Paper Menu >>
Journal Menu >>
 Pharmacology & Pharmacy, 2011, 2, 10-16 doi:10.4236/pp.2011.21002 Published Online January 2011 (http://www.SciRP.org/journal/pp) Copyright © 2011 SciRes. PP Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Mitsue Ishisaka1, Takashi Kudo2, Masamitsu Shimazawa1, Kenichi Kakefuda1, Atsushi Oyagi1, Kana Hyakkoku1, Kazuhiro Tsuruma1, Hideaki Hara1 1Molecular Pharmacology, Department of Biofunctional Evaluation, Gifu Pharmaceutical University; 2Department of Psychiatry, Osaka University Graduate School of Medicine. Email: hidehara@gifu-pu.ac.jp Received October 26th, 2010; revised November 23rd, 2010; accepted November 30th, 2010. ABSTRACT Depression is a significant public health concern but its pathology remains unclear. Previously, increases in an endo- plasmic reticulum (ER) stress-related protein were reported in the temporal cortex of subjects with major depressive disorder who had died by suicide. This finding suggests an association between depression and ER stress. The present study was designed to investigate whether acute stress co uld affect th e ER stress response. Mice were immobilized fo r a period of 6 hr and then expression of ER stress response-related genes was measured by real-time PCR. We also used enzyme-linked immunosorbent assay for concomitant measurement of the plasma corticosterone levels in the mice. The effect of corticosterone on ER stress proteins was fu rther investigated by treatin g mice with corticosterone for 2 weeks and then measuring ER protein expression by Western blotting. After a 6 hr restraint stress, mRNA levels of ER stress-related genes, such as the 78-kilodalton glucose regulated protein (GRP78), the 94-kilodalton glucose regulated protein (GRP94), and calreticulin, were increased in the cortex, hippocampus, and striatum of mouse brain. Blood plasma corticosterone level was also increased. In the corticosterone-treated mouse model, the expression of GRP78 and GRP94 was significan tly increased in the hippo campus. These re sults suggest tha t acute stress may affect ER func- tion and that ER stress may be involve d in the pathogenesis of restraint stress, including the development of depression. Keywords: Corticosteron e, Depression, Endoplasmic Reticulum Stress, Restraint Stress 1. Introduction Major depression, along with bipolar disorder, has be- come a common psychiatric disorder in modern society. About 1% of the population is estimated to be affected by major depression one or more times during their life- time [1]. Even though extensive studies have led to a variety of hypotheses regarding the molecular mecha- nism underlying depression, the pathogenesis of this dis- order remains to be fully elucidated. The endoplasmic reticulum (ER) is the cell organelle where secretory and membrane proteins are synthesized and folded. It also functions as a Ca2+ store and resource of calcium signals. The disturbance of ER functions through events such as disruption of Ca2+ homeostasis, inhibition of protein glycosylation or disulfide bond for- mation, hypoxia and viral or bacterial infection, can re- sult in the accumulation of unfolded or misfolded pro- teins and may trigger stress responses in the cell (ER stress). To overcome ER stress, an unfolded protein re- sponse (UPR) is invoked by the activation of several signaling pathways; this UPR promotes an adaptive re- sponse to ER stress and reestablishes homeostasis in the ER [2,3]. Molecular chaperones such as the 78-kilodalton glucose regulated protein (GRP78) and the 94-kilodalton glucose regulated protein are induced and promote cor- rect protein folding. If the damage is too severe to repair, C/EBP-homologous protein (CHOP) and other factors are activated and induce cell apoptosis [4]. On the other hand, if misfolded protein aggregates into insoluble higher-order structures, it can give rise to various dis- eases. For example, rhodopsin misfolding causes auto- somal dominant retinitis pigmentosa [5], while the ac- cumulation of amyloid β-peptide is associated with Alz- heimer’s disease [6].  Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 11 Some reports have also suggested a relationship be- tween mental disorder and ER stress. In bipolar disorder patients, DNA microarray analysis of cell derived from twins discordant with respect to the disease revealed a down-regulated expression of genes related to ER stress responses such as x-box binding protein 1 (XBP1) and GRP78 [7]. In schizophrenia patients, a similar abnor- mality of these genes was found [8]. In addition, mood-stabilizing drugs such as valproate and lithium have been reported to increase the expression of GRP78, GRP94, and calreticulin [9]. Similarly, olanzapine, one of the second-generation “atypical” anti-psychotic drugs, appears to potentiates neuronal survival and neural stem cell differentiation by regulation of ER stress response proteins [10]. A recent study reported that significantly increased levels of GRP78, GRP94, and calreticulin were found in the temporal cortex of subjects with major depressive disorder who had died by suicide compared with control subjects who had died of other causes [11]. In addition, hippocampal atrophy [12] and reduction of glial density in the subgenual prefrontal cortex [13] were found in patients with major depression. Stress, a risk factor for depression, has been shown to induce atrophy of the api- cal dendrites of the hippocampal neurons [14], and to promote neuronal apoptosis in the cerebral cortex [15] in animal depression models. These findings suggest that a stressful situation, which may increase the risk for sui- cide, serves as an ER stressor. To clarify the relationship between exogenous stress and ER stress, in the present study, we investigated the expression of ER stress-related genes after restraint stress. We also focused on the eleva- tion of corticosterone in the plasma and used a corticos- terone-treated depression model to clarify the relation- ship between chronic corticosterone elevation and ER stress. 2. Materials and Methods 2.1. Animals Male 9-week-old ddY mice and male 6-week-old ICR mice (Japan SLC, Hamamatsu, Japan) were used for all experiments. Mice were housed at 24 ± 2˚C under a 12 hr light-dark cycle (lights on from 8:00 to 20:00) and had ad libitum access to food and water when not under restraint. Animals were acclimatized to laboratory conditions be- fore the experiment. All procedures relating to animal care and treatment conformed to the animal care guide- lines of the Animal Experiment Committee of Gifu Pharmaceutical University. All efforts were made to minimize both suffering and the number of animal used. 2.2. Restraint Stress Male 9-week-old ddY mice (Japan SLC) weighing 30-40 g were used for real-time PCR studies. Mice were placed into 50-mL perforated plastic tubes, which prevented them from turning in any direction. Each mouse was maintained in the tube for 6 hr without any access to food or water. 2.3. Sampling After this restraint stress, a blood sample was collected from the tail and the mouse was decapitated. The brain was quickly removed from the skull, briefly washed in ice-cold saline, and laid on a cooled (4˚C) metal plate. The brain was rapidly dissected to separate the hippo- campus, striatum, and cortex and stored at -80°C until use. 2.4. RNA Isolation Total RNA was isolated from frozen brain using High Pure RNA Isolation Kit (Roche, Tokyo, Japan). RNA concentrations were determined spectrophotometrically at 260 nm. First-standed cDNA was synthesized in a 20-μl reaction volum using a random primer (Takara, Shiga, Japan) and Moloney murine leukemia virus re- verse transcriptase (Invitrogen, Carlsbad, CA, USA). 