated, 5.71 ± 0.13 nmol were eluted and 2.58 ± 0.12 nmol remained upon completion. 3.2. Effect of Native and Eluted Xemilofiban on Platelet Adhesion as Assessed Using a Microfluidic System Xemilofiban had a statistically significant effect on (a) (b) Figure 1. (a) Cumulative release of fluvastatin from fluvas- tatin-loaded 65/35 microgels embedded in an 85/15 copoly- mer matrix containing xemilofiban over a 14-day period. The microgels were pre-incubated with 5 mM fluvastatin while the matrix was pre-incubated with 2.5 mM xemilofi- ban and left overnight. Copolymer films were then cast in 24-well plates and after 24 h PBS was added to the wells. On the final day the copolymer films were dissolved and total drug incorporated was calculated. Cumulative release was subsequently expressed as a % of total drug incorpo- rated into the films. Data are representative of mean ± S.E.M. of 4 individual copolymer films. (b) Distribution graph of drug- eluting films releasing fluvastatin. The total amount of drug initially loaded into the copolymer films was determined after dissolving the copolymer films. Data are representative of mean ± S.E.M. of 4 individual co- polymer films. platelet adhesion at concentrations of 0.01 and 0.1 µM, with adhesion being reduced to 56.06 ± 7.8% and 3.44 ± 1.43% of controls, respectively (Figure 2). A group of controls were used in the experiment with a “high” xe- milofiban concentration (1 µM), a “low” xemilofiban concentration (0.01 µM) and a “low” concentration of fluvastatin (0.1 µM). All samples were mixed with whole blood and subjected to shear. Of the controls the “high” xemilofiban concentration significantly inhibited platelet adhesion, reducing it to 5.81 ± 1.59%. “Low” xemilofi- ban reduced platelet adhesion to 65.25 ± 9.91% and “low” fluvastatin to 57.83 ± 19.13%. The phase contrast images from which these results were extrapolated, by quantifying the number of adhered platelets, also show clearly the decrease in platelet adhesion using the high xemilofiban concentration (Figure 3). System A (the  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 22 Figure 2. Effect of xemilofiban on platelet adhesion as as- sessed using a microfluidic system. Whole blood which had been treated with increasing concentrations of xemilofiban was passed through microcapillary channels on a biochip under shear stress. Numbers of platelets adhered were counted using MetaMorph. Results were calculated as a % of control (no drug) and values are presented as mean ± S.E.M., n = 3, ***p < 0.001 w.r.t. control. Figure 3. The effects of xemilofiban and fluvastatin on whole blood assessed using the microfluidic system. Whole blood was mixed with either drug and then passed through the fibrinogen coated biochannels and subjected to shear rate, phase contrast images were captured using Meta- Morph. Images are as follows: (a) whole blood alone; (b) high xemilofiban, 1 µM; (c) low xemilofiban, 0.01 µM; (d) fluvastatin 0.1 µM. most hydrophilic system of the 3) showed significant decreases for day 1 and 3 with adhesion reduced to 7.18 ± 3.02% and 15.46 ± 5.62%, respectively; thereafter platelet adhesion increased to 62.43 ± 20.4% and 82.08 ± 24.97% respectively. System B (the more hydrophobic system) showed a significant decrease for day 1 with a reduction to 8.74 ± 4.57%. On days 3, 9 and 14, there was