64 Y. G. XIA ET AL.
2.2. Isolation of Polysaccharides from E. sinica
The dry stems of E. sinica were ground to powders, and
submitted to sequential extractions as follows: dry pow-
ders (1.0 kg) were extracted 3 times with 10 vol of 95%
EtOH under reflux for 3 h each time to remove lipids.
The residue was dried in air and then extracted 3 times
with 10 vol of distilled water for 24 h (each time) at 4°C.
The combined aqueous extracts were filtered, concen-
trated 10-fold, and 95% EtOH added to final concentra-
tion of 80%. The precipitate was dissolved in 600 mL of
water and deproteinated 15 times with 200 mL of 5:1
chloroform-n-butanol as described by Staub [5]. The
resulting aqueous fraction was extensively dialyzed
(cut-off Mw 3500 Da) against tap water for 48 h and dis-
tilled water for 48 h and precipitated again by adding a 5
fold volume of ethanol. After centrifugation, the precipi-
tate was washed with anhydrous ethanol and then dis-
solved in water and lyophilised to yield the crude poly-
saccharide A (8.5 g) was collected by centrifugation
(3000 rpm, 10 min, 20°C).
Crude polysaccharide A (3.0 g) was dissolved in dis-
tilled water and passed through two series connected
resin columns (Amberlite FPA90-Cl (Cl- form) and Am-
berlite IRC-84 (H+ form)) eluting with distilled water and
1.0 M NaCl to yield fractions Fr. A1 (1.2 g) and Fr. A2
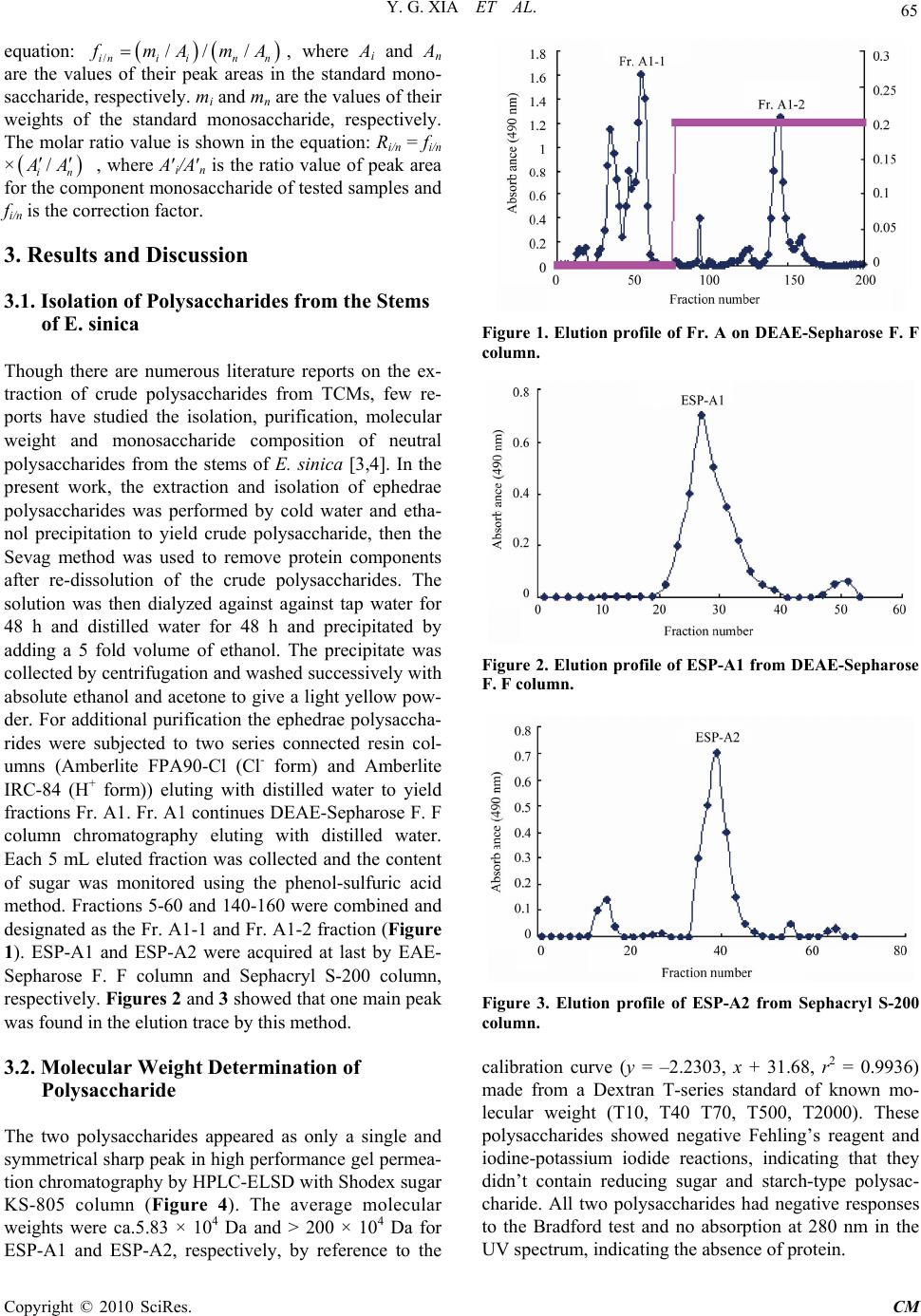
(0.9 g), respectively. Fr. A1 (1.0 g) was chromatogra-
phed over DEAE-Sepharose F. F eluting with distilled
water and 0.2 M NaCl to yield subfractions Fr. A1-1
(400.0 mg) and Fr. A1-2 (380.0 mg), respectively. Fr.