2.5. Reai-Time PCR Real-time PCR (TaqMan; Applied Biosystems, Foster City, CA, USA) was performed as described previously [16]. Single-standard cDNA was synthesized from total RNA using a high capacity cDNA archive kit (Applied Biosystems). Quantitative real-time PCR was performed using a sequence detection system (ABI PRISM 7900HT; Applied Biosystems) with a PCR master mix (TaqMan Universal PCR Master Mix; Applied Biosystems), ac- cording to the manufacturer’s protocol. A gene expression product (Assays-on-Demand Gene Expression Product; Applied Biosystems) was used for measurements of mRNA expression by real-time PCR. The primers used for amplification were as follows: GRP78: 5’-GTTTG CTGAGGAAGACAAAAAGCTC-3’ and 5’-CACTTCC ATAGAGTTTGCTGATAATTG-3’; CHOP: 5’-GGAG CTGGAAGCCTGGTATGAGG-3’ and 5’-TCCCTGGT CAGGCGCTCGATTTCC-3’; GRP94: 5’-CTCACCATT TGGATCCTGTGTG-3’ and 5’-CACATGACAAGATT TTACATCAAGA-3’; calreticulin: 5’-GCCAAGGACG AGCTGTAGAGAG-3’ and 5’-GGTGAGGGCTGAAG GAGAATC-3’; ERdj4: 5’-TCTAGAATGGCTACTCCC CAGTCAATTTTC-3’ and 5’-TCTAGACTACTGTCCT GAACAGTCAGTG-3’; EDEM: 5’-TGGGTTGGAAAG CAGAGTGGC-3’ and 5’-TCCATTCCTACATGGAGG TAG-3’; p58IPK 5’-GAGGTTTGTGTTTGGGATGCAG- 3’ and 5’-GCTCTTCAGCTGACTCAATCAG-3’; ASNS: 5’-AGGTTGATGATGCAATGATGG-3’ and 5’-TCCC CTATCTACCCACAGTCC-3’; β-actin: 5’-TCCTCCCT  Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 12 GGAGAAGAGCTAC-3’ and 5’-TCCTGCTTGCTGAT CCACAT-3’ The thermal cycler conditions were as fol- lows: 2 min at 50˚C and then 10 min at 95˚C, followed by two-step PCR for 50 cycles consisting of 95˚C for 15s followed by 60˚C for 1 min. For each PCR measurement, we checked the slope value, R2 value, and linear range of a standard curve of serial dilutions. All reactions were performed in duplicate. The results were expressed rela- tive to a β-actin internal control. 2.6. Measurement of Plasma Corticosterone Plasma was obtained as described previously [17] and the concentration of corticosterone was determined via a corticosterone EIA kit (Assay Designs, Inc., Ann Arbor, MI, USA) according to the manufacturer’s protocol. 2.7. Chronic Corticosterone Treatment Male 6-week-old ICR mice (Japan SLC) weighing 20-25 g were used for chronic oral corticosterone exposure as described in a previous report [18]. Briefly, corticoster- one (25 μg/mL free base; 4-pregnen-11β 21-DIOL-3 20-DIONE 21-hemisuccinate; Steraloids, Inc., RI, USA) was add to tap water and the pH was brought to 12-13 with 10 N NaOH (Kishidai Chemical, Osaka, Japan), followed by stirring at 4˚C until dissolved (3 to 7 hr). Following dissolution, the pH was brought to 7.0-7.4 with 10 N HCl (Wako, Osaka, Japan). Group-housed ICR mice were presented with this corticosterone solu- tion in place of normal drinking water for 14 days, re- sulting in a dose of approximately 8.7 mg/kg/day (p.o). Animals were weaned with 3 days of 12.5 μg/mL, and then 3 days with 6.25 μg/mL, to allow for gradual recov- ery of endogenous corticosterone secretion. 