no significant difference in reductions; however there was an apparent reduction in platelet adhesion to half that of the control on day 14. Finally, with system C in which xe- milofiban was incorporated into the microgels, there was a significant decrease seen for days 1, 3, 9 and 14, with reductions to 8.96 ± 5.14%, 47.97 ± 11.89%, 50.54 ± 15.37% and 51.11 ± 10.26% respectively (Figure 4). Figure 4. Effect of samples eluted from copolymer films on platelet adhesion assessed using the microfluidic system. System A composition was as follows 65/35 microgels con- taining fluvastatin embedded in a 85/15 matrix containing xemilofiban, System B was 65/35 microgels containing flu- vastatin embedded in an 85/15 matrix containing xemilofi- ban and System C was comprised of 65/35 microgels con- taining xemilofiban embedded in an 85/15 copolymer con- taining fluvastatin Copolymer films were cast into 24-well plates and overlaid with PBS and eluted samples were col- lected daily. Whole blood was treated with these eluted samples and passed through the microfluidic system. The amount of platelets adhered was counted using MetaMorph. Results were calculated as a % of control (no drug) and values are presented as mean ± S.E.M., n = 5, ***p < 0.001 w.r.t. cont r ol . 3.3. Effect of Eluted Fluvastatin on HCASMC Proliferation All samples eluted from each system significantly inhib- ited HCASMC proliferation (Figure 5). It could be seen that while proliferation was inhibited in the presence of each sample, a steady loss in anti-proliferative activity was seen over time. Samples eluted from System A de- creased proliferation to 12.86 ± 4.55% of control, using the Day 1 sample, with proliferation recovering to 62.29 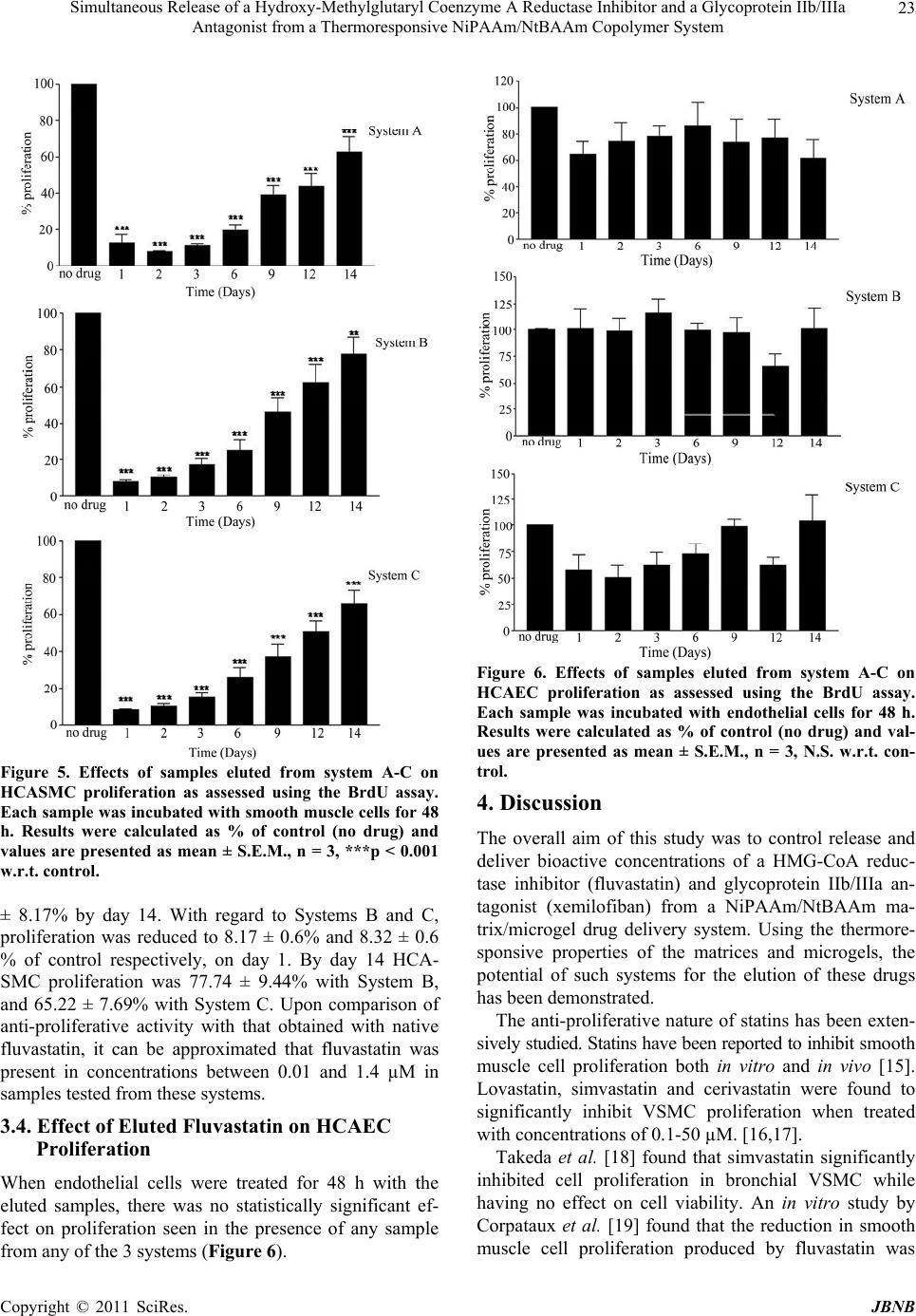 Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 23 Figure 5. Effects of samples eluted from system A-C on HCASMC proliferation as assessed using the BrdU assay. Each sample was incubated with smooth muscle cells for 48 h. Results were calculated as % of control (no drug) and values are presented as mean ± S.E.M., n = 3, ***p < 0.001 w.r.t. cont r ol. ± 8.17% by day 14. With regard to Systems B and C, proliferation was reduced to 8.17 ± 0.6% and 8.32 ± 0.6 % of control respectively, on day 1. By day 14 HCA- SMC proliferation was 77.74 ± 9.44% with System B, and 65.22 ± 7.69% with System C. Upon comparison of anti-proliferative activity with that obtained with native fluvastatin, it can be approximated that fluvastatin was present in concentrations between 0.01 and 1.4 µM in samples tested from these systems. 3.4. Effect of Eluted Fluvastatin on HCAEC Proliferation When endothelial cells were treated for 48 h with the eluted samples, there was no statistically significant ef- fect on proliferation seen in the presence of any sample from any of the 3 systems (Figure 6). Figure 6. Effects of samples eluted from system A-C on HCAEC proliferation as assessed using the BrdU assay. Each sample was incubated with endothelial cells for 48 h. Results were calculated as % of control (no drug) and val- ues are presented as mean ± S.E.M., n = 3, N.S. w.r.t. con- trol. 4. Discussion The overall aim of this study was to control release and deliver bioactive concentrations of a HMG-CoA reduc- tase inhibitor (fluvastatin) and glycoprotein IIb/IIIa an- tagonist (xemilofiban) from a