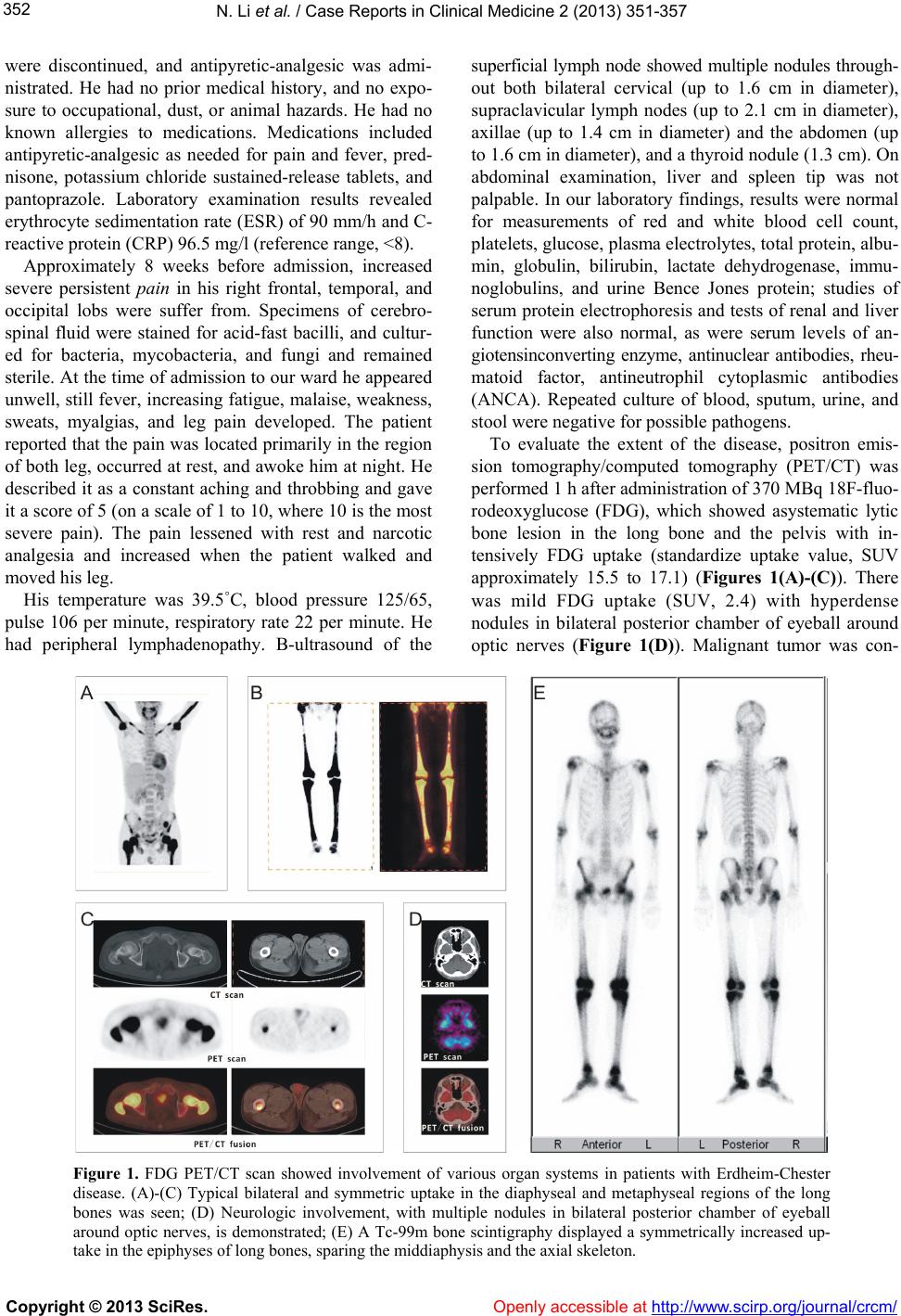
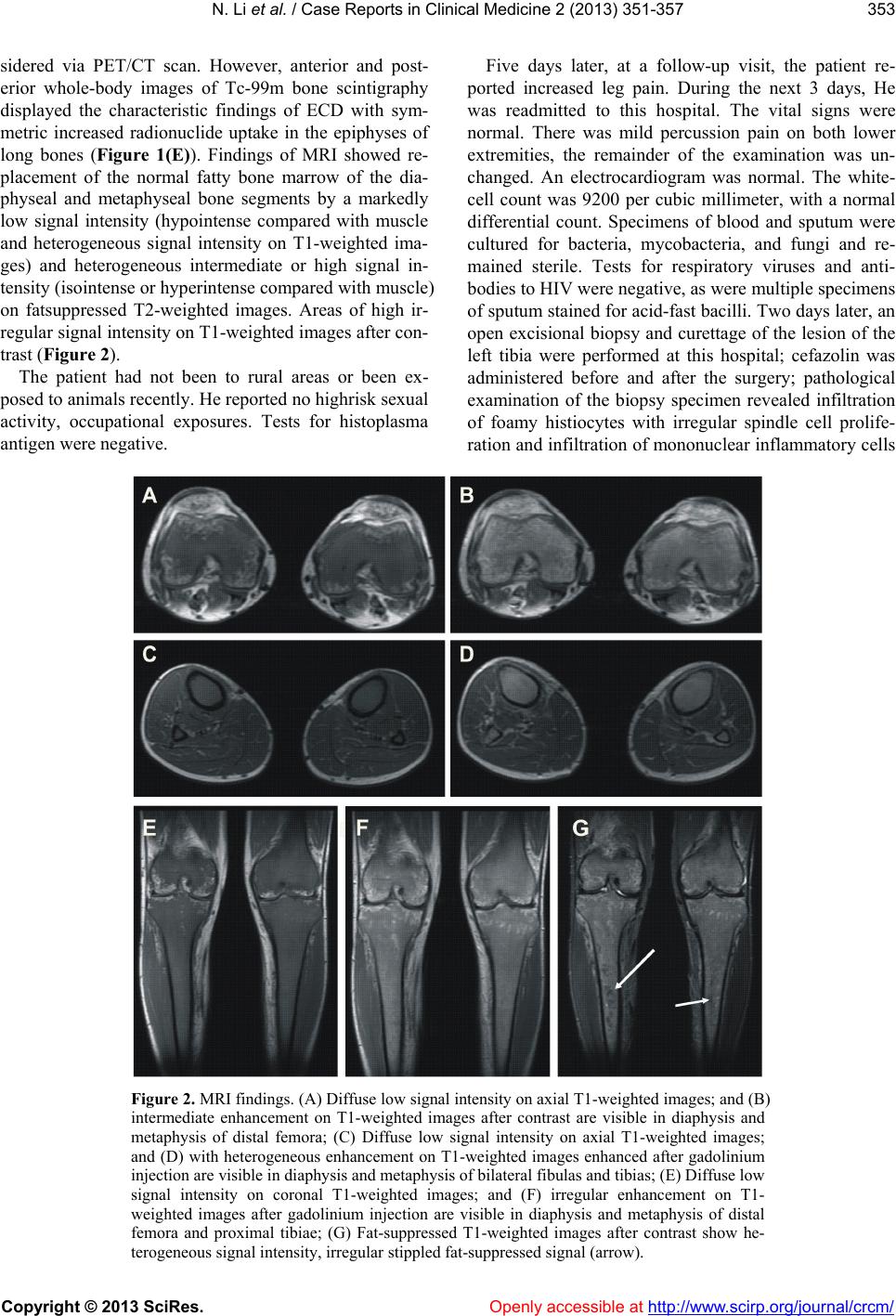
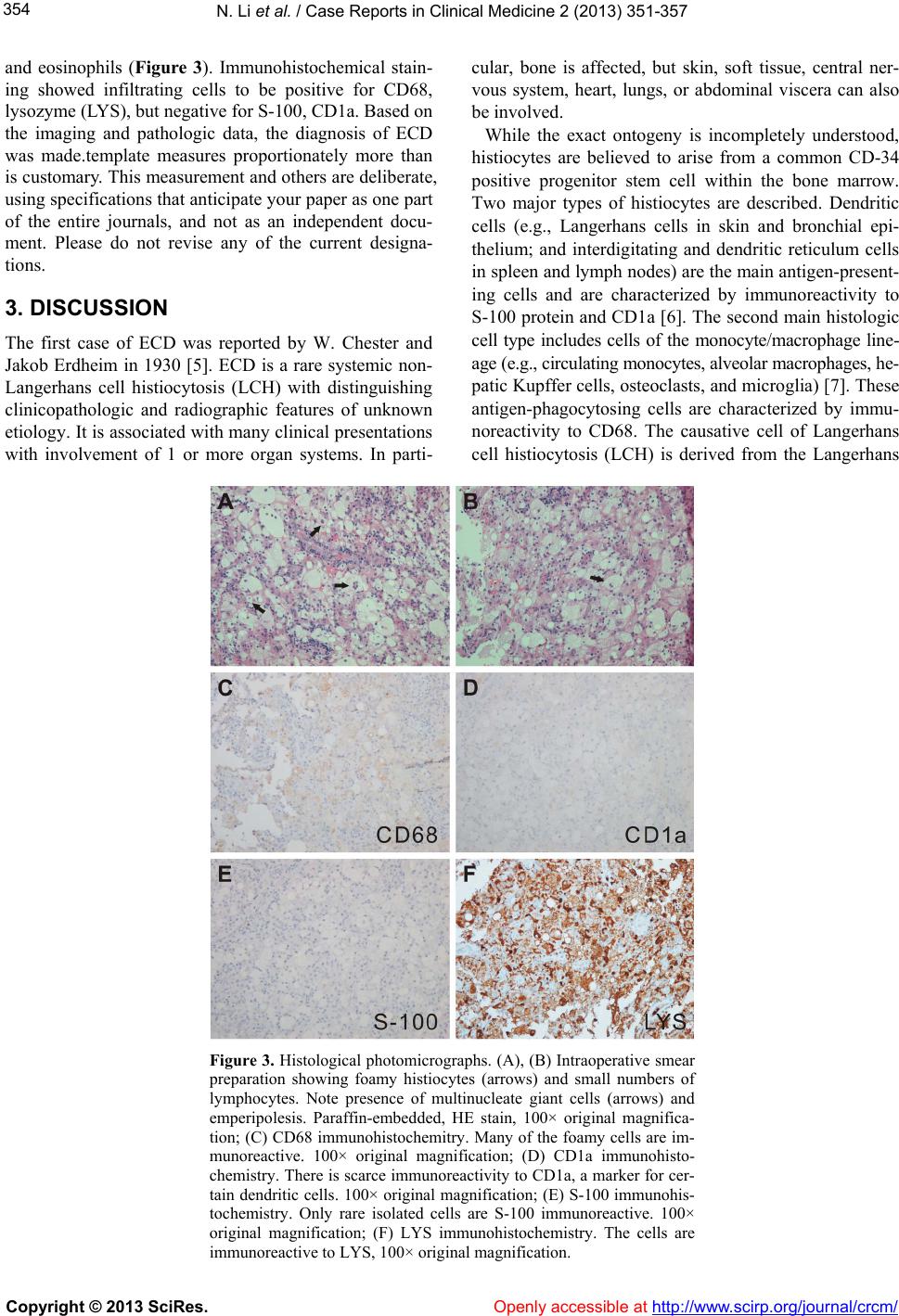
N. Li et al. / Case Reports in Clinical Medicine 2 (2013) 35 1-35 7
356
kine/chemokine levels in ECD patients compared with
controls, with principal component analysis identifying
an ECD-specific signature based on increased expression
of IFN-α, IL-1/IL-1RA, IL-6, IL-12, monocyte chemo-
tactic protein-1, and decreased expression of IL-4 and
IL-7. The authors conclude that the systemic immune
Th-1-oriented perturbation associated with this condition,
and provides clues for the choice of more focused
therapeutic agents.
4. CONCLUSION
ECD is a rare form of non-Langerhans’ cell histiocy-
tosis. ECD appears to be the pathognomonic clinical,
radiographic and histological features. Diagnosis of ECD
is based on the identification in tissue biopsy of histio-
