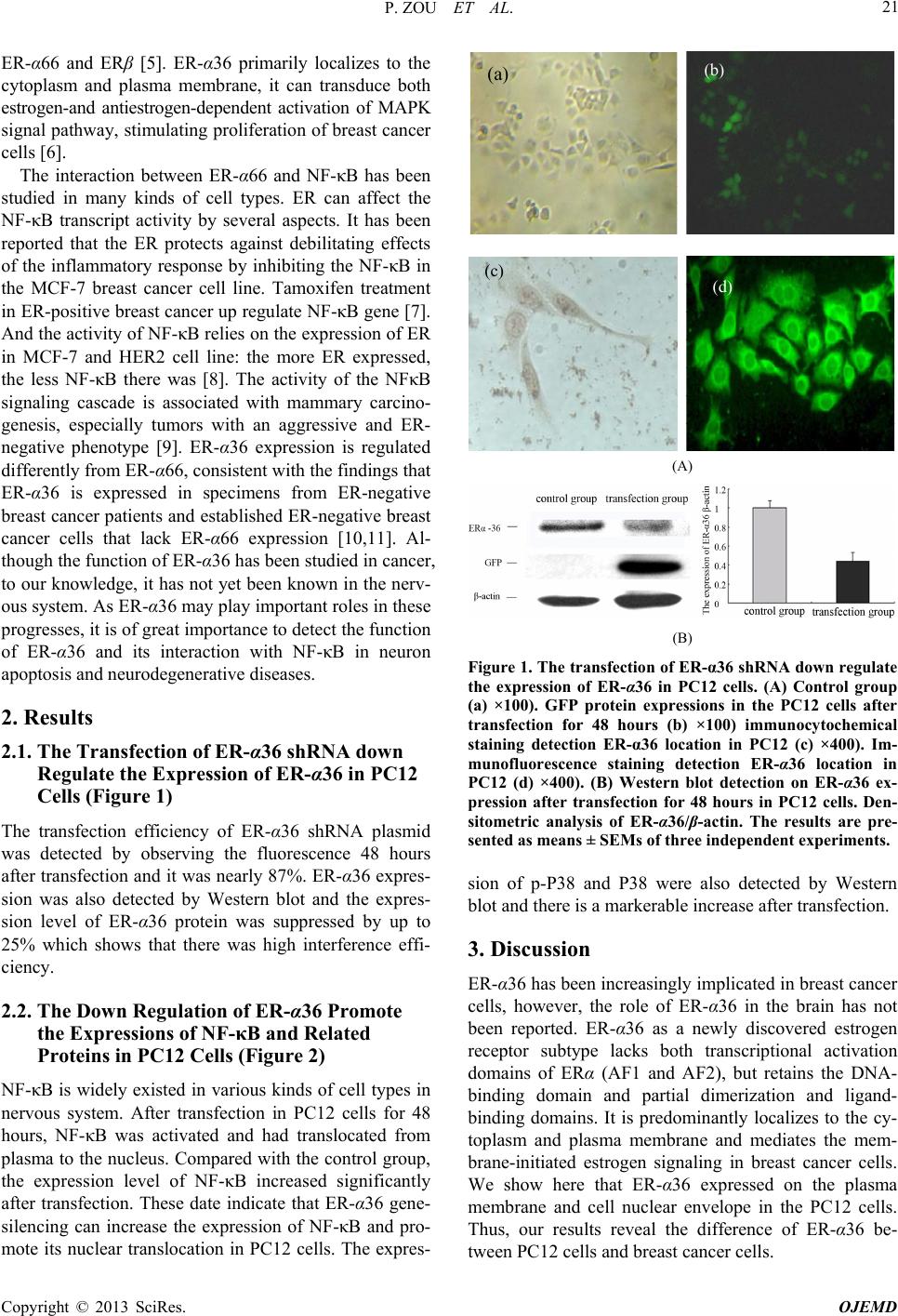
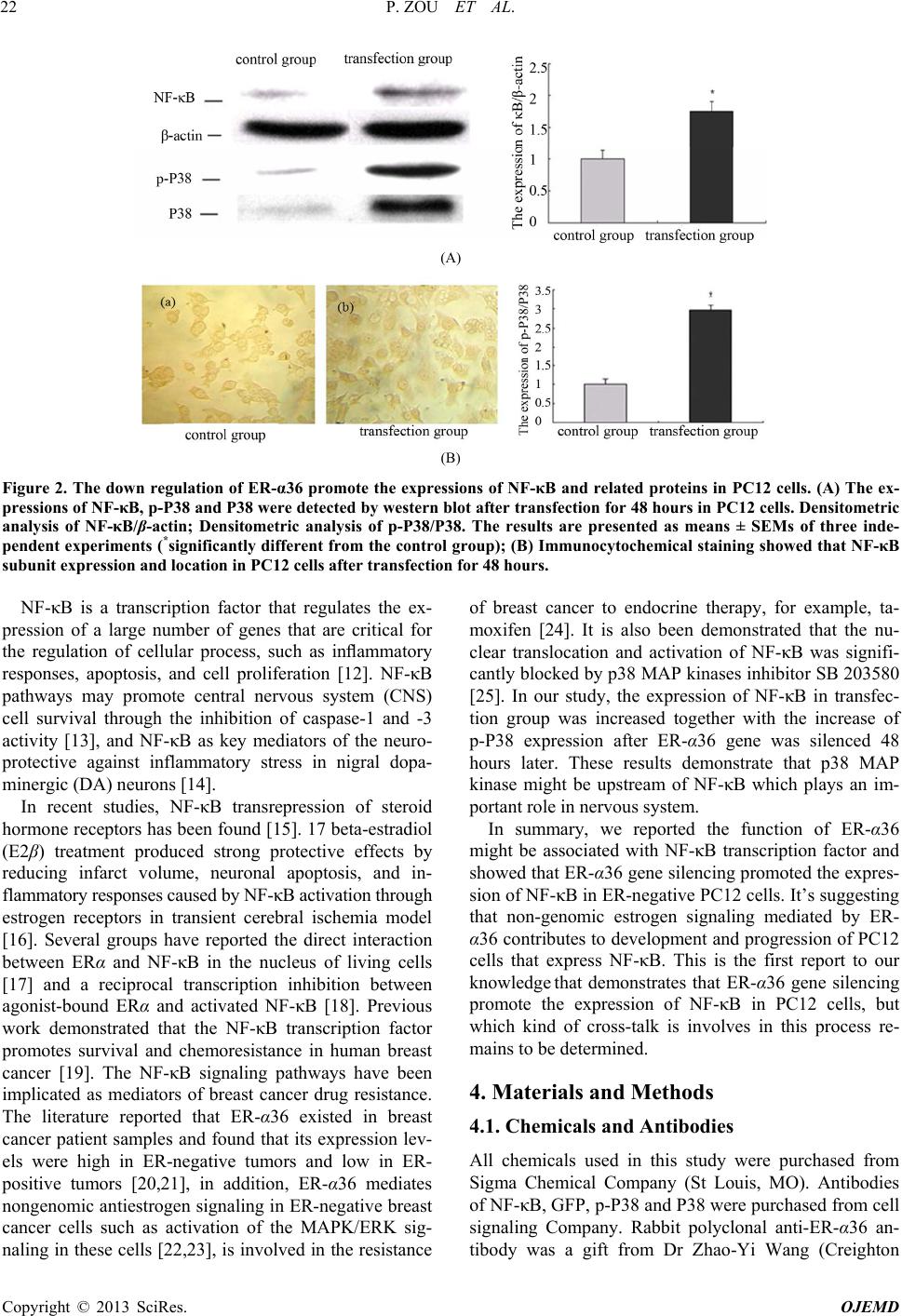
P. ZOU ET AL.
Copyright © 2013 SciRes. OJEMD
24
Sharma and G. L. Greene, “CBP Is a Dosage-Dependent
Regulator of Nuclear Factor—KappaB Suppression by
the Estrogen Receptor,” Molecular Endocrinology, Vol.
22, No. 2, 2008, pp. 263-272. doi:10.1210/me.2007-0324
[9] J. W. Antoon, M. D. White, E. M. Slaughter, J. L. Driver,
H. S. Khalili, S. Elliott, C. D. Smith, M. E. Burow and
B.S. Beckman, “Targeting NFκB Mediated Breast Cancer
Chemoresistance through Selective Inhibition of Sphin-
gosine Kinase-2,” Cancer Biology & Therapy, Vol. 11,
No. 7, 2011, pp. 678-689. doi:10.4161/cbt.11.7.14903
[10] Y. Zhou, S. Eppenberger-Castori, C. Marx, C. Yau, G. K.
Scott, U. Eppenberger and C. C. Benz, “Activation of Nu-
clear Faetor-KappaB (NFkappaB) Identifies a High-Risk
Subset of Hormone-Dependent Breast Cancers,” The In-
ternational Journal of Biochemistry & Cell Biology, Vol.
37, No. 5, 2005, pp. l130-l144.
doi:10.1016/j.biocel.2004.09.006