A1-1 (400.0 mg) was further purified by gel-permeation
chromatography on a high resolution Sephacryl S-400
eluting with distilled water to afford Fr.-A1-1-1 (160.0
mg) and Fr.-A1-1-2 (180.0 mg). Fr.-A1-1-1 (160.0 mg)
was further purified by DEAE-Sepharose F. F eluting
with distilled water to afford ESP-A1 (130.0 mg). Fr.
-A1-1-2 was further purified by Sephacryl S-200 eluting
with distilled water to afford ESP-A2 (145.0 mg).
2.3. Identification on Purity of Polysaccharides
and Molecular Weight Determination
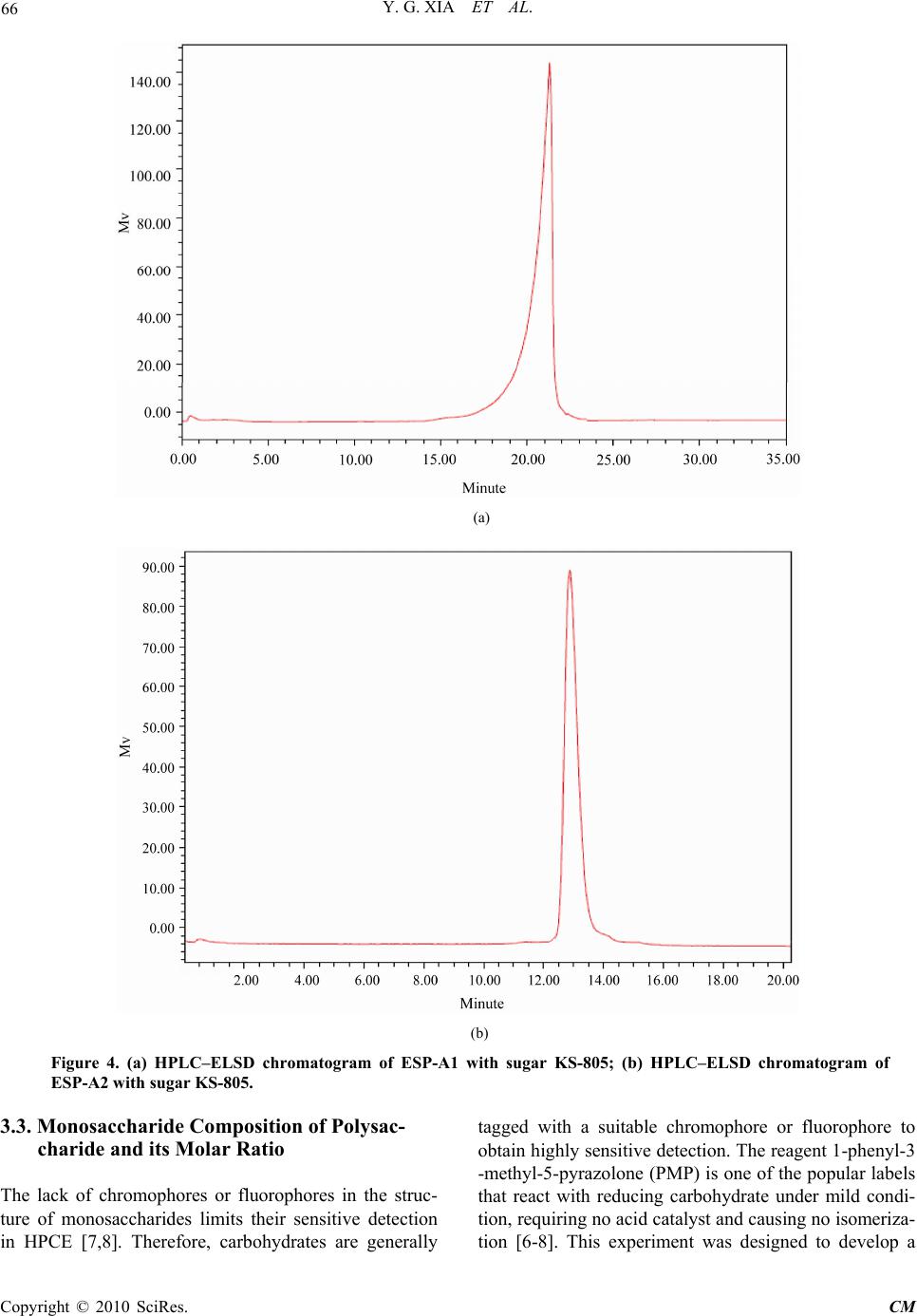
The molecular mass of the polysaccharide (5 mg/mL)
was determined by high performance liquid chromatog-
raphy (HPLC), using Waters 2695 HPLC and Alltech
ELSD 2000 detector. The separation was carried out on a
Shodex sugar KS-805 column (8.0 mm × 300 mm, 17
μm) coupled with a Shodex KS-G guard column (6 mm
× 50 mm, 7 μm). The Dextran standards (T-10, T-40,
T-70, T-500, T-2000) were used for the calibration curve.
The isocratic elution was employed using water with 0.5
mL/min at 30°C and the injection volume was 10 μL.
While the drift tube temperature for ELSD was set at 116
°C, the nitrogen flow rate was 3.3 L/min for the deter-
mination of polysaccharides. Their purities were over
98% by HPLC analysis. Total carbohydrate contents in
purified samples were determined by phenol-sulfuric
acid colorimetric method using glucose as the standard
(Dubois, Gilles, Hamilton, Rebers, & Smith 1956). Pro-
teins in the polysaccharides were detected by the method
of UV absorption on a TU-1800PC spectrophotometer
(Beijing Purkinje General Instrument Co., Ltd., China).
2.4. Analysis of Monosaccharide Composition
Monosaccharide composition was analyzed according to
the following procedure: Each polysaccharide sample
(20 mg) was dissolved in 2 ml of 2.0 M H2SO4 in an
ampoule (5 ml). The ampoule was sealed under a nitro-
gen atmosphere and kept in 110°C to hydrolyze the