cytes, which are typically foamy and immunostain for
CD68+ CD1a−. Central nervous system involvement is a
major prognostic factor in ECD. Because the incidence
of ECD is low, the data from literature remains limited.
More case data for this histiocytosis need therefore be
obtained for a better understanding of its characteristics
and an exploration of appropriate therapy.
REFERENCES
[1] Aouba, A., Georgin-Lavialle, S., Pagnoux, C., Silva,
N.M., Renand, A., Galateau-Salle, F., et al. (2010) Ra-
tionale and efficacy of interleukin-1 targeting in Erdheim-
Chester disease. Blood, 116, 4070-4076.
doi:10.1182/blood-2010-04-279240
[2] Haroche, J., Arnaud, L. and Amoura, Z. (2012) Erdheim-
Chester disease. Current Opinion in Rheumatology, 24,
53-59. doi:10.1097/BOR.0b013e32834d861d
[3] Veyssier Belot, C., Cacoub, P., Caparros Lefebvre, D.,
Wechsler, J., Brun, B., Remy, M., et al. (1996) Erdheim-
Chester disease—Clinical and radiologic characteristics
of 59 cases. Medicine, 75, 157-169.
doi:10.1097/00005792-199605000-00005
[4] Wimpissinger, T.F., Schernthaner, G., Feichtinger, H. and
Stackl, W. (2005) Compression of kidneys in Erdheim-
Chester disease of retroperitoneum: Open surgical ap-
proach. Urology, 65, E29-E31.
doi:10.1016/j.urology.2004.10.051
[5] Chester, W. (1930) The lipogranulomatosis. Virchows
Archiv für Pathologische Anatomie und Physiologie und
für Klinische Medizin, 279, 561-602.
doi:10.1007/BF01942684
[6] Kairouz, S., Hashash, J., Kabbara, W., McHayleh, W. and
Tabbara, I.A. (2007) Dendritic cell neoplasms: An over-
view. American Journal of Hematology, 82, 924-928.
doi:10.1002/ajh.20857
[7] Zelger, B.W.H., Sidoroff, A., Orchard, G. and Cerio, R.