[11] L. Shi, B. Dong, Z. Li, Y. Lu, T. Ouyang, J. Li, T. Wang,
Z. Fan, T. Fan, B. Lin, Z. Wang and Y. Xie, “Expression
of ER-(α)36, a Novel Variant of Estrogen Receptor-(α),
and Resistance to Tamoxifen Treatment in Breast Can-
cer,” Journal of Clinical Oncology, Vol. 27, No. 21, 2009,
pp. 3423-3429. doi:10.1200/JCO.2008.17.2254
[12] G. Zhang and S. Ghosh, “Toll-Like Receptor-Mediated
NFκB Activation: A Phylogenetically Conserved Para-
digm in Innate Immunity,” Journal of Clinical Investiga-
tion, Vol. 107, No. 1, 2001, pp. 13-19.
doi:10.1172/JCI11837
[13] A. L. Bernardino, T. A. Myers, X. Alvarez, A. Hasegawa
and M. T. Philipp, “Toll-Like Receptors: Insights into
Their Possible Role in the Pathogenesis of Lyme Neuro-
borreliosis,” Infection and Immunity, Vol. 76, No. 10,
2008, pp. 4385-4395. doi:10.1128/IAI.00394-08
[14] J. K. Lee, J. Chung, K. M. Druey and M. G. Tansey,
“RGS10 Exerts a Neuroprotective Role through the PKA/
c-AMP Response- Element (CREB) Pathway in Dopami-
nergic Neuron-Like Cells,” Journal of Neurochemistry,
Vol. 122, No. 2, 2012, pp. 333-343.