NiPAAm/NtBAAm ma- trix/microgel drug delivery system. Using the thermore- sponsive properties of the matrices and microgels, the potential of such systems for the elution of these drugs has been demonstrated. The anti-proliferative nature of statins has been exten- sively studied. Statins have been reported to inhibit smooth muscle cell proliferation both in vitro and in vivo [15]. Lovastatin, simvastatin and cerivastatin were found to significantly inhibit VSMC proliferation when treated with concentrations of 0.1-50 µM. [16,17]. Takeda et al. [18] found that simvastatin significantly inhibited cell proliferation in bronchial VSMC while having no effect on cell viability. An in vitro study by Corpataux et al. [19] found that the reduction in smooth muscle cell proliferation produced by fluvastatin was  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 24 significantly greater than that with other statins such as simvastatin, lovastatin, atorvastatin, cerivastatin and pra- vastatin. Jaschke et al. [17] showed that after treatment with cerivastatin (0-100 nM) only the highest concentra- tion had a significant effect on endothelial cell prolifera- tion. Thus, although fluvastatin was used to treat both HCAEC and HCASMC in this study, the latter were much more sensitive to these anti-proliferative effects. Therefore fluvastatin was selected as the statin of choice in the present project for local drug delivery due to its active and lipophilic nature, both essential requirements for a potential candidate for local drug delivery. We have previously reported that NiPAAm/NtBAAm copolymers can elute fluvastatin for up to 60 days while retaining its bioactivity, as assessed under both static and perfusion conditions [20]. The present study has further shown that it is possible to co-elute bioactive fluvastatin with a second drug type incorporated in the system. In System A fluvastatin was eluted from 65/35 microgels and then released through a hydrophilic 85/15 matrix. Upon completion of this release experiment there was < 20% of the drug remaining in the system. It is important to note here that the pH value of blood may have an ef- fect on drug elution in vivo; however such an investiga- tion in vitro was beyond the scope of the present work. Also, while every step in the preparation of the mi- crogel/copolymer systems was carefully carried out, there is the possibility that small quantities of drug were lost. However, such losses were deemed minimal, since subsequent experiments showed comparable amounts of activity eluted in replicate experiments. While the inhibi- tion of vSMC proliferation steadily decreased with each of the daily samples used, proliferation at 48 h was still inhibited. Samples eluted from the other two systems showed an inhibition of proliferation resembling that of System A. In Systems A and B, fluvastatin was incorporated into the microgels and then eluted through hydrophilic (Sys- tem A) or hydrophobic (System B) matrices. This may explain the slight differences between these systems i.e. System A samples showed slightly greater inhibiton of proliferation and the drug was eluted faster. With regard to System C fluvastatin was dispersed in the matrix and thus only had one barrier to pass through; the results here were similar to those for System A. This could be due to the fact that