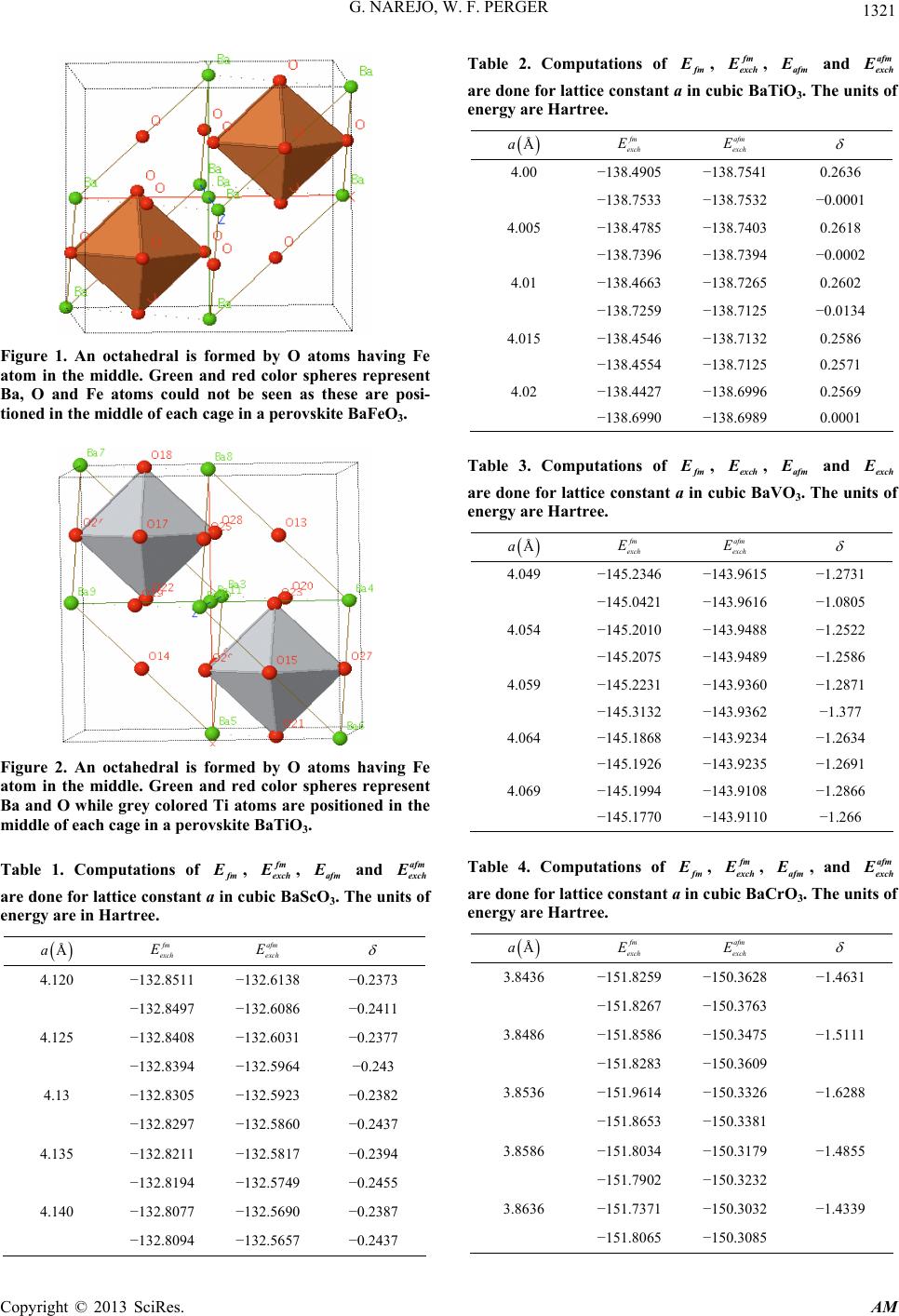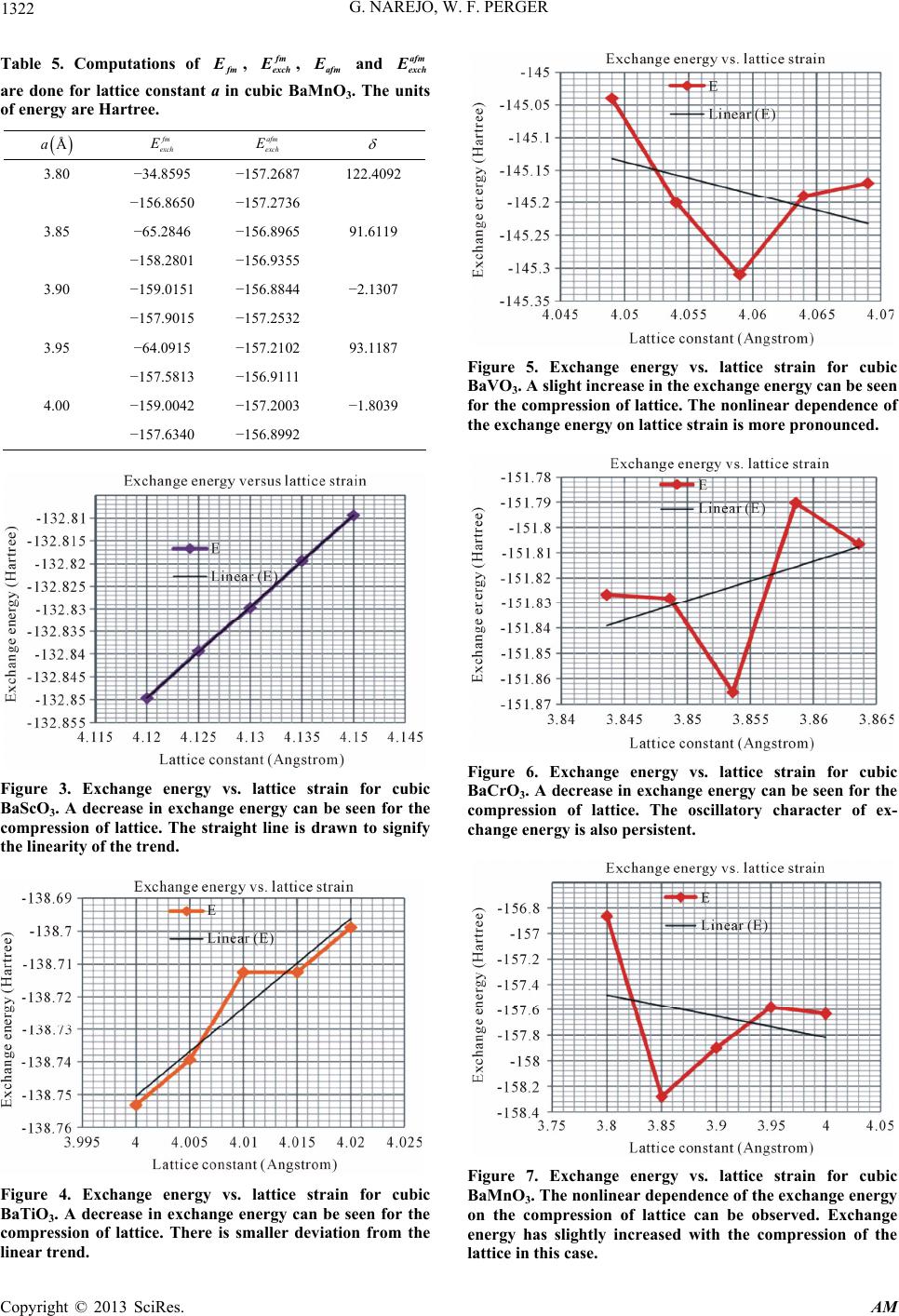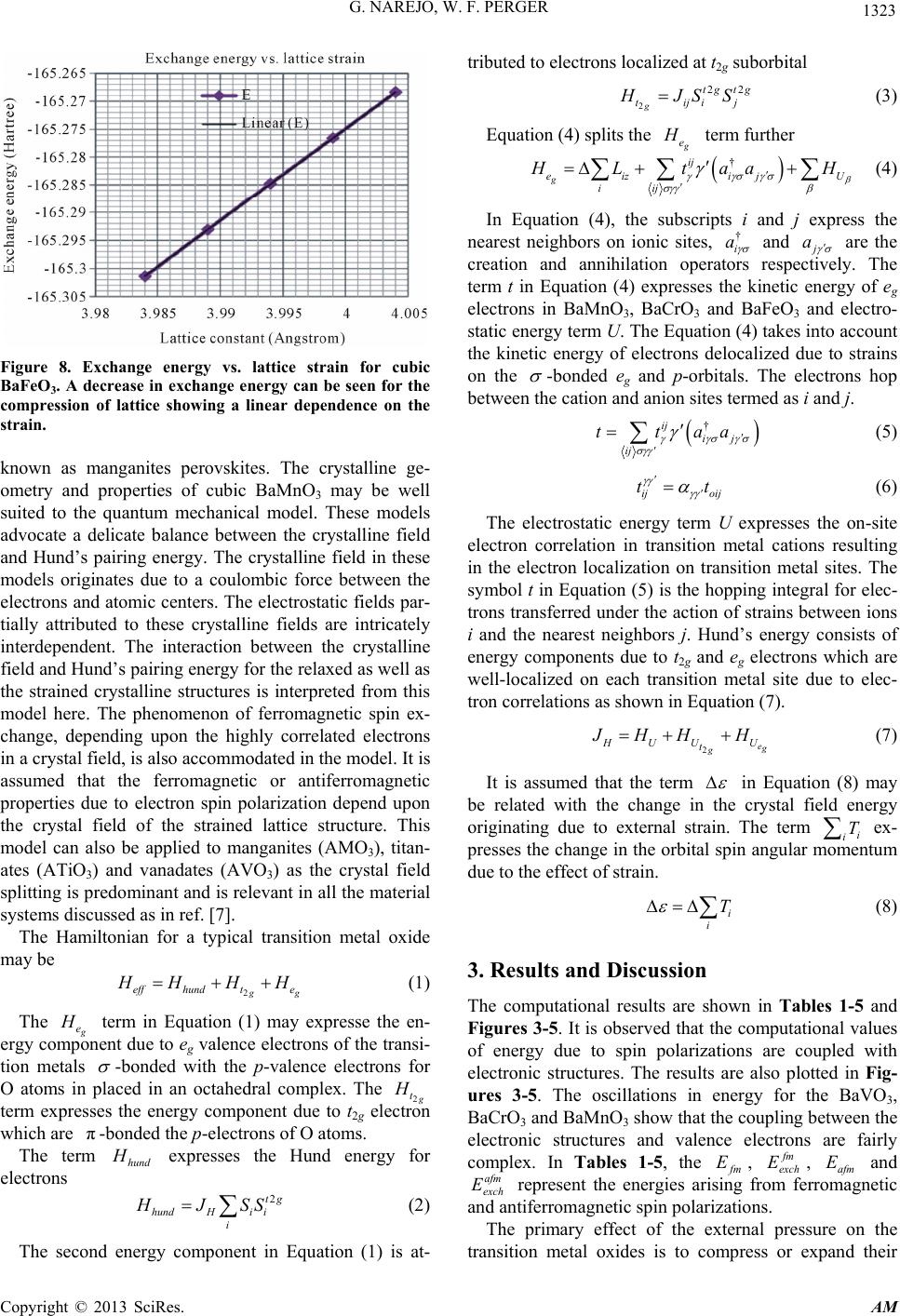
G. NAREJO, W. F. PERGER
1324
bond lengths connecting the transition metal and oxygen
atoms in a perovskite. The expansion and contraction of
the bond length result in the weakening or strengthening
of the electron interactions within crystal structures
among the transition metal eg electrons and oxygen p
electrons. The interactions between electrons and ions
couple them in a complicated manner.
Moreover, these interactions are facilitated by the
strains only if there are enough numbers of electrons in
eg valence orbitals. This phenomenon can be observed in
the computational results obtained for BaCrO3, BaMnO3
and BaFeO3. Less variations in energy as a function of
lattice strains for some oxides is a function of the local-
ized nature of the t2g and eg electron orbitals.
