Paper Menu >>
Journal Menu >>
 Advances in Bioscience and Biotechnology, 2010, 1, 417-425 ABB doi:10.4236/abb.2010.15055 Published Online December 2010 (http://www.SciRP.org/journal/abb/). Published Online December 2010 in SciRes. http://www.scirp.org/journal/ABB Cloning, expression, purification and characterization of replication protein from plasmid pGP2 from Acetobacter estunensis Peter Grones, Jozef Grones Comenius University, Department of Molecular Biology, Mlynska dolina B2, 842 15 Bratislava 4, Slovak Republic. Email: grones@fns.uniba.sk Received 10 September 2010; revised 22 October 2010; accepted 8 November 2010. ABSTRACT The Acetobacter estunensis Rep34 protein participates in the replication of bacterial plasmid pGP2. The Rep34 protein of the A. estunensis, was cloned to the expression vector, that ensure fusion with a His-tag sequence (Rep34 His-tagged), over-expressed in Es- cherichia coli and purified by metal-affinity chroma- tography to yield a highly purified and active protein. On this purified protein number different activities and motifs were detected. DNA band-shift assays showed that the Rep34 His-tagged protein bound to the regulation region for replication on the linear double-stranded DNA. In the protein was determined phosphatase activity, ATPase activity and protein is possible to unwind double strand DNA. Keywords: Acetobacter Estunensis; Rep34 Protein; DNA-Binding Activity; ATPase Activity; Phosphatase Activity; Unwinding Activity The replication of eukaryotic and prokaryotic chromo- somes, bacteriophages and bacterial plasmids DNA in- volves several analogous events and similarities in replisome architecture. Many systems have specific ini- tiation proteins, including bacterial DnaA protein, phage lambda O protein, plasmid replication initiation proteins (Rep) and the eukaryotic origin recognition complex (ORC). The specific mechanism for replication initiation of a given replicon is dependent on both the structure of the replication origin and the nature of the replication initiation protein. The replication of bacterial ex- tra-chromosomal replicons, such as plasmids or phages is generally limited to a single host or a few closely re- lated host´s proteins [1]. Replication proteins generally initiates and regulates bacterial chromosome or plasmid replication and serves as a transcription factor [2,3]. The origins of prokaryotic and some eukaryotic repli- cons possess characteristic functional elements, include- ing specific binding sites for the appropriate initiation protein and an AT-rich region where DNA duplex desta- bilization occurs. Plasmid origins usually contain multi- ple binding sites (iterons) for the plasmid-specific repli- cation initiation protein as well as one or more binding sites for the host replication initiation protein, DnaA (DnaA boxes) [1]. These proteins interact with repeated regulation sequences. They also interact with Rep pro- tein or DnaA protein on regulation boxes, which are lo- cated within the ori regions or the promoter regions or intergenic regions of many bacterial chromosomes and plasmids [4]. The structural elements of the origin are employed for broad-host-range plasmid replication and maintenance in different host bacteria species. For example, the minimal origin of the broad-host-range plasmid RK2 possesses five iterons and is functional in E. coli. However, the presence of three iterons stabilizes RK2 plasmid main- tenance in Pseudomonas putida [5]. In addition, the re- gion with four DnaA boxes is essential for RK2 replica- tion in E. coli, but is dispensable for replication of the plasmid in Pseudomonas aeruginosa [6,7]. In the E. coli chromo s o me, the replication origin (oriC) contains five DnaA box sequences. The binding of mul- tiple DnaA molecules in the presence of the histone-like HU protein and the site-specific DNA-binding protein IHF (integration host factor) results in destabilization of the duplex DNA within the nearby AT-rich sequences of the oriC of E. coli [3]. Origin opening of the narrow- host-range plasmids pSC101, F, P1, and R6K requires, in addition to E. coli DnaA, HU and/or IHF proteins, the binding of plasmid-encoded replication initiation pro- teins [8-12]. Similarly, the formation of an open complex at the replication origin of the broad-host-range plasmid