polysaccharide into component monosaccharides for 6 h,
then cooled to room temperature and neutralized with 2
ml of 4.0 M sodium hydroxide. The reaction mixture was
diluted to 5 ml with deionized water and was centrifu-
galized at 1000 rpm for 5 min. Then the supernatant was
ready for the following experiments.
PMP derivatization of monosaccharides was carried
out as described previously [6]. 200 μl of individual
standard monosaccharide, or mix standard monosaccha-
ride solutions, or the hydrolyzed polysaccharide samples
were placed in the 2.0 mL centrifuge tubes, respectively,
then 0.5 M methanol solution (100 μl) of PMP and 0.3 M
aqueous sodium hydroxide (100 μl) were added to each.
Each mixture was allowed to react for 30 min at 70°C
water bath, then cooled to room temperature and neu-
tralized with 100 μl of 0.3 M HCl. The resulting solution
was performed on liquid-liquid extraction with same
volume of isoamyl acetate (two times) and chloroform
(one time), respectively. After being shaken vigorously
and centrifuged, the organic phase was carefully dis-
carded to remove the excess reagents. Then the aqueous
layer was filtered through a 0.45 μm membrane and di-
luted with water before HPCE analysis.
The analysis of PMP-labeled monosaccharides was
carried out on a P/ACE MDQ capillary electrophoresis
instrument (Beckman Coulter, Fullerton, CA, USA). An
integrated P/ACE 32 Karat Station (software version 4.0)
was used to perform the data collection and to control the
operational variables of the system. Separation was car-
ried out in an unmodified fused silica capillary (48.5 cm
× 50 μm i.d., effective length 40 cm) with direct UV
monitoring using a photodiode array detector at wave-
length 254 nm including 35 mM borate at pH 10.02, cap-
illary temperature 25°C and applied voltage 20 kV. The
molar ratio of the component monosaccharides is calcu-
lated as follows. The correction factor is shown in the
Copyright © 2010 SciRes. CM