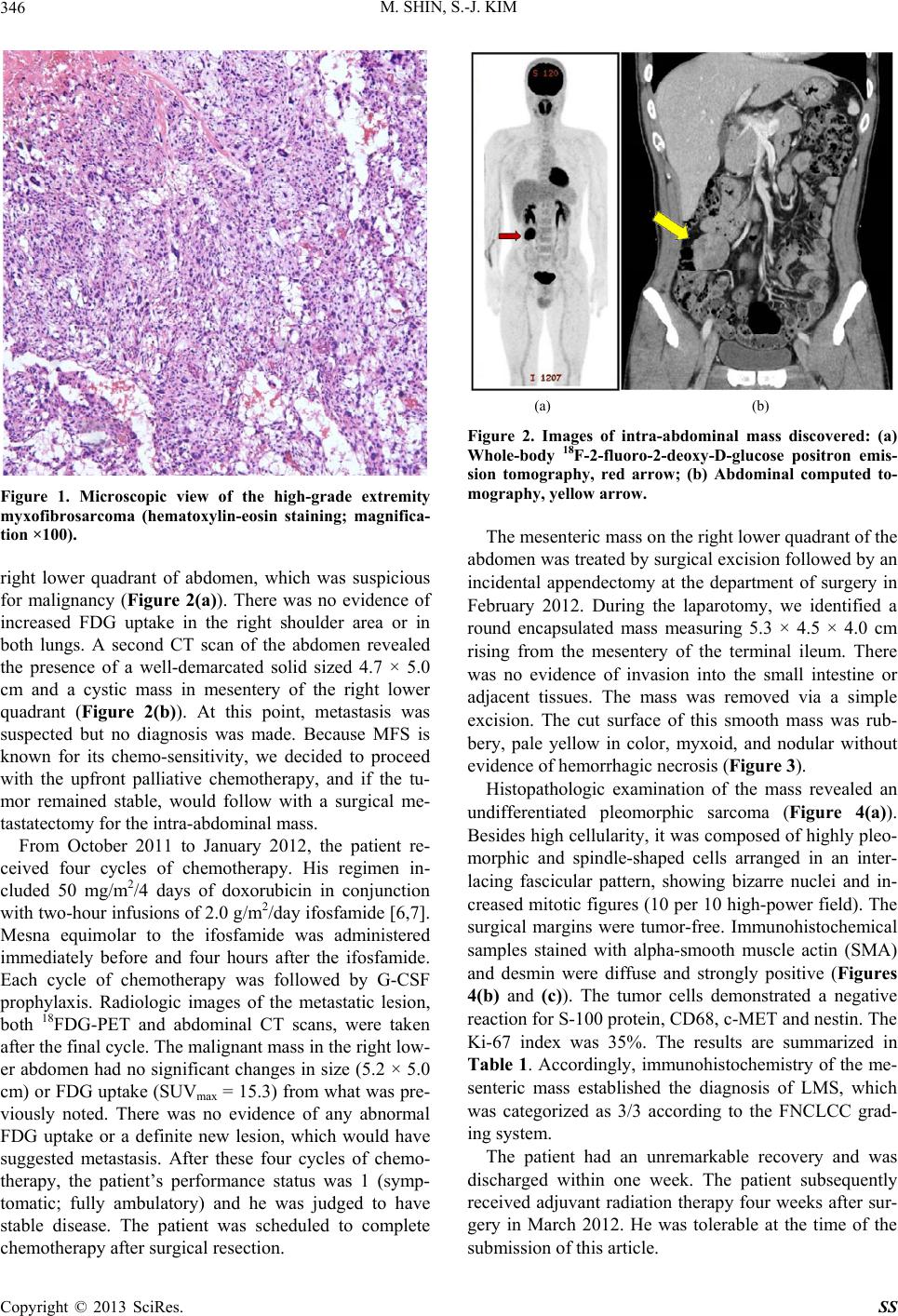
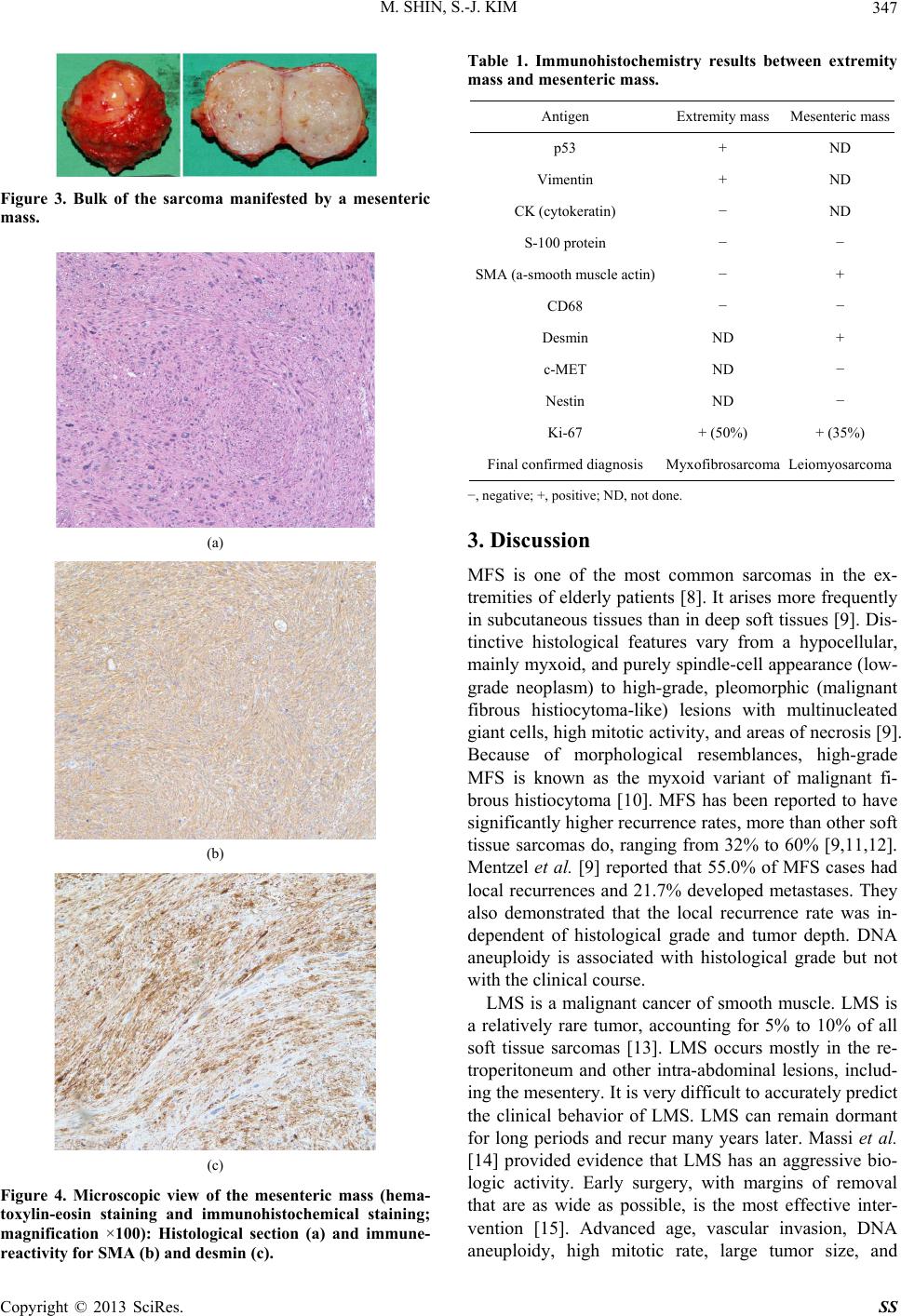
M. SHIN, S.-J. KIM
Copyright © 2013 SciRes. SS
349
doi:10.1097/00000478-199604000-00001
[10] S. W. Weiss and F. M. Enzinger, “Myxoid Variant of
Malignant Fibrous Histiocytoma,” Cancer, Vol. 39, No. 4,
1977, pp. 1672-1685.
doi:10.1002/1097-0142(197704)39:4<1672::AID-CNCR2
820390442>3.0.CO;2-C
[11] L. Angervall, L. G. Kindblom and C. Merck, “Myxofi-
brosarcoma. A Study of 30 Cases,” Acta Pathologica Mi-
crobiologica Scandinavica Section A Pathology, Vol.