hydrophobic drug was eluted from microgels into a hydrophilic matrix in one instance and straight from a hydrophobic matrix in the other. It is possible that the time course of elution from a hydrophobic matrix is comparable to that of elution from microgels and a hy- drophilic matrix together. However it is also important to note that major differences may only be seen with exten- sion of the 14-day period (Figure 6). The comparable levels of overall inhibition seen between systems could also be due to the fact that each of the systems is eluting a similar concentration of fluvastatin on the relevant days. None of the samples eluted from system A-C had a significant effect on HCAEC proliferation after 48 h, which suggests that re-endothelialisation in vivo would not be impeded up to this point. The rationale for using the GpIIb/IIIa antagonist xe- milofiban in this study was based on the fact that GpIIb/ IIIa antagonists have been used successfully as intrave- nous anti-platelet agents post-surgery (PTCA) [21] fol- lowing their development of oral agents, mixed results coronary syndromes and long-term management of pa- tients [22]. However with 5 large-scale trials completed by 2001, which included over 42,000 patients (EXCITE, [23] OPUS, [24] SYMPHONY 1 and 2, [25,26] BRAVO [27]), it was consistently found that the GpIIb/IIIa agents xemilofiban, orbofiban, sibrafiban and lotrafiban were no more effective than aspirin when given post-surgery. However, another study showed that their use during PCA procedures was effective and in fact improved in-hospital survival rates [28]. Heer et al. [29] have also offered evidence of their effectiveness during primary angioplasty. Therefore their effectiveness as an intrave- nous treatment in conjunction with PCA gives an indica- tion that these agents could be used for local delivery from a stent platform. The rationale for elution of an anti-platelet agent has been strengthened by reports demonstrating elution of GpIIb/IIIa receptor monoclonal antibody from polymer- coated stents. Yin et al. [30] showed that the GpIIb/IIIa receptor antibody eluted from I-PLA polymer-coated stents inhibited platelet aggregation. Aggarwal et al. [31] showed that GpIIb/IIIa antibody could be eluted for up to 14 days, with antibody still remaining after that time un- der conditions of flow. Our study firstly showed that xemilofiban concentra- tions of 0.01 and 0.1 µM had a statistically significant effect on platelet adhesion (Figure 3). This gives a good indication that xemilofiban is capable of exerting its ef- fects under conditions of flow and that platelets in circu- lating blood are more susceptible to the anti-adhesive action of xemilofiban in this model. Under conditions of flow System A showed that on day 1 and 3, samples decreased platelet adhesion signifi- cantly, and the results were in fact on a comparable level with the control “high xemilofiban” concentration; how- ever day 9 and 14 samples had only a minor effect on adhesion equivalent to that of the “low xemilofiban” concentration (Figures 4 and 5). As this was a hydro-  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 25 philic matrix it could be inferred that the hydrophobic xemilofiban was expelled rapidly and therefore only had a significant effect at day 1 and 3. It may be possible to prolong the elution