2.8. Western Blot Analysis At 35 days, each mouse was decapitated and its brain was quickly removed from the skull, briefly washed in ice-cold saline, and laid on a cooled (4˚C) metal plate. The brain was rapidly dissected to separate the hippo- campus and stored at -80˚C until use. Brain samples were homogenized in 10 mL/g tissue ice-cold lysis buffer [50 mM Tris-HCl (pH 8.0) containing 159 mM NaCl, 50 mM EDTA, 1% Triton X-100, and protease/phosphatase in- hibitor mixture] using a homogenizer (Physcotron; Mi- crotec Co. Ltd., Chiba, Japan). Lysates were centrifuged at 12,000×g for 15 min at 4˚C. Supernatants were col- lected and boiled for 5 min in SDS sample buffer (Wako). Equal amounts of protein were subjected to 10% SDS- PAGE gradient gel and then transferred to poly (vi- nylidene difluoride) membranes (Immobilon-P; Millipore, MA, USA). After blocking with Block Ace (Snow Brand Milk Products Co. Ltd., Tokyo, Japan) for 30 min, the membranes were incubated with primary antibody. The primary antibodies used were as follows: mouse anti-BiP antibody (BD Bioscience, CA, USA) for GRP78, mouse anti-KDEL antibody (Stressgen Bioreagents Limited Partnership, B.C., Canada) for GRP94, and mouse anti- actin antibody (Sigma-Aldrich, St. Louis, MO, USA). Subsequently, the membrane was incubated with the secondary antibody [goat anti-mouse (Pierce Biotech- nology, IL, USA)]. The immunoreactive bands were visualized using Super Signal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology) and then measured using LAS-4000 mini (Fujifilm, Tokyo, Ja- pan). 2.9. Statistical Analysis Statistical comparisons were made by Student’s t-test using Statview version 5.0 (SAS Institute Inc., NC, USA), with p < 0.05 being considered statistically significant. 3. Results and Discussion Real-time PCR was carried out to investigate whether the expression of ER stress response-related genes in the brain was changed by 6-hr restraint stress. In this study, we investigated the expression of GRP94, carleticulin, ER degradation-enhancing α-mannosidase-like protein (EDEM), protein kinase inhibitor of 58 kDa (p58IPK), asparagines synthetase (ASNS), GRP78, ER-localized DnaJ 4 (ERdj4), and C/EBP homologous protein (CHOP). The expression of GRP78, GRP94, and calreticulin mRNA was significantly increased in the hippocampus, striatum, and cortex (Figure 1). In addition, there was significantly increased expression of p58IPK mRNA in the cortex, but not in the hippocampus or striatum. We next investigated whether restraint stress affected the plasma concentrations of corticosterone, as previ- ously reported. Immediately following the 6-hr restraint stress, significantly higher plasma corticosterone concen- trations were found in stressed mice compared to un- stressed mice. Seven days after the restraint stress, the plasma corticosterone recovered to the normal control level (Figure 2). To clarify the mechanism of ER stress-related mRNA elevation, we artificially elevated the plasma concentra- tions of corticosterone in mice for 2 weeks and then measured the levels of ER stress-related proteins. In the corticosterone-treated animal model, the expression of GRP78 and GRP94 in the hippocampus was significantly increased compared to control levels (Figure 3). Restraint stress is used widely to induce stress re- sponses in animals, and it is known that a number of stresses, including restraint stress, can cause depression in animals. In the present study, we found that several ER stress-related genes were increased in the mouse hippocampus, striatum, and cortex after restraint stress. 