RK2 by the plasmid encoded TrfA initiation protein re- quires E. coli HU, and is stabilized by E. coli DnaA [13]. In contrast to the chromosomal oriC, plasmid origins  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 418 do not require ATP for open complex formation [8-10]. A basis for this lack of dependence on ATP, induced in an ATP-independent mode, by the complex of the plas- mid-encoded Rep protein and the host HU or IHF [9,14,15]. Plasmids encoded systems that control their replica- tion such that fairly precise, steady-state copy numbers are maintained [16]. Plasmid replicons from Gram- negative bacteria always seem to encode a negative feedback control system. Two basic mechanisms for the regulation of plasmid replication have been recognized so far: one operates via an antisense RNA transcript that negatively regulates the replication; the other operates via iterons, a series of direct repeat sequence located within ori that intereract with iteron-binding Rep pro- teins are responsible for both the initiation of replication and its control [16,17]. From acetic acid bacteria were purified and character- ised several DNA plasmids which encoded rep gene which product is able to regulate replication process. The first identified cryptic plasmid from Acetobacter encoding Rep protein had been used for the construction of cloning vectors [18,19]. Later from Acetobacter pas- teurianus was identified large plasmid pAC1 [20], and pAP12875 [21]. From Gluconobacter was isolated plasmid pJK2-1 [22], and from A. aceti plasmid pAG20 [23]. In this paper we presented replication protein of plas- mid pGP2 isolated from Acetobacter estunensis GP2 strain and characterise main activities that belong to the bacterial replication proteins. 1. MATERIALS AND METHODS 1.1. Bacterial Strains and Cultivation Media Escherichia coli strain XL1 Blue (tetracyclineR) [24] was used for plasmid isolation and for cloning DNA fragments and strain BL21 (DE3) [F– ompT gal dcm lon hsdSB(rB - mB -) λ(DE3 [lacI lacUV5-T7 gene1 ind1 sam7 nin5]) was used as a host for protein expression. The pGP2 plasmid isolated from Acetobacter estunensis GP2 used as template for rep34 gene. Acetobacter strain was cultivated in YPG medium (5% yeast extract, 3% pep- tone, and 1% manitol) and E. coli strain on LB medium (10% Tryptone, 5% yeast extract and 5% NaCl pH 7.4) supplemented with 50 µg/ml kanamycin and 100 µg/ml ampicillin. 1.2. Biochemical Material The reagents for PCR and oligonucleotide primers were obtained from Invitrogen Life Technologies (Carlsbad, CA, USA). The bacterial vector pGEM-T Easy (ampicil- linR; Promega, Madison, WI, USA) expression pET- 28a+ vector (T7 promoter, kanamycinR; Novagen, Madi- son, WI, USA), was used for cloning and expression rep gene. Restriction endonucleases, isopropylthio-β-D- galactopyranoside (IPTG), and T4 DNA ligase were ob- tained from BioLabs. p-nitrophenyl phosphate (pNPP) and protein standard used as SDS–PAGE marker were from Sigma Chemical (St. Louis, MO, USA). 1.3. Cloning and Expression Vector Construction Recombinant DNA techniques were performed using conventional protocols. The rep34 gene of Acetobacter estunensis (ORF2) was amplified using: forward 5´-GGA TCC ATG TGG TAT CAA AAG ACG CT–3 and reverse primer 5´-AAG CTT TTA TTC AGA TGG CGG CTT G–3´. The amplified DNA encoding the rep34 gene produced a fragment of around 627 bp was cloned into pGEM-T Easy vector and transformed in E. coli XL1. Selected construct pGEM-rep34 was sequenced by dideoxy chain termination method [25] using an ABI Prism 3200 automated DNA sequencer (Applied Bio- systems, Foster City, CA, USA). Sequenced data were analyzed using BLAST-Basic local alignment search tools program [26] through the network service of the National Center for Biotechnology Information (http:// ww.ncbi.nlm.nih.gov). The DNA encoding the rep34 was then sub-cloned into vector pET-28a- in BamHI and HindIII sites which was used to transform E. coli XL1. The new vector construct was named pET–rep34. Sequencing of the cloned vectors revealed the open reading frame (ORF2) of the rep34 gene plus the expected 32 additional amino acid residues derived from pET-28a- vector at its amino terminus (MGSSHHHHHHSSGLVPRGSH MASMTGGEEMGR), including the cluster of six histidine residues for protein purification by metal affinity chromatography. The ex- pression vector pET-rep34 was extracted from the trans- formants using JETQuick Plasmid kit (Genomed), and used to transform to E. coli BL21 (DE3) competent cells. Selected transformant pET-28-rep34 was used for protein expression and purification. 