of the drug from this matrix type by incubating the matrix or microgels with a higher concen- tration of xemilofiban. Alternatively using a more hy- drophobic matrix could also potentially prolong the re- lease of xemilofiban. It should be noted here that meas- urement of the exact concentration of xemilofiban re- leased was beyond the scope of the present study. System B had 65/35 microgels containing fluvastatin embedded in a 50/50 matrix containing xemilofiban; this is the more hydrophobic system and therefore it could be anticipated that elution would be prolonged. With this system a statistically significant effect was seen on day 1 with a reduction to a level similar to “high xemilofiban”. This correlated well with the burst release that is usually seen on Day 1 with all systems. While the remaining samples did not elicit a statistically significant effect it could be seen that there was a trend towards continued inhibition of platelet adhesion (Figure 5). Thus this sys- tem gave an indication that the inhibitory effect would have continued beyond 14 days. System C saw the incorporation of xemilofiban into 65/35 microgels, which were then embedded in a 50/50 matrix. This final system showed release of bioactive drug over an extended time interval. In this case drug would have firstly been released from the microgels and then passed through the hydrophobic matrix. Each of the daily samples had a significant effect on platelet adhe- sion, with the day 1 sample having the greatest effect (Figure 5). The contrasts between the profiles of each of the de- livery systems can be explained in terms of their compo- sition. With regard to Systems A and B, xemilofiban is contained in a hydrophilic and hydrophobic matrix re- spectively. The drug appears to be eluted faster from System A as a greater effect can be seen on Days 1 and 3, after which the anti-adhesive effect is reduced. The hy- drophobic matrix of System B retards the release of xe- milofiban slighty, thereby resulting in a greater anti-ad- hesive effect being seen at Day 14 compared with Sys- tem A. Another point to note with these two systems is that as xemilofiban is contained in the matrix, it only has one diffusion barrier prior to release, thereby resulting in faster elution. With System C however, xemilofiban is contained in the microgel component, and therefore has two diffusion barriers prior to release. Comparing Sys- tem C with Systems A and B, it can be noted that a steady elution rate appears to be maintained, which may reflect more tightly controlled release. With all three sys- tems the Day 1 eluate had the most significant anti-ad- hesive activity, which is characteristic of the burst release seen with these systems [20]. 5. Conclusions Overall, this study showed that it is possible to elute two bioactive drug types from one copolymer system, result- ing in three points of information. Firstly alteration of the composition of the copolymer systems i.e. the hydropho- bic or hydrophilic nature of the matrix or microgels can affect elution. Also placement in either matrix or mi- crogels can alter the period of elution i.e. the incorpora- tion of drug into the microgels can prolong the effect of the drug. The study finally shows that xemilofiban re- mains bioactive and can affect platelet adhesion under flow conditions. The use of