[15] L. I. McKay and J. A. Cidlowski, “Molecular Control of
Immune/Inflammatory Responses: Interactions between
Nuclear Factor-Kappa B and Steroid Receptor-Signaling
Pathways,” Endocrine Reviews, Vol. 20, No. 4, 1999, pp.
435-459. doi:10.1210/er.20.4.435
[16] N. Maulik, M. Sato, B. D. Price and D. K. Das, “An Es-
sential Role of NFkappaB in Tyrosine Kinase Signaling
of p38 MAP Kinase Regulation of Myocardial Adaptation
to Ischemia,” FEBS Letters, Vol. 429, No. 3, 1998, pp.
365-369. doi:10.1016/S0014-5793(98)00632-2
[17] I. Sarnico, A. Lanzillotta, M. Benarese, M. Alghisi, C.
Baiguera, L. Battistin, P. Spano and M. Pizzi, “NF-Kap-
paB Dimers in the Regulation of Neuronal Survival,” In-
ternational Review of Neurobiology, Vol. 85, 2009, pp.
351-362. doi:10.1016/S0074-7742(09)85024-1
[18] C. H. Nijboer, C. J Heijnen, F. Groenendaal, M. J. May, F.
Bel and A. Kavelaars, “Strong Neuroprotection by Inhibi-
tion of NF-KappaB after Neonatal Hypoxia-Ischemia In-
volves Apoptotic Mechanisms but Is Independent of Cy-
tokines,” Stroke, Vol. 39, No. 7, 2008, pp. 2129-2137.
doi:10.1161/STROKEAHA.107.504175
[19] C. B. Weldon, A. P. Parker, D. Patten, S. Elliott, Y. Tang,
D. E. Frigo, C. M. Dugan, E. L. Coakley, N. N. Butler, J.
L. Clayton, J. Alam, T. J. Curiel, B. S. Beckman, B. M.
Jaffe and M. E. Burow, “Sensitization of Apoptotically-
Resistant Breast Carcinoma Cells to TNF and TRAIL by
Inhibition of p38 Mitogen-Activated Protein Kinase Sig-
naling,” International Journal of Oncology, Vol. 24, No.
6, 2004, pp. 1473-1480.
[20] J. Zhang, G. Li, Z. Li, X. Yu, Y. Zheng, K. Jin, H. Wang,
Y. Gong, X. Sun, X. Teng, J. Cao and L. Teng, “Estro-
gen-Independent Effects of ER-a36 in ER-Negative Breast
Cancer,” Steroids, Vol. 77, No. 6, 2012, pp. 666-673.
doi:10.1016/j.steroids.2012.02.013
[21] X. T. Zhang, L. Ding, L. G. Kang and Z. Y. Wang, “In-
volvement of ER-α36, Src, EGFR and STAT5 in the Bi-
phasic Estrogen Signaling of ER-Negative Breast Cancer
Cells,” Oncology Reports, Vol. 27, No. 6, 2012, pp. 2057-
2065.
[22] X. Zhang, L. Ding, L. Kang and Z. Y. Wang, “Estrogen
Receptor-Alpha 36 Mediates Mitogenic Antiestrogen
Signaling in ER-Negative Breast Cancer Cells,” PLOS
One, Vol. 7, No. 1, 2012, pp. e30174-30186.
[23] L. Kang, Y. Guo, X. Zhang, J. Meng and Z. Y. Wang, “A
Positive Cross-Regulation of HER2 and ER-α36 Controls
ALDH1 Positive Breast Cancer Cells,” Molecular Biol-
ogy, Vol. 127, No. 3-5, 2011, pp. 262-268.
[24] J. Rao, X. Jiang, Y. Wang and B. Chen, “Advances in the
Understanding of the Structure and Function of ER-α36, a
Novel Variant of Human Estrogen Receptor-Alpha,” Mo-
lecular Biology, Vol. 127, No. 3-5, 2011, pp. 231-237.
[25] M. E. Quaedackers, C. E. van den Brink, P. T. van der
Saag and L. G. Tertoolen, “Tertoolen LG Direct Interac-
tion between Estrogen Receptor Alpha and NF-KappaB
in the Nucleus of Living Cells,” Molecular and Cellular
Endocrinology, Vol. 273, No. 1-2, 2007, pp. 42-50.
doi:10.1016/j.mce.2007.05.002