(1996) Non-Langerhans cell histiocytoses—A new uni-
fying concept. American Journal of Dermatopathology,
18, 490-504. doi:10.1097/00000372-199610000-00008
[8] Dion, E., Graef, C., Miquel, A., Haroche, J., Wechsler, B.,
Amoura, Z., et al. (2006) Bone involvement in Erdheirn-
Chester disease: Imaging findings including periostitis
and partial epiphyseal involvement. Radiology, 238, 632-
639. doi:10.1148/radiol.2382041525
[9] Allen, C.E. and McClain, K.L. (2011) Erdheim-Chester:
Beyond the lesion. Blood, 11 7, 2745-2746.
doi:10.1182/blood-2011-01-330233
[10] Wright, R.A., Hermann, R.C. and Parisi, J.E. (1999) Neu-
rological manifestations of Erdheim-Chester disease. Jour-
nal of Neurology, Neurosurgery & Psychiatry, 66, 72-75.
doi:10.1136/jnnp.66.1.72
[11] Perras, B., Petersen, D., Lorch, H. and Fehm, H.L. (2002)
Psychoneuroendocrine disturbances in a patient with a
rare granulomatous disease. Experimental and Clinical
Endocrinology & Diabetes, 110, 248-252.
doi:10.1055/s-2002-33075
[12] Naqi, R., Azeemuddin, M., Idrees, R. and Wasay, M.
(2010) Meningioma-like lesions in Erdheim Chester dis-
ease. Acta Neurochirurgica, 152, 1619-1621.
doi:10.1007/s00701-010-0655-0
[13] Donaldson, G., Bullock, P. and Monson, J.P. (2010) Erd-
heim-Chester disease mimicking multiple meningiomas.
British Journal of Neurosurgery, 24, 296-297.
doi:10.3109/02688691003624612
[14] Arnaud, L., Pierre, I., Beigelman-Aubry, C., Capron, F.,
Brun, A.L., Rigolet, A., et al. (2010) Pulmonary in-
volvement in Erdheim-Chester disease: A single-center
study of thirty-four patients and a review of the literature.
Arthritis & Rheumatism, 62, 3504-3512.
doi:10.1002/art.27672
[15] Haroche, J., Amoura, Z., Touraine, P., Seilhean, D., Graef,
C., Birmele, B., et al. (2007) Bilateral adrenal infiltration
in Erdheim-Chester disease. Report of seven cases and
literature review. Journal of Clinical Endocrinology &
Metabolism, 92, 2007-2012. doi:10.1210/jc.2006-2018
[16] Stoppacciaro, A., Ferrarini, M., Salmaggi, C., Colarossi,
C., Praderio, L., Tresoldi, M., et al. (2006) Immunohis-
tochemical evidence of a cytokine and chemokine net-
work in three patients with Erdheim-Chester disease—
Implications for pathogenesis. Arthritis and Rheumatism,
54, 4018-4022. doi:10.1002/art.22280
[17] Dagna, L., Girlanda, S., Langheim, S., et al. (2010) Erd-
heim-Chester disease: Report on a case and new insights
on its immunopathogenesis. Rheumatology, 49, 1203-
1206. doi:10.1093/rheumatology/kep461
[18] Braiteh, F., Boxrud, C., Esmaeli, B. and Kurzrock, R.
(2005) Successful treatment of Erdheim-Chester disease,
a non-Langerhans-cell histiocytosis, with interferon-alpha.
Blood, 106, 2992-2994. doi:10.1182/blood-2005-06-2238
[19] Jeon, I.-S., Lee, S.S. and Lee, M.K. (2010) Chemother-
apy and Interferon-alpha treatment of Erdheim-Chester
disease. Pediatric Blood & Cancer, 55, 745-747.
doi:10.1002/pbc.22636
[20] Suzuki, H.I., Hosoya, N., Miyagawa, K., Ota, S., Naka-
shima, H., Makita, N., et al. (2010) Erdheim-Chester dis-
ease: Multisystem involvement and management with in-
terferon-alpha. Leukemia Research, 34, E21-E24.
doi:10.1016/j.leukres.2009.07.026
Copyright © 2013 SciRes. Openly accessible at http://www.sc irp.or g/journal/crcm/