thermoresponsive copoly- mers is relevant as they allow for the incorporation of drug into a soluble mixture followed by precipitation above the LCST onto, potentially, a stent. Therefore the thermoresponsive properties coupled with the relative hydrophobicities and hydrophilicities of the NiPAAm/ NtBAAm copolymers adds to their potential for the de- livery of numerous drugs for extended periods. Therefore this study has shown that two drugs eliciting different actions can be eluted for up to 14 days in bioactive con- centrations with the potential for extended release. Also fluvastatin did not alter platelet adhesion at concentra- tions eluted nor did xemilofiban affect cell proliferation at estimated concentrations eluted. 6. Acknowledgements We would like to acknowledgement the financial support provided by the Irish Heart Foundation without whom this work would not have been possible. REFERENCES [1] U. Sigwart, J. Puel, V. Mirkovitch, F. Joffre and L. Kap- penberger, “Intravascular Stents to Prevent Occlusion and Restenosis after Transluminal Angioplasty,” New Eng- land Journal of Medicine, Vol. 316, No. 12, 1987, pp. 701-706. doi:10.1056/NEJM198703193161201 [2] V. A. Voudris, J. S. Skoularigis, Y. K. Dimitriou, G. N. Grapsa, J. S. Malakos, G. S. Pavlides, et al., “Diabetes Mellitus and Unstable Coronary Artery Disease: Im- proved Clinical Outcome of Coronary Artery Stenting in an Era of Glycoprotein IIb/IIIa Inhibitors and Lipid- Lowering Therapy,” Coronary Artery Disease, Vol. 15, No. 6, 2004, pp. 353-359. doi:10.1097/00019501-200409000-00009 [3] A. T. Ong and P. W. Serruys, “Drug-Eluting Stents: Cur- rent Issues,” Texas Heart Institute Journal, Vol. 32, No. 3, 2005, pp. 372-377. [4] B. L. Hiatt, F. Ikeno, A. C. Yeung and A. J. Carter, “Drug-Eluting Stents for the Prevention of Restenosis: In  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 26 Quest for the Holy Grail,” Catheterization and Cardio- vascular Interventions, Vol. 55, No. 3, 2002, pp. 409-417. doi:10.1002/ccd.10161 [5] S. Park and S. Lee, “Optimal Management of Platelet Function after Coronary Stenting,” Current Treatment Options in Cardiovascular Medicine, Vol. 9, No. 1, 2007, pp. 37-45. doi:10.1007/s11936-007-0049-7 [6] R. M. da Silva, J. F. Mano and R. L. Reis, “Smart Ther- moresponsive Coatings and Surfaces for Tissue Engi- neering: Switching Cell-Material Boundaries,” Trends in Biotechnology, Vol. 25, No. 12, 2007, pp. 577-583. doi:10.1016/j.tibtech.2007.08.014 [7] E. S. Ron and L. E. Bromberg, “Temperature-Responsive Gels and Thermogelling Polymer Matrices for Protein and Peptide Delivery,” Advanced Drug Delivery Reviews, Vol. 31, No. 3, 1998, pp. 197-221. doi:10.1016/S0169-409X(97)00121-X [8] F. Eeckman, K. Amighi and A. J. Moes, “Effect of Some Physiological and Non-Physiological Compounds on the Phase Transition Temperature of Thermoresponsive Poly- mers Intended for Oral Controlled-Drug Delivery,” In- ternational Journal of Pharmaceutics, Vol. 222, No. 2, 2001, pp. 259-270. [9] K. B. Doorty, T. A. Golubeva, A. V. Gorelov, Y. A. Rochev, L. T. Allen, K. A. Dawson, W. M. Gallagher and A. K. Keenan, “Poly(N-isopropylacrylamide) Co-Poly- mer Films as Potential Vehicles for Delivery of an An- timitotic Agent to Vascular Smooth Muscle Cells,” Car- diovascular Pa t hology, Vol. 12, No. 2, pp. 105-110. doi:10.1016/S1054-8807(02)00165-5 [10] S. J. Wilson, A. V. Gorelov, Y. A. Rochev, F. C. McGil- licuddy, K. A. Dawson, W. M. Gallagher and A. K. Keenan, “Extended Delivery of the Antimitotic Agent Colchicine from Thermoresponsive N-isopropylacryla- mide-Based Copolymer Films to Human Vascular Smooth Muscle Cells,” Journal of Biomedical Materials Research, Vol. 67, No. 2, 2003, pp. 667-673. [11] C. A. Kavanagh, T. A. Gorelova, I. I. Selezneva, Y. A. Rochev, K. A. Dawson, W. M. Gallagher, A. V. Gorelov and A. K. Keenan, “Poly(N-isopropylacrylamide) Co- polymer Films as Vehicles for the Sustained Delivery of Proteins to Vascular Endothelial Cells,” Journal of Bio- medical Materials Research, Vol. 72, No. 1, 2005, pp. 25-35. doi:10.1002/jbm.a.30192 [12] I. Lynch and K. A. Dawson, “Synthesis and Characteriza- tion of an Extremely Versatile Structural Motif Called the “Plum-Pudding” Gel,” Journal of Physical Chemistry, Vol. 107, No. 36, 2003, pp. 9629-9637. [13] F. C. McGillicuddy, “Evaluation of N-isopropylacryla- mide/N-tert-butylacrylamide Copolymer Microgel/Matrix Systems as Anti-Restenotic Drug Delivery Vehicles,” Ph.D. Thesis, 2006. [14] Y. Li and Y. Bae, “Volume Phase Transition of Submi- cron-Sized NIPAM/BAm Particles by Photon Correlation Spectroscopy,” Journal of Applied Polymer Science, Vol. 67, 1998, pp. 2088-2092. [15] B. R. Kwak, F. Mulhaupt and F. Mach, “Atherosclerosis: Anti-Inflammatory and Immunomodulatory Activities of Statins,” Autoimmunity Reviews, Vol. 2, No. 6, 2003, pp. 332-338. doi:10.1016/S1568-9972(03)00049-1 [16] R. Riessen, D. I. Axel, M. Fenchel, U. U. Herzog, H. Rossmann and K. R. Karsch, “Effect of HMG-CoA Re- ductase Inhibitors on Extracellular Matrix Expression in Human Vascular Smooth Muscle Cells,” Basic Research in Cardiology, Vol. 94, No. 5, 1999, pp. 322-332. [17] B. Jaschke, C. Michaelis, S. Milz, M. Vogeser, T. Mund, L. Hengst, et al., “Local Statin Therapy Differentially In- terferes with Smooth Muscle and Endothelial Cell Prolif- eration and Reduces Neointima on a Drug-Eluting Stent Platform,” Cardiovascular Research, Vol. 68, No. 3, 2005, pp. 483-492. doi:10.1016/j.cardiores.2005.06.029 [18] N. Takeda, M. Kondo, S. Ito, Y. Ito, K. Shimokata and H. Kume, “Role of RhoA Inactivation in Reduced Cell Pro- liferation of Human Airway Smooth Muscle by Simvas- tatin,” American Journal of Respiratory Cell and Mo- lecular Biology, Vol. 35, No. 6, 2006, pp. 722-729. doi:10.1165/rcmb.2006-0034OC [19] J. M. Corpataux, J. Naik, K. E. Porter and N. J. London, “The Effect of Six Different Statins on the Proliferation, Migration, and Invasion of Human Smooth Muscle Cells,” Journal of Surgery Research, Vol. 129, No. 1, 2005, pp. 52-56. doi:10.1016/j.jss.2005.05.016 [20] F. C. McGillicuddy, I. Lynch, Y. A. Rochev, M. Burke, K. A. Dawson, W. M. Gallagher and A. K. Keenan, “Novel “Plum Pudding” Gels as Potential Drug-Eluting Stent Coatings: Controlled Release of Fluvastatin,” Journal of Biomedical Materials Research, Vol. 79, No. 4, 2006, pp. 923-933. doi:10.1002/jbm.a.30839 [21] J. F. Granada and N. S. Kleiman, “Therapeutic Use of Intravenous Eptifibatide in Patients Undergoing Percuta- neous Coronary Intervention: Acute Coronary Syndromes and Elective Stenting,” American Journal of Cardiovas- cular Drugs, Vol. 4, No. 1, 2004, pp. 31-41. [22] C. Patrono, F. Bachmann, C. Baigent, C. Bode, R. De Caterina, B. Charbonnier, et