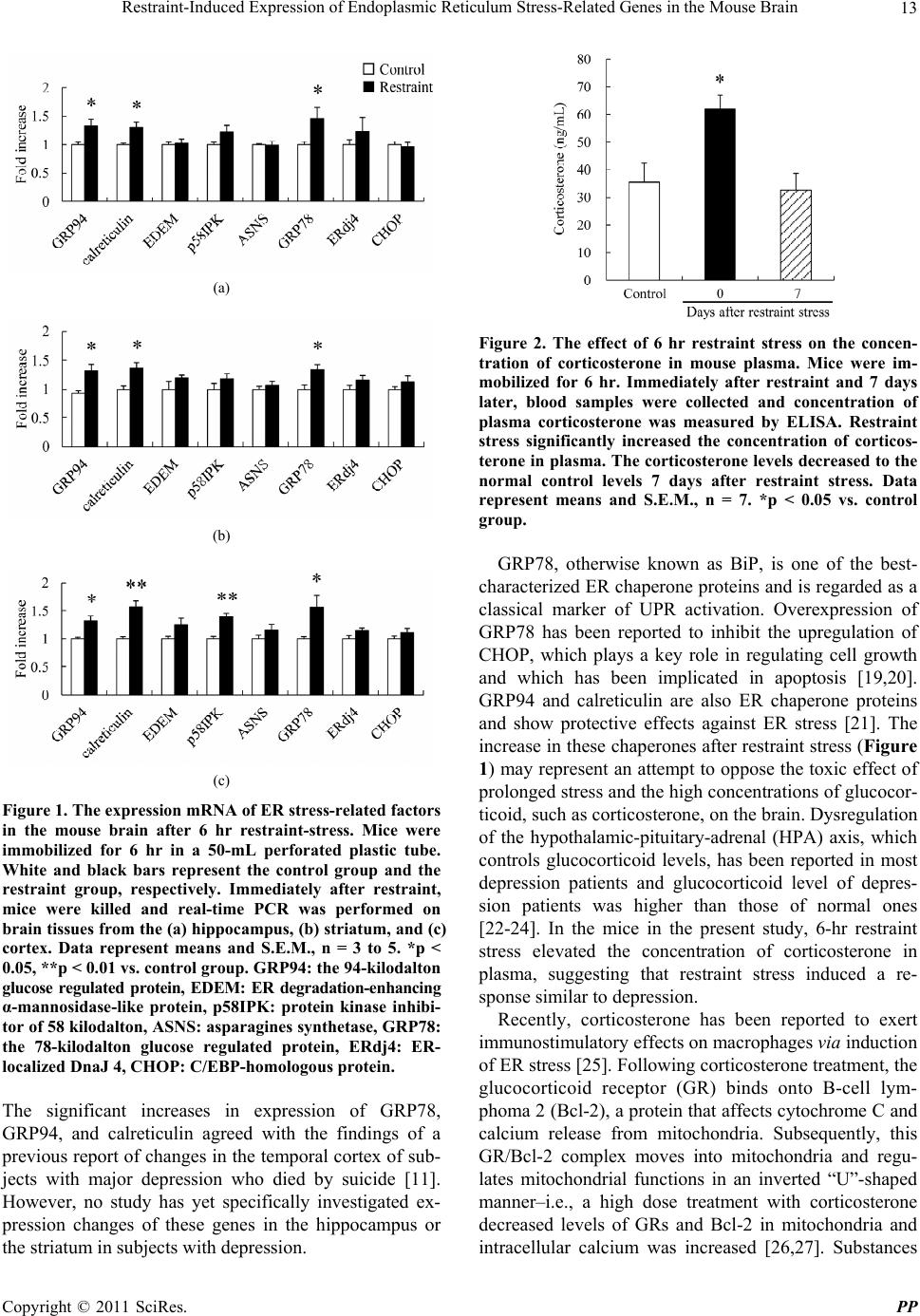 Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 13 (a) (b) (c) Figure 1. The expression mRNA of ER stress-related factors in the mouse brain after 6 hr restraint-stress. Mice were immobilized for 6 hr in a 50-mL perforated plastic tube. White and black bars represent the control group and the restraint group, respectively. Immediately after restraint, mice were killed and real-time PCR was performed on brain tissues from the (a) hippocampus, (b) striatum, and (c) cortex. Data represent means and S.E.M., n = 3 to 5. *p < 0.05, **p < 0.01 vs. control group. GRP94: the 94-kilodalton glucose regulated protein, EDEM: ER degradation-enhancing α-mannosidase-like protein, p58IPK: protein kinase inhibi- tor of 58 kilodalton, ASNS: asparagines synthetase, GRP78: the 78-kilodalton glucose regulated protein, ERdj4: ER- localized DnaJ 4, CHOP: C/EBP-homologous protein. The significant increases in expression of GRP78, GRP94, and calreticulin agreed with the findings of a previous report of changes in the temporal cortex of sub- jects with major depression who died by suicide [11]. However, no study has yet specifically investigated ex- pression changes of these genes in the hippocampus or the striatum in subjects with depression. Figure 2. The effect of 6 hr restraint stress on the concen- tration of corticosterone in mouse plasma. Mice were im- mobilized for 6 hr. Immediately after restraint and 7 days later, blood samples were collected and concentration of plasma corticosterone was measured by ELISA. Restraint stress significantly increased the concentration of corticos- terone in plasma. The corticosterone levels decreased to the normal control levels 7 days after restraint stress. Data represent means and S.E.M., n = 7. *p < 0.05 vs. control group. GRP78, otherwise known as BiP, is one of the best- characterized ER chaperone proteins and is regarded as a classical marker of UPR activation. Overexpression of GRP78 has been reported to inhibit the upregulation of CHOP, which plays a key role in regulating cell growth and which has been implicated in apoptosis [19,20]. GRP94 and calreticulin are also ER chaperone proteins and show protective effects against ER stress [21]. The increase in these chaperones after restraint stress (Figure 1) may represent an attempt to oppose the toxic effect of prolonged stress and the high concentrations of glucocor- ticoid, such as corticosterone, on the brain. Dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, which controls glucocorticoid levels, has been reported in most depression patients and glucocorticoid level of depres- sion patients was higher than those of normal ones [22-24]. In the mice in the present study, 6-hr restraint stress elevated the concentration of corticosterone in plasma, suggesting that restraint stress induced a re- sponse similar to depression. Recently, corticosterone has been reported to exert immunostimulatory effects on macrophages via induction of ER stress [25]. Following corticosterone treatment, the glucocorticoid receptor (GR) binds onto B-cell lym- phoma 2 (Bcl-2), a protein that affects cytochrome C and calcium release from mitochondria. Subsequently, this GR/Bcl-2 complex moves into mitochondria and regu- lates mitochondrial functions in an inverted “U”-shaped manner–i.e., a high dose treatment with corticosterone decreased levels of GRs and Bcl-2 in mitochondria and intracellular calcium was increased [26,27]. Substances 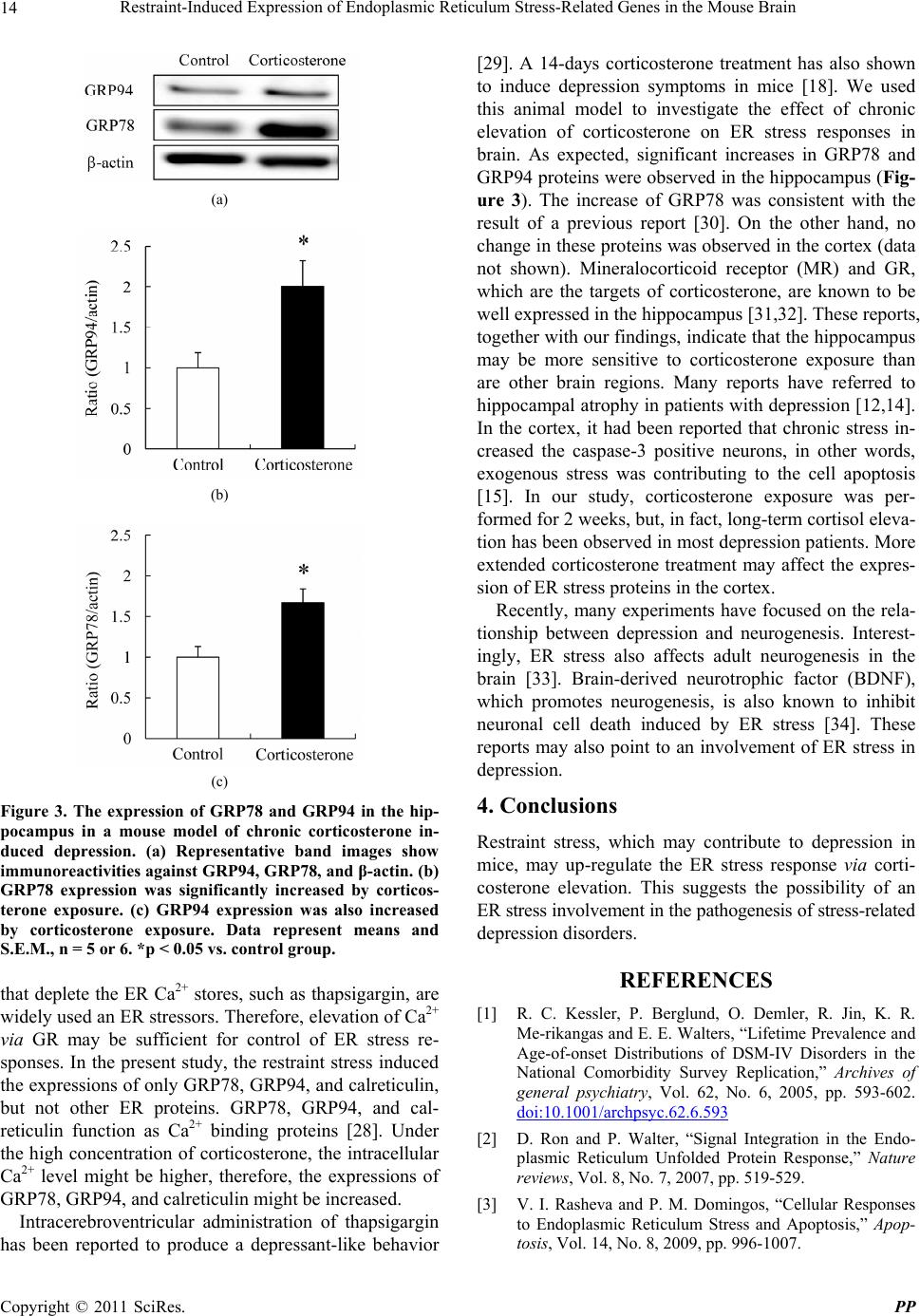 Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 14 (a) (b) (c) Figure 3. The expression of GRP78 and GRP94 in the hip- pocampus in a mouse model of chronic corticosterone in- duced depression. (a) Representative band images show immunoreactivities against GRP94, GRP78, and β-actin. (b) GRP78 expression was significantly increased by corticos- terone exposure. (c) GRP94 expression was also increased by corticosterone exposure. Data represent means and S.E.M., n = 5 or 6. *p < 0.05 vs. control group. that deplete the ER Ca2+ stores, such as thapsigargin, are widely used an ER stressors. Therefore, elevation of Ca2+ via GR may be sufficient