1.4. Expression and Purification of Recombinant Rep34 A total of 100 µl of an overnight culture of E. coli BL21 (DE3) with pET-28-rep34 was diluted into 100 ml of LB medium containing kanamycin (50 µg/ml). The culture was grown at 37℃ until optical density at 590 nm reached 0.6 and was induced with 0.5 mM IPTG. The incubation continued for an additional 2 h in the same temperature. After incubation, the culture was harvested by centrifugation at 8,000 g for 10 min at 4℃. The pellet cells were suspended in 25 mM Tris.HCl, pH 8.0 buffer  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 419 containing 20 mM NaCl, and 5% glycerol and disrupted for 10 min at 4℃ followed by sonication. The suspend- sion was then centrifuged at 16,000 g for 10 min at 4℃ to separate the cell debris, and the solubility of the re- combinant fusion protein was analyzed by 12% SDS– PAGE. The soluble fraction containing recombinant pET-rep34 was loaded onto a Ni–NTA column (Qiagen, Hilden, Germany) pre-equilibrated with 50 mM sodium phosphate, pH 8.0 buffer containing 300 mM NaCl and 10 mM imidasole. The unbound proteins were washed out with the same buffer used for equilibration of the column. Subsequently, the recombinant proteins were eluted with the same buffer containing 50 to 250 mM imidazole. The resulting pET-rep34 was exhaustively dialyzed in 50 mM Tris.HCl, pH 8.0 buffer containing 50 mM NaCl for elimination of imidazole. The protein purity was confirmed by the presence of a single band on SDS–PAGE 12% of molecular weight predicted for the Rep34 (about 25.5 kDa, with His-tag). For protein visu- alization, the gels were stained with Coomassie Brilliant Blue G-250 (Bio-Rad Laboratories) and distained with 10% acetic acid and 20% methanol. The purified protein was frozen and stored at –80℃. Protein concentration recombinant soluble protein concentration was deter- mined by UV absorbance at 280 nm (Spectrophotometer ND-1000 UV-Vis, NanoDrop Technologies). 1.5. Computational Methods for Secondary Structure Prediction The DNA nucleotide sequence was translated into a pro- tein sequence and the deduced amino acid sequence was analyzed using the Expasy SwissProt Web server (http://www.expasy.ch). The sequence of amino acids from Rep34 was aligned with 45 sequences of GST en- zymes using ClustalW [27]. The methods used for general secondary- structure prediction were Jpred [28], PHD [29], PSIPRED [30], and SSpro [31]. The predict- tion of secondary structure and analysion of protein mo- tifs were made using program CLC Main Workbench 5.1. 1.6. Assay of A TPase Activity The ATPase activity was assayed by using non radioac- tive modified method [32]. With interaction of free anorganic phosphate with fresh prepared 0.045% (w/v) malachit green and 4.2% (w/v) molibden ammonium in 4 M HCl in the rate of 3:1. Reaction mixture contained in 50 µl (1 µg protein, 50 mM Tris-HCl pH 7.9, 5 mM MgCl2 and 1 mM ATP) at 37℃ in times volume 0, 5, 10, 15, 20 min. After reference times was added 800 µl malachit-molybdene solution and after 1 min added 100 µl of 34% (v/v) sodium citrate and the absorbance change was measured spectrophotometrically at 660 nm. As a standard was used reaction with different con- centration of Na3PO4. 1.7. Helicase Assay Helicase activity was detected by the release of a Fam3- labeled oligonucleotide annealed to M13mp18 single- stranded (ss) DNA [33]. The oligonucleotide, which consisted of 20 bases (5´ – fam3-GTT GTA AAA CGA CGG CCA GT – 3´) was complementary to the M13 DNA. Labeled oligonucleotide was annealed with M13 ss DNA (New England BioLabs) in 10 mM Tris.HCl (pH 8.0), 1 mM EDTA, and 100 mM NaCl, heat for 5 min at 65℃ and slowly cooling down for 30 min to room tem- perature. The double-strand (ds) substrate was purified by ethanol precipitation to remove unannealed oligonu- cleotide. The helicase reaction mixture contained 5 nM substrate in 20 mM Tris.HCl, 2 mM DTT, 5 mM MgCl2, 5% glycerol, 5 mM ATP, 0.1 mg/ml BSA, and the solu- tion was adjusted to pH 7.5. The reactions were incu- bated at 37℃ for 30 min and stopped adding 1/10 vol- ume of 3 M sodium acetate and ethanol precipitate. After precipitate pellet was suspended in 100 µl of sterile water and unwound ds DNA substrate was quantified by fluorometric analysis at 492 nm on fluorescence ana- lyzer (Tecan Safire 2 Microplate Fluorescence Reader). DNA unwinding was calculated as a percentage of the total counts in each reaction. 