The oxides of transition metal have varied number of
electrons in their highly correlated d-orbitals. The con-
tracted wavefunctions of d electrons in BaScO3, BaTiO3,
BaVO3, BaMnO3 and BaFeO3 experience the varied de-
gree of competitive forces of the coulomb repulsion ver-
sus hybridization. The former tries to localize the elec-
trons at atomic lattice sites while the latter favors the
overlaps with p- and d-orbitals of O and transition metal
to delocalize these electrons. The forces of coulomb re-
pulsion and hybridization are varied by lattice strain. A
trend can be seen in all computations as there is a con-
sistent decrease in energy for the compression and in-
crease in energy for expansion of lattice volume. The
chemical bond in transition metal oxides is a combina-
tion of covalent and ionic parts. The covalent and ionic
parts vary as the transition metal ionic radius increases
from Sc to Fe. The contribution of ionic bonding is in-
creased as the number of electrons in transition metals
are increased with more impact on the energy as a func-
tion of lattice strain. The computational results of
BaScO3, BaTiO3, BaVO3, BaCrO3, BaFeO3 show sig-
nificant variations in chemical bonding from strongly
covalent to moderately ionic in nature for the materials
tested.
From the computed results shown in Tables 1-6, an
increase in the energy is observed for spins polarized in
same direction for all crystalline systems tested confirm-
ing the coupling between the crystalline structure and
electron spin polarization.
4. Concluding Remarks
We have employed first principles computations to ex-
tract the coupling between the crystalline structure and
electron spin polarization. The optimized crystalline
structures are computed by a variety of methods for each
of the transition metal oxides. Later on, the coupling be-
tween the electronic structure and electron spin polariza-
tion is determined by computing the energy for the spins
aligned in the parallel as well as antiparallel polariza-