al., “Expert Consensus Document on the Use of Antiplatelet Agents. The Task Force on the Use of Antiplatelet Agents in Patients with Atherosclerotic Cardiovascular Disease of the European Society of Cardiology,” European Heart Journal, Vol. 25, No. 2, 2004, pp. 166-181. doi:10.1016/j.ehj.2003.10.013 [23] W. W. O'Neill, P. Serruys, M. Knudtson, G. A. van Es, G. C. Timmis, C. van der Zwaan, et al., “Long-Term Treat- ment with a Platelet Glycoprotein-Receptor Antagonist after Percutaneous Coronary Revascularization. EXCITE Trial Investigators. Evaluation of Oral Xemilofiban in Controlling Thrombotic Events,” New England Journal of Medicine, Vol. 342, No. 18, 2000, pp. 1316-1324. [24] C. P. Cannon, C. H. McCabe, R. G. Wilcox, A. Langer, A. Caspi, P. Berink, et al., “Oral Glycoprotein IIb/IIIa Inhi- bition with Orbofiban in Patients With Unstable Coronary Syndromes (OPUS-TIMI 16) Trial,” Circulation, Vol. 102, No. 2, 2000, pp. 149-156.  Simultaneous Release of a Hydroxy-Methylglutaryl Coenzyme A Reductase Inhibitor and a Glycoprotein IIb/IIIa Antagonist from a Thermoresponsive NiPAAm/NtBAAm Copolymer System Copyright © 2011 SciRes. JBNB 27 [25] L. K. Newby, “Long-Term Oral Platelet Glycoprotein IIb/IIIa Receptor Antagonism with Sibrafiban after Acute Coronary Syndromes: Study Design of the Sibrafiban Versus Aspirin to Yield Maximum Protection from Ischemic Heart Events Post-Acute Coronary Syndromes (SYMPHONY) Trial,” Symphony Steering Committee, American Heart Journal, Vol. 138, 1999, pp. 210-218. doi:10.1016/S0002-8703(99)70104-3 [26] SYMPHONY, “Comparison of Sibrafiban with Aspirin for Prevention of Cardiovascular Events after Acute Coronary Syndromes: A Randomised Trial. The SYM- PHONY Investigators. Sibrafiban Versus Aspirin to Yield Maximum Protection from Ischemic Heart Events Post- acute Coronary Syndromes,” Lancet, Vol. 355, 2000, pp. 337-345. doi:10.1016/S0140-6736(99)11179-6 [27] E. J. Topol, J. D. Easton, P. Amarenco, R. Califf, R. Har- rington, C. Graffagnino, et al., “Design of the Blockade of The Glycoprotein IIb/IIa Receptor to Avoid Vascular Occlusion (BRAVO) Trial,” American Heart Journal, Vol. 139, 2000, pp. 927-933. [28] V. S. Srinivas, B. Skeif, A. Negassa, J. Y. Bang, H. Shaqra and E. S. Monrad, “Effectiveness of Glycoprotein IIb/IIa Inhibitor Use during Primary Coronary Angio- plasty: Results of Propensity Analysis Using the New York State Percutaneous Coronary Intervention Reporting Sys te m, ” American Journal of Cardiology, Vol. 99, 2007, pp. 482-485. doi:10.1016/j.amjcard.2006.08.061 [29] T. Heer, U. Zeymer, C. Juenger, A. K. Gitt, H. Wienber- gen, R. Zahn, et al., “Beneficial Effects of Abciximab in Patients with Primary Percutaneous Intervention for Acute ST Segment Elevation Myocardial Infarction in Clinical Practice,” Heart, Vol. 92, 2006, pp. 484-489. [30] T. Yin, G. Wang, C. Ruan, R. Guzman and R. Guidoin, “In-Vitro Assays of Polymer-Coated Stents Eluting Platelet Glycoprotein IIb/IIIa Receptor Monoclonal An- tibody,” Journal of Biomedical Materials Research, Vol. 83, 2007, pp. 861-867. doi:10.1002/jbm.a.31369 [31] R. K. Aggarwal, D. C. Ireland, M. A. Azrin, M. D. Eze- kowitz, D. P. de Bono and A. H. Gershlick, “Antithrom- botic Potential of Polymer-Coated Stents Eluting Platelet Glycoprotein IIb/IIIa Receptor Antibody,” Circulation, Vol. 94, No. 12, 1996, pp. 3311-3317. |