1.8. Gel mobility Shift Assays The DNA probe for the electrophoretic mobility shift assay (EMSA) was the plasmid pGP2 transcription and replication region in position 1671-2761 bp. The probe was synthesized by PCR, using the primers 5’ – GAG CTC ATG CAT GTA CGC CGC GGT – 3’ and 5’ – AAG CTT TTA TTC AGA TGG CGG CTT G – 3’. After am- plification, the DNA probe was cleaved by PvuII which created two fragments 539 and 475 bp. Mobility shifts were performed as previously described [34], with some modifications. The reaction mixtures (15 µl) contained 50 mM Tris.HCl (pH 8.0), 50 mM NaCl, 4.8 mM di- thiothreitol, 1.0 mg/ml bovine serum albumin per ml and 20% (w/v) glycerol. DNA probe, reaction mixture and recombinant Rep34 protein (0.4 pmol) were incubated at 30℃ for 15 min. The separation take place on a 4% polyacrylamide gel containing 0.09% bis-acrylamide, 2.5% glycerol, and 1x TBE at 5-10 V/cm. The complexes were visualized after colored ethidium bromide [35]. 1.9. Phosphatase Activity The enzymatic activity of Rep34 towards p-nitrophenyl phosphate (pNPP) substrate was assayed at 37℃ by spectrophotometric detection of the absorbance at 405 nm due to the release of p-nitrophenol (pNP) [36].  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 420 Kinetic measurements assigned to determine substrate kinetic parameters were performed in reaction mixtures of a total volume of 400 μl. The mixtures contained 50 mM Tris–acetate, pH 5.5, 50-100 nM Rep34, in the presence or absence of 10 mM MgCl2, and different concentrations of pNPP. The reactions were initiated by the addition of the enzyme, and quenched after 10 min by the addition of 100 μl of 2 M NaOH followed by centrifugation at 14,000g for 5 min. The absorbance at 405 nm was read for supernatants of reaction mixtures (containing enzyme) and controls (the same substrate concentration omitting the enzyme), and the difference between these measurements represented the real rate of product formation. 1.10. Protein Analysis Proteins were analyzed by SDS–PAGE [37] stained ei- ther by Coomassie Brilliant Blue R-250 method. Protein concentrations were determined by the Bradford proce- dure [38] or by densitometric analyses of Coomassie Brilliant Blue-stained SDS–PAGE gels. Bovine serum albumin (BSA) was used as the standard. 2. RESULTS AND DISCUSSION Bacterial plasmid pGP2 purified from Acetobacter es- tunensis GP2 (2 797 bp) encoded three proteins. ORF2 encoded 209 aa large protein belonging to group of rep- lication protein designed as Rep34 (Figure 1). By the analysis of amino acid sequence were determined the number of alpha helixes (4 larger than 4 aa) and beta structures (4 larger than 4 aa) and two domains: Helicase conserved C-terminal domain (137-175 aa) and HTH motive (169-203 aa). Isoelectric point of this protein, determined at pI 9.25, is similar to the isoelectric point determined in replication proteins of plasmid pAG20 from A. aceti 3620 (pI 8.2) [23], but a bit distinct to isoelectric point of protein from pAP12875 plasmid from A. pasteurianus (pI 12.1) [21]. The rep gene was amplified using PCR amplification and this product was cloned into pGEM-T easy vector (pGEM-rep34 recombinant). Re-cloning gene in pET28a- expression vector was in E. coli BL21 (DE3) cells am- plified protein under T7 promoter and was constructed pET28-rep34. 