85A, No. 2, 1977, pp. 127-140.
doi:10.1111/j.1699-0463.1977.tb00410.x
[12] A. Gronchi, S. Lo Vullo, C. Colombo, et al., “Extremity
Soft Tissue Sarcoma in a Series of Patients TREATED at
a Single Institution: Local Control Directly Impacts Sur-
vival,” Annals of Surgery, Vol. 251, No. 3, 2010, pp.
506-511. doi:10.1097/SLA.0b013e3181cf87fa
[13] P. Gustafson, H. Willen, B. Baldetorp, et al., “Soft Tissue
Leiomyosarcoma. A Population-Based Epidemiologic and
Prognostic Study of 48 Patients, Including Cellular DNA
Content,” Cancer, Vol. 70, No. 1, 1992, pp. 114-119.
doi:10.1002/1097-0142(19920701)70:1<114::AID-CNCR
2820700119>3.0.CO;2-U
[14] D. Massi, G. Beltrami, M. M. Mela, et al., “Prognostic
Factors in Soft Tissue Leiomyosarcoma of the Extremi-
ties: A Retrospective Analysis of 42 Cases,” European
Journal of Surgical Oncology, Vol. 30, No. 5, 2004, pp.
565-572. doi:10.1016/j.ejso.2004.03.002
[15] G. D. Demetri, S. Antonia, R. S. Benjamin, et al., “Soft
Tissue Sarcoma,” Journal of the National Comprehensive
Cancer Network, Vol. 8, No. 6, 2010, pp. 630-674.
[16] K. Miyajima, Y. Oda, Y. Oshiro, et al., “Clinicopatho-
logical Prognostic Factors in Soft Tissue Leiomyosar-
coma: A Multivariate Analysis,” Histopathology, Vol. 40,
No. 4, 2002, pp. 353-359.
doi:10.1046/j.1365-2559.2002.01361.x
[17] A. Tsang, J. R. Nash, M. V. Fordham, M. N. Hartley and
G. J. Poston, “The Management of Retroperitoneal Li-
posarcoma with Synchronous Intra-Duodenal Sarcoma,”
European Journal of Surgical Oncology, Vol. 29, No. 6,
2003, pp. 515-518. doi:10.1016/S0748-7983(03)00050-7
[18] A. Hadidy, A. Alsharif, R. Sheikh-Ali, et al., “Odonto-
genic Myxofibroma Synchronous with Primary Angio-
sarcoma of the Spleen,” British Journal of Radiology, Vol.
83, No. 985, 2010, pp. e10-13. doi:10.1259/bjr/14078580
[19] K. Tsutsumi, Y. Aida, T. Ohno and M. Ookura, “Primary
cardiac Rhabdomyosarcoma Following a Uterine Leio-
myosarcoma: Double Primary Sarcomas,” Japanese Jour-
nal of Thoracic and Cardiovascular Surgery, Vol. 53, No.