for control of ER stress re- sponses. In the present study, the restraint stress induced the expressions of only GRP78, GRP94, and calreticulin, but not other ER proteins. GRP78, GRP94, and cal- reticulin function as Ca2+ binding proteins [28]. Under the high concentration of corticosterone, the intracellular Ca2+ level might be higher, therefore, the expressions of GRP78, GRP94, and calreticulin might be increased. Intracerebroventricular administration of thapsigargin has been reported to produce a depressant-like behavior [29]. A 14-days corticosterone treatment has also shown to induce depression symptoms in mice [18]. We used this animal model to investigate the effect of chronic elevation of corticosterone on ER stress responses in brain. As expected, significant increases in GRP78 and GRP94 proteins were observed in the hippocampus (Fig- ure 3). The increase of GRP78 was consistent with the result of a previous report [30]. On the other hand, no change in these proteins was observed in the cortex (data not shown). Mineralocorticoid receptor (MR) and GR, which are the targets of corticosterone, are known to be well expressed in the hippocampus [31,32]. These reports, together with our findings, indicate that the hippocampus may be more sensitive to corticosterone exposure than are other brain regions. Many reports have referred to hippocampal atrophy in patients with depression [12,14]. In the cortex, it had been reported that chronic stress in- creased the caspase-3 positive neurons, in other words, exogenous stress was contributing to the cell apoptosis [15]. In our study, corticosterone exposure was per- formed for 2 weeks, but, in fact, long-term cortisol eleva- tion has been observed in most depression patients. More extended corticosterone treatment may affect the expres- sion of ER stress proteins in the cortex. Recently, many experiments have focused on the rela- tionship between depression and neurogenesis. Interest- ingly, ER stress also affects adult neurogenesis in the brain [33]. Brain-derived neurotrophic factor (BDNF), which promotes neurogenesis, is also known to inhibit neuronal cell death induced by ER stress [34]. These reports may also point to an involvement of ER stress in depression. 4. Conclusions Restraint stress, which may contribute to depression in mice, may up-regulate the ER stress response via corti- costerone elevation. This suggests the possibility of an ER stress involvement in the pathogenesis of stress-related depression disorders. REFERENCES [1] R. C. Kessler, P. Berglund, O. Demler, R. Jin, K. R. Me-rikangas and E. E. Walters, “Lifetime Prevalence and Age-of-onset Distributions of DSM-IV Disorders in the National Comorbidity Survey Replication,” Archives of general psychiatry, Vol. 62, No. 6, 2005, pp. 593-602. doi:10.1001/archpsyc.62.6.593 [2] D. Ron and P. Walter, “Signal Integration in the Endo- plasmic Reticulum Unfolded Protein Response,” Nature reviews, Vol. 8, No. 7, 2007, pp. 519-529. [3] V. I. Rasheva and P. M. Domingos, “Cellular Responses to Endoplasmic Reticulum Stress and Apoptosis,” Apop- tosis, Vol. 14, No. 8, 2009, pp. 996-1007.  Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 15 doi:10.1007/s10495-009-0341-y [4] S. Oyadomari and M. Mori, “Roles of CHOP/GADD153 in Endoplasmic Reticulum Stress,” Cell Death and Dif- ferentiation, Vol. 11, No. 4, 2004, pp. 381-389. doi:10.1038/sj.cdd.4401373 [5] R. S. Saliba, P. M. Munro, P. J. Luthert and M. E. Chee-tham, “The Cellular Fate of Mutant Rhodopsin: Quality Control, Degradation and Aggresome Forma- tion,” Journal of cell science, Vol. 115, No. Pt 14, 2002, pp. 2907-2918. [6] E. H. Koo, P. T. Lansbury, Jr. and J. W. Kelly, “Amyloid Diseases: Abnormal Protein Aggregation in Neurodegen- era-tion,” Proceedings of the National Academy of Sci- ences of the United States of America, Vol. 96, No. 18, 1999, pp. 9989-9990. doi:10.1073/pnas.96.18.9989 [7] C. Kakiuchi, K. Iwamoto, M. Ishiwata, M. Bundo, T. Kasahara, I. Kusumi, T. Tsujita, Y. Okazaki, S. Nanko, H. Kunugi, T. Sasaki and T. Kato, “Impaired Feedback Regulation of XBP1 as a Genetic Risk Factor for Bipolar disorder,”Nature Genetics, Vol. 35, No. 2, 2003, pp. 171-175. doi:10.1038/ng1235 [8] C. Kakiuchi, M. Ishiwata, T. Umekage, M. Tochigi, K. Kohda, T. Sasaki and T. Kato, “Association of the XBP1- 116C/G Polymorphism with Schizophrenia in the Japa- nese Population,” Psychiatry and Clinical Neurosciences, Vol. 58, No. 4, 2004, pp. 438-440. doi:10.1111/j.1440-1819.2004.01280.x [9] L. Shao, X. Sun, L. Xu, L. T. Young and J. F. Wang, “Mood Stabilizing Drug Lithium Increases Expression of Endoplasmic Reticulum Stress Proteins in Primary Cul- tured Rat Cerebral Cortical Cells,” Life Sciences, Vol. 78, No. 12, 2006, pp. 1317-1323. doi:10.1016/j.lfs.2005.07.007 [10] S. Kurosawa, E. Hashimoto, W. Ukai, S. Toki, S. Saito and T. Saito, “Olanzapine Potentiates Neuronal Survival and Neural Stem Cell Differentiation: Regulation of Endo-plasmic Reticulum Stress Response Proteins,” Journal of Neural Transmission, Vol. 114, No. 9, 2007, pp. 1121- 1128. doi:10.1007/s00702-007-0747-z [11] C. Bown, J. F. Wang, G. MacQueen and L. T. Young, “Increased Temporal Cortex ER Stress Proteins in De- pressed Subjects Who Died by Suicide,” Neuropsycho- pharmacology, Vol. 22, No. 3, 2000, pp. 327-332. doi:10.1016/S0893-133X(99)00091-3 [12] Y. I. Sheline, P. W. Wang, M. H. Gado, J. G. Csernansky and M. W. Vannier, “Hippocampal Atrophy in Recurrent Major Depression,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, No. 9, 1996, pp. 3908-3913. doi:10.1073/pnas.93.9.3908 [13] D. Ongur, W. C. Drevets and J. L. Price, “Glial Reduction in the Subgenual Prefrontal Cortex in Mood Disorders,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 95, No. 22, 1998, pp. 13290-13295. doi:10.1073/pnas.95.22.13290 [14] Y. Watanabe, E. Gould and B. S. McEwen, “Stress In- duces Atrophy of Apical Dendrites of Hippocampal CA3 Pyramidal Neurons,” Brain research, Vol. 588, No. 2, 1992, pp. 341-345. doi:10.1016/0006-8993(92)91597-8 [15] A. Bachis, M. I. Cruz, R. L. Nosheny and I. Mocchetti, “Chronic Unpredictable Stress Promotes Neuronal Apop- tosis in the Cerebral Cortex,” Neuroscience Letters, Vol. 442, No. 2, 2008, pp. 104-108. doi:10.1016/j.neulet.2008.06.081 [16] D. Chen, E. Padiernos, F. Ding, I. S. Lossos and C. D. Lopez, “Apoptosis-stimulating Protein of P53-2 (ASPP2/ 53BP2L) is an E2F Target Gene,” Cell Death and Dif- ferentiation, Vol. 12, No. 4, 2005, pp. 358-368. doi:10.1038/sj.cdd.4401536 [17] O. I. Abatan, K. B. Welch and J. A. Nemzek, “Evaluation of Saphenous Venipuncture and Modified Tail-clip Blood Collection in Mice,” Journal of the American Association for Laboratory Animal Science,, Vol. 47, No. 3, 2008, pp. 8-15. [18] S. L. Gourley and J. R. Taylor, “Recapitulation and Re- versal of a Persistent Depression-like Syndrome in Ro- dents,” Current Protocols in Neuroscience Chapter 9, 2009, Unit-9.32. [19] X. Z. Wang, B. Lawson, J. W. Brewer, H. Zinszner, A. Sanjay, L. J. Mi, R. Boorstein, G. Kreibich, L. M. Hend- ershot and D. Ron, “Signals from the Stressed Endoplas- mic Reticulum Induce C/EBP-homologous Protein (CHOP/GADD153),” Molecular and Cellular Biology, Vol. 16, No. 8, 1996, pp. 4273-4280. [20] H. Zinszner, M. Kuroda, X. Wang, N. Batchvarova, R. T. Lightfoot, H. Remotti, J. L. Stevens and D. Ron, “CHOP is Implicated in Programmed Cell