2.1. Protein Purification The expression vector containing the encoding rep34 gene was used to transform to E. coli BL21 (DE3) cells, which over expressed a Rep34 protein after IPTG induc- tion. An over-expressed band on SDS–PAGE corre- sponding to a protein of approximately 25.55 kDa (with His-tag) was observed in the crude bacterial lysates, consistent with that expected for the recombinant protein (Figure 2(a)). The greater part of Rep34 was found in the supernatant after lysis and after using affinity chroma- tography, the recombinant protein was quickly purified to apparent homogeneity (Figure 2(b)). More detailed data and purification steps were presented in Table 1. The total protein yield at the last purification step was approximately 71 mg of Rep34 per liter of bacterial cul- ture. Upon SDS–PAGE under reducing conditions, the iso- lated protein migrated as a large homogenous band with a molecular weight between 20 and 30 kDa, consistent with that expected for a Rep34 monomer. A monomer of the recombinant protein has a predicted molecular weight of approximately 23.31 kDa. The molecular mass of Rep34 is lower than the molecular mass of RepA monomer protein described in plasmid pRSF1010 that was determined on 31 kDa [39]. The results of the sec- ondary-structure prediction indicate that overall Rep34 is composed of the same amount of α-helical and β-sheet conformation. 2.2. In Vitro DNA-Binding Activity DNA band-shift assays showed that the purified Rep34 His-tagged protein was able to bind specifically to linear double-stranded amplified 1091 bp fragment from repli- cation region of plasmid pGP2 in position 1670-2761 bp. PCR product was cleaved by PvuII and afford two frag- ments 475 bp and 539 bp which were used as substrate for gel shift assay. Incubation of increasing concentra- tions of Rep34 with a fixed amount of DNA progress- sively altered the mobility of the DNA, indicating the formation of protein–DNA complexes. Since the Rep34 apparently has sequence specificity. Furthermore, the increase in DNA band retardation dependent on the Rep34 protein concentration is probably due to the pro- gressive occupation of the DNA molecule by Rep34 (Figure 3 lane 2-6). Interaction Rep34 protein with substrate DNA was specific on the 539 bp large fragment encoded regulation region for transcription of replication protein as well as ori region of plasmid pGP2. The second smaller DNA fragment 475 bp did not change its position during experiment and showed that it is not a specific substrate for analyzed protein. 2.3. Assay of A TPase Activity The most of enzymes, that catalyse biomolecular reac- tions involving binding to the DNA and modificate its structure, need energy from ATP dissociation for their right functioning. ATP molecule can be dissociated by the supporting protein or by the enzyme itself. Rep34 protein belongs to the second group and the ATPase ac- 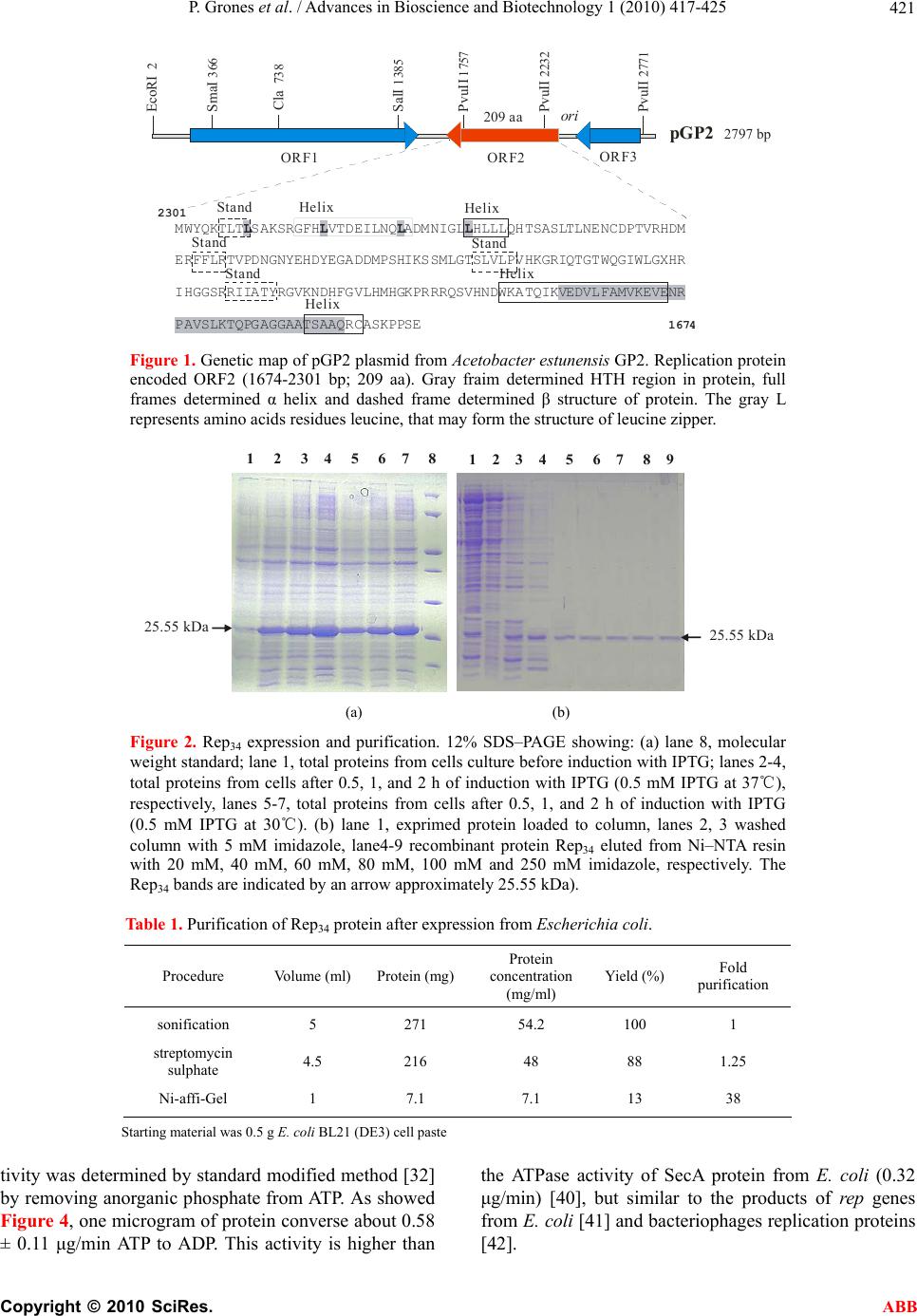 P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 421 ORF1 ORF2 pGP2 2797 bp or i ORF3 PvuII 1757 PvuII 2232 PvuII 2771 SalI 1385 Cla 738 SmaI 366 EcoRI 2 2301 MWYQKTLTLSA KSRGFH LVTDEILNQLADMNIGLLHLLLQHTSASLTLNENCDPTV RHDM ERFFLRTVPDNGNYEHDYEGADD MPSHIKSSMLGTSLVLPVHKGRIQTGTWQGIWL GXHR IHGGSRRIIATYRGVKNDHFGVL HMHGKPRRRQSVHNDWKATQIKVEDVLFAMVKE VENR PAVSLKTQPGAGGAATSAAQRCA SKPPSE 1674 HelixStand Helix Stand St and Stand He lix Helix 209 aa Figure 1. Genetic map of pGP2 plasmid from Acetobacter estunensis GP2. Replication protein encoded ORF2 (1674-2301 bp; 209 aa). Gray fraim determined HTH region in protein, full frames determined α helix and dashed frame determined β structure of protein. The gray L represents amino acids residues leucine, that may form the structure of leucine zipper. 1 2 3 4 5 6 7 8 1 2 3 4 5 6 7 8 9 25.55 kDa25.55 kDa (a) (b) Figure 2. Rep34 expression and purification. 12% SDS–PAGE showing: (a) lane 8, molecular weight standard; lane 1, total proteins from cells culture before induction with IPTG; lanes 2-4, total proteins from cells after 0.5, 1, and 2 h of induction with IPTG (0.5 mM IPTG at 37℃), respectively, lanes 5-7, total proteins from cells after 0.5, 1, and 2 h of induction with IPTG (0.5 mM IPTG at 30℃). (b) lane 1, exprimed protein loaded to column, lanes 2, 3 washed column with 5 mM imidazole, lane4-9 recombinant protein Rep34 eluted from Ni–NTA resin with 20 mM, 40 mM, 60 mM, 80 mM, 100 mM and 250 mM imidazole, respectively. The Rep34 bands are indicated by an arrow approximately 25.55 kDa). Table 1. Purification of Rep34 protein after expression from Escherichia coli. Procedure Volume (ml) Protein (mg) Protein concentration (mg/ml) Yield (%)Fold purification sonification 5 271 54.2 100 1 streptomycin sulphate 4.5 216 48 88 1.25 Ni-affi-Gel 1 7.1 7.1 13 38 Starting material was 0.5 g E. coli BL21 (DE3) cell paste tivity was determined by standard modified method [32] by removing anorganic phosphate from ATP. As showed Figure 4, one microgram of protein converse about 0.58 ± 0.11 μg/min ATP to ADP. This activity is higher than the ATPase activity of SecA protein from E. coli (0.32 μg/min) [40], but similar to the products of rep genes from E. coli [41] and bacteriophages replication proteins [42]. 