8, 2005, pp. 458-462. doi:10.1007/s11748-005-0086-7
[20] S. R. Grobmyer, N. Luther, C. R. Antonescu, S. Singer
and M. F. Brennan, “Multiple Primary Soft Tissue Sar-
comas,” Cancer, Vol. 101, No. 11, 2004, pp. 2633-2635.
doi:10.1002/cncr.20679
[21] L. Congyang, W. Xinggui, L. Hao and H. Weihua, “Syn-
chronous Histiocytic Sarcoma and Diffuse Large B Cell
Lymphoma Involving the Stomach: A Case Report and
Review of the Literature,” International Journal of He-
matology, Vol. 93, No. 2, 2011, pp. 247-252.
doi:10.1007/s12185-011-0773-3
[22] A. Cossu, A. Deiana, A. Lissia, et al., “Synchronous In-
terdigitating Dendritic Cell Sarcoma and B-Cell Small
Lymphocytic Lymphoma in a Lymph Node,” Archives of
Pathology & Laboratory Medicine, Vol. 130, No. 4, 2006,
pp. 544-547.
[23] M. Rettenmaier, H. D. Epstein, L. N. Abaid, K. A. Bech-
tol and B. H. Goldstein, “Leiomyosarcoma with Syn-
chronous Clear Cell Ovarian Carcinoma,” Onkologie, Vol.
33, No. 12, 2010, pp. 695-697. doi:10.1159/000322216
[24] Y. Takemoto, M. Kawahara, S. Yamamoto, et al., “Syn-
chronous Primary Adenocarcinoma of the Lung and
Leiomyosarcoma of the Small Intestine,” Internal Medi-
cine, Vol. 39, No. 8, 2000, pp. 655-658.
doi:10.2169/internalmedicine.39.655
[25] O. Merimsky, Y. Kollender, J. Issakov, et al., “Multiple
Primary Malignancies in Association with Soft Tissue
Sarcomas,” Cancer, Vol. 91, No. 7, 2001, pp. 1363-1371.
doi:10.1002/1097-0142(20010401)91:7<1363::AID-CNC
R1140>3.0.CO;2-F
[26] R. Geva, I. Jiveliouk, M. Inbar, et al., “The Co-Occur-
rence of Breast Cancer and Soft Tissue Sarcoma in a Sin-
gle Cohort Series,” American Journal of Clinical Oncol-
ogy, Vol. 32, No. 1, 2009, pp. 34-37.
doi:10.1097/COC.0b013e31817b6087
[27] A. Fon, L. Mileshkin, J. Slavin, et al., “Synchronous Di-
agnosis of Hodgkin Lymphoma and Osteosarcoma,” In-
ternal Medicine Journal, Vol. 37, No. 10, 2007, pp. 731-
732. doi:10.1111/j.1445-5994.2007.01465.x
[28] C. V. Wa, S. DeVries, Y. Y. Chen, F. M. Waldman and E.
S. Hwang, “Clinical Application of Array-Based Com-
parative Genomic Hybridization to Define the Relation-
ship between Multiple Synchronous Tumors,” Modern
Pathology, Vol. 18, No. 4, 2005, pp. 591-597.
doi:10.1038/modpathol.3800332
[29] E. Hernando, E. Charytonowicz, M. E. Dudas, et al., “The
AKT-mTOR Pathway Plays a Critical Role in the Devel-
opment of Leiomyosarcomas,” Nature Medicine, Vol. 13,
No. 6, 2007, pp. 748-753. doi:10.1038/nm1560
[30] V. de Giorgi, R. Santi, M. Grazzini, et al., “Synchronous
Angiosarcoma, Melanoma and Morphea of the Breast
Skin 14 Years after Radiotherapy for Mammary Carci-
noma,” Acta Dermato-Venereologica, Vol. 90, No. 3,
2010, pp. 283-286.
[31] N. Ally, D. Kumar and F. M. Wodajo, “Synchronous
Fibrosarcoma and Medullary Thyroid Cancer in a Man
with AIDS,” American Journal of Clinical Oncology, Vol.
29, No. 5, 2006, pp. 532-533.
doi:10.1097/01.coc.0000177913.82468.ba
[32] J. H. Park, C. H. Kang, C. H. Kim and I. J. Chae, “Highly
Malignant Soft Tissue Sarcoma of the Extremity with a
Delayed Diagnosis,” World Journal of Surgical Oncology,
Vol. 8, 2010, p. 84. doi:10.1186/1477-7819-8-84