Death in Response to Impaired Function of the Endoplasmic Reticulum,” Genes & Development, Vol. 12, No. 7, 1998, pp. 982-995. doi:10.1101/gad.12.7.982 [21] M. Cechowska-Pasko, “Endoplasmic Reticulum Chaper- ons,” Postepy Biochemii, Vol. 55, No. 4, 2009, pp. 416- 424. [22] C. A. Sandman, J. L. Barron and L. Parker, “Disregula- tion of Hypothalamic-pituitary-adrenal Axis in the Men- tally Retarded,” Pharmacology, Biochemistry, and Be- havior, Vol. 23, No. 1, 1985, pp. 21-26. doi:10.1016/0091-3057(85)90124-8 [23] A. Roy, “Hypothalamic-pituitary-adrenal Axis Function and Suicidal Behavior in Depression,” Biological psy- chiatry, Vol. 32, No. 9, 1992, pp. 812-816. doi:10.1016/0006-3223(92)90084-D [24] J. F. Lopez, D. M. Vazquez, D. T. Chalmers and S. J. Watson, “Regulation of 5-HT Receptors and the Hypotha- lamic-pituitary-adrenal Axis. Implications for the Neuro- biology of Suicide,” Annals of the New York Academy of Sciences, Vol. 836, No. 1, 1997, pp. 106-134. [25] J. Y. Zhou, H. J. Zhong, C. Yang, J. Yan, H. Y. Wang and J. X. Jiang, “Corticosterone Exerts Immunostimula- tory Effects on Macrophages via Endoplasmic Reticulum Stress,” The British Journal of Surgery, Vol. 97, No. 2, 2010, pp. 281-293. doi:10.1002/bjs.6820 [26] J. Du, B. McEwen and H. K. Manji, “Glucocorticoid Receptors Modulate Mitochondrial Function: A Novel Mechanism for Neuroprotection,” Communicative & In-  Restraint-Induced Expression of Endoplasmic Reticulum Stress-Related Genes in the Mouse Brain Copyright © 2011 SciRes. PP 16 tegrative Biology, Vol. 2, No. 4, 2009, pp. 350-352. [27] J. Du, Y. Wang, R. Hunter, Y. Wei, R. Blumenthal, C. Falke, R. Khairova, R. Zhou, P. Yuan, R. Machado- Vieira, B. S. McEwen and H. K. Manji, “Dynamic Regu- lation of Mitochondrial Function by Glucocorticoids,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 106, No. 9, 2009, pp. 3543-3548. doi:10.1073/pnas.0812671106 [28] H. Coe and M. Michalak, “Calcium Binding Chaperones of the Endoplasmic Reticulum,” General Physiology and Biophysics, Vol. 28 Spec No Focus, 2009, pp. 96- 103. [29] N. Galeotti, A. Bartolini and C. Ghelardini, “Blockade of Intracellular Calcium Release Induces an Antidepressant- like Effect in the Mouse Forced Swimming Test,” Neu- ropharmacology, Vol. 50, No. 3, 2006, pp. 309-316. doi:10.1016/j.neuropharm.2005.09.005 [30] S. L. Gourley, F. J. Wu, D. D. Kiraly, J. E. Ploski, A. T. Kedves, R. S. Duman and J. R. Taylor, “Regionally Spe- cific Regulation of ERK MAP Kinase in a Model of An- tidepressant-sensitive Chronic Depression,” Biological psychiatry, Vol. 63, No. 4, 2008, pp. 353-359. doi:10.1016/j.biopsych.2007.07.016 [31] T. Ito, N. Morita, M. Nishi and M. Kawata, “In Vitro and in Vivo Immunocytochemistry for the Distribution of Mineralocorticoid Receptor with the Use of Specific An- tibody,” Neuroscience Research, Vol. 37, No. 3, 2000, pp. 173-182. doi:10.1016/S0168-0102(00)00112-7 [32] F. Han, H. Ozawa, K. Matsuda, M. Nishi and M. Kawata, “Colocalization of Mineralocorticoid Receptor and Glu- cocorticoid Receptor in the Hippocampus and Hypothala- mus,” Neuroscience Research, Vol. 51, No. 4, 2005, pp. 371-381. doi:10.1016/j.neures.2004.12.013 [33] P. J. Lucassen, W. Scheper and E. J. Van Someren, “Adult Neurogenesis and the Unfolded Protein Response; New Cellular and Molecular Avenues in Sleep Research,” Sleep Medicine Reviews, Vol. 13, No. 3, 2009, pp. 183-186. doi:10.1016/j.smrv.2008.12.004 [34] G. Chen, Z. Fan, X. Wang, C. Ma, K. A. Bower, X. Shi, Z. J. Ke and J. Luo, “Brain-derived Neurotrophic Factor Suppresses Tunicamycin-induced Upregulation of CHOP in Neurons,” Journal of Neuroscience Research, Vol. 85, No. 8, 2007, pp. 1674-1684. doi:10.1002/jnr.21292 |