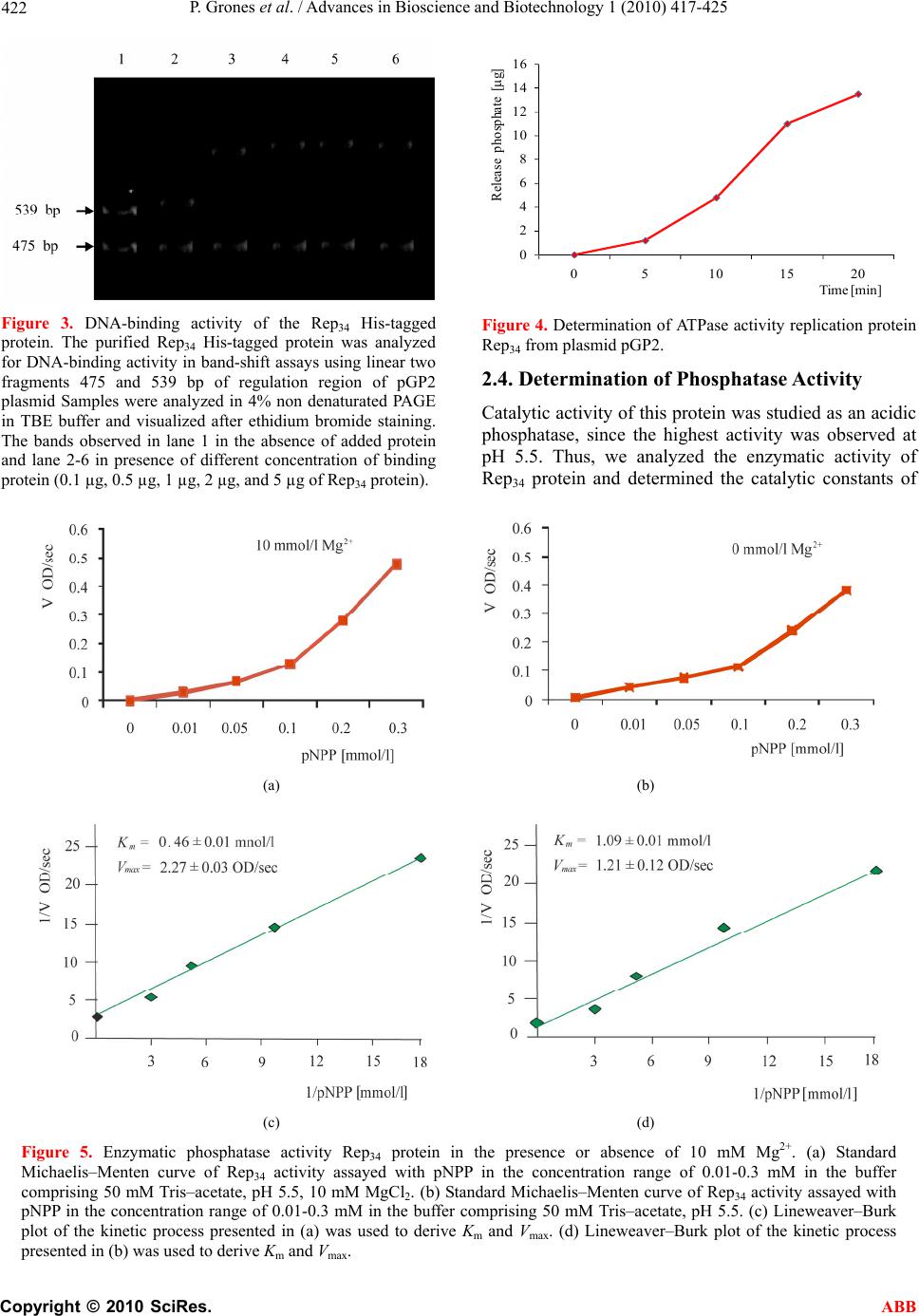 P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 422 Figure 3. DNA-binding activity of the Rep34 His-tagged protein. The purified Rep34 His-tagged protein was analyzed for DNA-binding activity in band-shift assays using linear two fragments 475 and 539 bp of regulation region of pGP2 plasmid Samples were analyzed in 4% non denaturated PAGE in TBE buffer and visualized after ethidium bromide staining. The bands observed in lane 1 in the absence of added protein and lane 2-6 in presence of different concentration of binding protein (0.1 µg, 0.5 µg, 1 µg, 2 µg, and 5 µg of Rep34 protein). 0 2 4 6 8 10 12 14 16 0510 15 20 Release phosphate [g] Time [min] Figure 4. Determination of ATPase activity replication protein Rep34 from plasmid pGP2. 2.4. Determination of Phosphatase Activity Catalytic activity of this protein was studied as an acidic phosphatase, since the highest activity was observed at pH 5.5. Thus, we analyzed the enzymatic activity of Rep34 protein and determined the catalytic constants of (a) (b) (c) (d) Figure 5. Enzymatic phosphatase activity Rep34 protein in the presence or absence of 10 mM Mg2+. (a) Standard Michaelis–Menten curve of Rep34 activity assayed with pNPP in the concentration range of 0.01-0.3 mM in the buffer comprising 50 mM Tris–acetate, pH 5.5, 10 mM MgCl2. (b) Standard Michaelis–Menten curve of Rep34 activity assayed with pNPP in the concentration range of 0.01-0.3 mM in the buffer comprising 50 mM Tris–acetate, pH 5.5. (c) Lineweaver–Burk plot of the kinetic process presented in (a) was used to derive Km and Vmax. (d) Lineweaver–Burk plot of the kinetic process presented in (b) was used to derive Km and Vmax.  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 423 (a) (b) Figure 6. A representative DNA helicase assay in the presence of ATP. (a) Effect of decrease fam-labeled ds DNA substrate after inteaction with Rep34 protein associed helicase activity. Helicase reactions were incubated at 37℃ for 30 min and the substrate and product were quantified by fluorometric analysis. (b) Unwinding super coiled form of plasmid pGP2 by Rep34 helicase activity. Lane 1 standard plasmid pGP2, lane 2-8 activity measured after every 10 minutes of incubation with rep34 protein in optimal reaction condition (OC – open circular, SC – super coiled form). the protein, using the cleavage of synthetic substrate p-nitrophenyl phosphate. The Rep34 was able to cleave pNPP in the presence and in the absence of Mg2+ ions (Figure 5). Enzyme activity is higher about 20% in the presence than absence Mg2+ ions. However phosphatase activity of this protein is about hundred times lower than is described in bacterial acid phosphatases [43]. Assum- ing a Michaelis–Menten model of the enzymatic activity, we used Lineweaver–Burk plots to derive catalytic pa- rameters Km and Vmax. The Km value calculated in the presence of Mg2+ for Rep34 is 0.46 ± 0.01 mM and in the absence of Mg2+ is 1.09 ± 0.01. The phospthatase active- ity is lower than described in bacterial C acid phos- phatase of Helicobacter pylori (1.20 ± 0.25 mM) [44]. 2.5. Helicase Assay To demonstrate DNA helicase activity, unwinding was monitored by the release of a Fam-labeled 32-mer from a partially double-stranded circular M13 DNA substrate. The release labeled primer from double strand DNA was separated by ethanol precipitation. The pellet was dis- solved in water and used for quantification on a fluores- cent reader (Figure 6(a)). Unwinding activity Rep34 pro- tein showed continuously decrease labeled dsDNA after 30 min incubation at 37℃ with 0.5 µg of protein (Fig- ure 6(b)). Although a concentration-dependent increase in helicase activity was observed at lower protein con- centrations, the amount of unwinding was not signifi- cantly increased at greater than 0.5 µg of protein (data not shown). Finally, new replication protein from plasmid isolated from Acetobacter strain was cloned and exprimed in E. coli expression systems. Small protein with basic isoelectric