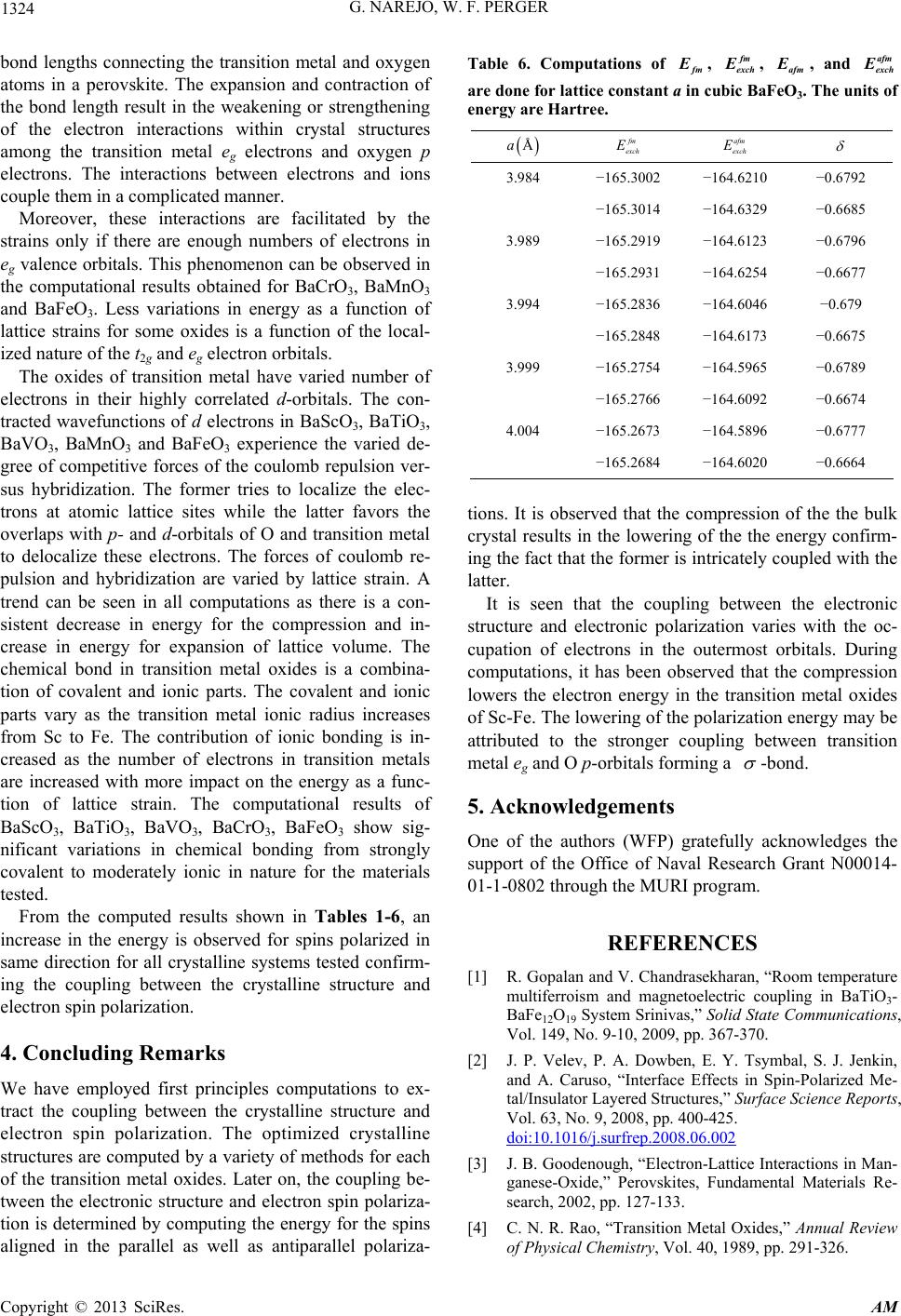
Table 6. Computations of fm
, fm
exch
, afm
, and afm
exch
are done for lattice constant a in c ubic BaFeO3. The units of
energy are Hartree.
Åa fm
exch
E afm
exch
E
3.984 −165.3002 −164.6210 −0.6792
−165.3014 −164.6329 −0.6685
3.989 −165.2919 −164.6123 −0.6796
−165.2931 −164.6254 −0.6677
3.994 −165.2836 −164.6046 −0.679
−165.2848 −164.6173 −0.6675
3.999 −165.2754 −164.5965 −0.6789
−165.2766 −164.6092 −0.6674
4.004 −165.2673 −164.5896 −0.6777
−165.2684 −164.6020 −0.6664
tions. It is observed that the compression of the the bulk
crystal results in the lowering of the the energy confirm-
ing the fact that the former is intricately coupled with the
latter.
It is seen that the coupling between the electronic
structure and electronic polarization varies with the oc-
cupation of electrons in the outermost orbitals. During
computations, it has been observed that the compression
lowers the electron energy in the transition metal oxides
of Sc-Fe. The lowering of the polarization energy may be
attributed to the stronger coupling between transition
metal eg and O p-orbitals forming a
-bond.
5. Acknowledgements
One of the authors (WFP) gratefully acknowledges the
support of the Office of Naval Research Grant N00014-
01-1-0802 through the MURI program.
REFERENCES
[1] R. Gopalan and V. Chandrasekharan, “Room temperature
multiferroism and magnetoelectric coupling in BaTiO3-
BaFe12O19 System Srinivas,” Solid State Communications,
Vol. 149, No. 9-10, 2009, pp. 367-370.
[2] J. P. Velev, P. A. Dowben, E. Y. Tsymbal, S. J. Jenkin,
and A. Caruso, “Interface Effects in Spin-Polarized Me-
tal/Insulator Layered Structures,” Surface Science Reports,
Vol. 63, No. 9, 2008, pp. 400-425.
doi:10.1016/j.surfrep.2008.06.002
[3] J. B. Goodenough, “Electron-Lattice Interactions in Man-
ganese-Oxide,” Perovskites, Fundamental Materials Re-
search, 2002, pp. 127-133.
[4] C. N. R. Rao, “Transition Metal Oxides,” Annual Review
of Physical Chemistry, Vol. 40, 1989, pp. 291-326.
Copyright © 2013 SciRes. AM