point has two domains one for interaction with other replication protein with leucine zipper and second HTH domain for interaction with DNA. Purified replication protein has phosphatase activity, ATP-ase activity and is able to unwind double strand DNA molecule. HTH domain specific recognise boundig region for iniciation replication and transcription of plasmid pGP2. Fusion of this replication protein with GFP protein was used to monitor protein expression by fluorometric microscopy. REFERENCES [1] Konieczny, I. (2003) Strategies for helicase recruitment and loading in bacteria. EMBO Report, 4(1), 37-41. [2] Messer, W. (2002) The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiology Review, 26(4), 355-374. [3] Messer, W. and Weigel, C. (2003) DnaA as a transcript- tion regulator. Methods in Enzymology, 370, 338-349. [4] Mackiewicz, P., Zakrzewska-Czerwinska, J., Zawilak, A., Dudek, M.R. and Cebrat, S. (2004) Where does bacterial replication start? Rules for predicting the oriC region. Nucleic Acids Research 32(13), 3781-3791. [5] Schmidhauser, T.J., Filutowicz, M. and Helinski, D.R. (1983) Replication of derivatives of the broad host range plasmid RK2 in two distantly related bacteria. Plasmid, 9(3), 325-330. [6] Shah, D.S., Cross, M.A., Porter, D., Thomas, C.M. (1995) Dissection of the core and auxiliary sequences in the vegetative replication origin of promiscuous plasmid RK2. Journal of Molecular Biology, 254(4), 608-622. [7] Doran, K.S., Helinski, D.R. and Konieczny, I. (1999) Host-dependent requirement for specific DnaA boxes for plasmid RK2 replication. Molecular Microbiology, 33(3), 490-498. [8] Kawasaki, Y., Matsunaga, F., Kano, Y., Yura, T. and Wada, C. (1996) The localized melting of mini-F origin by the combined action of the mini-F initiator protein (RepE) and HU and DnaA of Escherichia coli. Molecular  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 424 and General Genetic, 253(1-2), 42-49. [9] Lu, Y.B., Datta, H.J. and Bastia D. (1998) Mechanistic studies of initiator–initiator interaction and replication initiation. EMBO Journal, 17(17), 5192-5200. [10] Park, K., Mukhopadhyay, S. and Chattoraj, D.K. (1998) Requirements for and regulation of origin opening of plasmid P1. Journal of Biological Chemistry, 273(38), 24906-24911. [11] Kruger, R., Konieczny, I. and Filutowicz, M. (2001) Monomer/dimer ratios of replication protein modulate the DNA strand-opening in a replication origin. Journal of Molecular Bioliology, 306(5), 945-955. [12] Sharma, R., Kachroo, A. and Bastia, D. (2001) Mecha- nistic aspects of DnaA–RepA interaction as revealed by yeast forward and reverse twohybrid analysis. EMBO Journal, 20(16), 4577-4587. [13] Konieczny, I. and Helinski, D.R. (1997) Helicase deliv- ery and activation by DnaA and TrfA proteins during the initiation of replication of the broad host range plasmid RK2. Journal of Biological Chemistry, 272(52), 33312-33318. [14] Doran, K.S., Konieczny, I. and Helinski, D.R. (1998) Replication origin of the broad host range plasmid RK2: positioning of various motifs is critical for initiation of replication. Journal of Biological Chemistry, 273(14), 8447-8453. [15] Sharma, R., Kachroo, A. and Bastia, D. (2001) Mecha- nistic aspects of DnaA–RepA interaction as revealed by yeast forward and reverse twohybrid analysis. EMBO Journal, 20(16), 4577-4587. [16] Kues, U. and Stahl, U. (1989) Replication of plasmids in gram-negative bacteria. Microbiology Review, 53(4), 332-343. [17] del Solar, G., Giraldo, R., Ruiz-Echevarria, M.J., Espi- nosa, M. and Diaz-Orejas, R. (1998) Replication and control of circular bacterial plasmids. Microbiology Mo- lecular Biology Review, 62(2), 434-464. [18] Okumura, H., Uozumi, T. and Beppu, T. (1985) Con- struction of plasmid vectors and genetic transformation system for Acetobacter aceti. Agricultural and Biological Chemistry, 49(4), 1011-1017. [19] Fukaya, M., Okumura, T., Masai, H., Uozumi, T. and Beppu, T. (1985) Construction of new shuttle vector for Acetobacte. Agricultural and Biological Chemistry, 49(7), 2083-2090. [20] Grones, J., Škereňová, M., Bederková, K. and Turňa, J. (1989) Isolation and characterisation of plasmid pAC1 from Acetobacter pasteurianus. Biologia, 44(12), 1181-1186. [21] Fomenkov, A., Xiao, J. and Xu, S. (1995) Nucleotide sequence of a small plasmid isolated from Acetobacter pasteurianus. Gene, 158(1), 143-144. [22] Trček, J., Raspor, P. and Teuber, M. (2000) Molecular identification of Acetobacter isolates from submerged vinegar production, sequence analysis of plasmid pJK2-1 and application in the development of a cloning vector. Applied. Microbiology and Biotechnology, 53(3), 289-295. [23] Kretová, M., Szemes, T., Laco, J., Gronesová, P. and Grones, J. (2005) Analysis of replication region of the cryptic plasmid pAG20 from Acetobacter aceti 3620. Biochemical and Biophysical Research Commununica- tion, 328(1), 27-31. [24] Bullock, W.O., Fernandez, J.M. and Short J.M. (1987) XL1-Blue - a high-efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection, Biotechniques 5(3), 376-379. [25] Sanger, F., Nicklen, S. and Coulson, A.R. (1977) DNA sequencing with chainterminating inhibitors, Proceedings of the National Academy of Sciences of the U.S.A., 74(12), 5463-5467. [26] Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search Programs. Nucleic Acids Research, 25(17), 3389-3402. [27] Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22(22), 4673-4680. [28] Cuff, J.A., Clamp, M.E., Siddiqui, A.S., Finlay, M. and Barton, G.J. (1998) Jpred: a consensus secondary struc- ture prediction server. Bioinformatics, 14(10), 892-893. [29] Rost, B., Yachdav, G. and Liu, J. (2004) The PredictPro- tein Server. Nucleic Acids Research, 32, W321-W326. [30] Bryson, K., McGuffin, L.J., Marsden, R.L., Ward, J.J., Sodhi, J.S. and Jones, D.T. (2005) Protein structure pre- diction servers at University College London. Nucleic Acids Research, 33, W36-W38. [31] Cheng, J., Randall, A., Sweredoski, M. and Baldi, P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Research, 33, W72-W76. [32] Lanzetta, P.A., Alvarez, L.J., Reinach, P.S. and Candia, O.A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Analytical Biochemistry, 100(1), 95-97. [33] Matson, S.W., Tabor, S. and Richardson, C.C. (1983) The gene 4 protein of bacteriophage T7: Characterization of helicase activity. Journal of Biological Chemistry, 258(22), 14017-14024. [34] Wiley, S.R., Kraus, R.J. and Mertz, J.E. (1992) Func- tional binding of the TATA box binding component of transcription factor TFIID to the -30 region of TATA-less promoters. Proceedings of the National Academy of Sci- ences of the U.S.A., 89(13), 5814-5818. [35] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Mo- lecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [36] Kamenski, T., Heilmeier, S., Meinhart, A. and Cramer, P. (2004) Structure and mechanism of RNA polymerase II CTD phosphatase. Molecular Cell, 15(3), 399-407. [37] Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T7. Nature, 227(5259), 680-685. [38] Bradford, M.M. (1976) A rapid and sensitive for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding, Analytical Bio- chemistry, 72, 248-254. [39] Scherzinger, E., Haring, V., Lurz, R. and Otto, S. (1991) Plasmid RSF1010 DNA replication in vitro promoted by purified RSF1010 RepA, RepB and RepC proteins. Nu- cleic Acids Research, 19(6), 1203-1211. [40] Robson, A., Gold, V.A., Hodson, S., Clarke, A.R. and  P. Grones et al. / Advances in Bioscience and Biotechnology 1 (2010) 417-425 Copyright © 2010 SciRes. ABB 425 Collinson, I. (2009) Energy transduction in protein transport and the ATP hydrolytic cycle of SecA. Pro- ceedings of the National Academy of Sciences of the U.S.A., 106(13), 5111-5116. [41] Scott, J.F. and Kornberg, A. (1978) Purification of the re p protein of Escherichia coli. Journal of Biological Chem- istry, 253(9), 2392-2397. [42] Ebisuzaki, K., Behme, M.T., Senior, C., Shannon, D. and Dunn, D. (1972) An Alternative Approach to the Study of New Enzymatic Reactions Involving DNA (DNA-de- pendent ATPases-purification-properties-E. coli). Pro- ceedings of the National Academy of Sciences of the U.S.A., 69(2), 515-519. [43] Reilly, T.J., Chance, D.L., Calcutt, M.J., Tanner, J.J., Felts, R.L., Waller, S.C., Henzl, M.T., Mawhinney, T.P., Ganjam, I.K. and Fales, W.H., (2009) Characterization of a Unique Class C Acid Phosphatase from Clostridium perfringens. Applied of Environmental Microbiology, 75(11), 3745-3754. [44] Reillya, T.J. and Calcuttb, M.J. (2004) The class C acid phosphatase of Helicobacter pylori is a 50 nucleotidase. Protein Expression and Purification, 33(1), 48-56. |