Paper Menu >>
Journal Menu >>
 Neuroscience & Medicine, 2010, 1, 50-59 doi:10.4236/nm.2010.12008 Published Online December 2010 (http://www.SciRP.org/journal/nm) Copyright © 2010 SciRes. NM Added after Anoxia-Reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Carmen Arce 1, José Luis Arteaga 1,Eduardo Sánchez-Mendoza 1, María Jesús Oset-Gasque 1, Sixta Cañadas 1, María Pilar González 1,2 1Department of Biochemistry and Molecular Biology, Faculty of Pharmacy, Universidad Complutense, Madrid, Spain; 2Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu, Madrid, Spain Email: pilarg@fa r m.ucm.es Received September 7th, 2010; revised September 17th, 2010; accepted November 8th, 2010 ABSTRACT Estrogens and phytoestrogens have neuroprotective effect against neuronal damage induced by cerebral ischemia /reperfusion (I/R) injury. In preceding studies, the phytoestrogen effects have been assessed by administration previous to the ischemic period, conditions which are unusual to apply to the treatment of human stroke. Here we present a study on neuroprotection afforded by genistein on rat embryo cortical neurons subjected to oxygen and glucose deprivation (OGD) followed by re-oxigenation, when added after the stress stimulus. At 1 and 2 h of OGD times and after 24 h of reperfusion, cell viability, necrotic, apoptotic and autophagic cell death and different parameters related to oxidative stress and mitochondrial dysfunction were measured in the absence and presence of 1 µM genisteine. We found an in- creasing loss of neuronal viability after 1-5 h of OGD which was only reversed in part by 24 h of reperfusion. These changes were preceded by increases in ROS generation, caspase-3 activation, LDH release and increase in LC3B lipi- dation, indicative of autophagia. Treatment with 1 µM genistein during the 24 h reperfusion significantly attenuated neuronal necrosis and autophagia induced by 1 and 2 h of OGD exposure. Genistein also decreased ROS generation and lipid-peroxidation induced by 2 h of OGD. These results suggest an important neuroprotective effect of genistein against transient post-isch emic-like conditions. Keywords: Cortical Neurons, Oxygen-glucose Deprivation, Brain Ischemia, Neuronal Death, Necrosis, Apoptosis, Autophagy, Neuroprotection, N eurorepair,Phytoestrogens, Genistei n 1. Introduction Global cerebral ischemia is a clinical outcome that occurs as consequence of different conditions like cardiac arrest, coronary artery bypass surgery and reversible severe hy- pertension. All these events are the cause of a dra matically reduced blood supply of oxygen and nutrients, as glucose, which are the responsible for cellular damage. Studies performed during the last 20 years have identified several biochemical and cellular events that lead to ischemic neu- ronal degeneration [1,2]. Although the management of stroke has improved remarkably over the last decade due mainly to the use of thrombolysis, moreover, nowadays there is a growing interest to develop treatments aimed to promote repair and regeneration of brain tissue damaged by ischemia, which could be more effective than neuro- protection on the brain recovery after ischemia. Our increasing knowledge concerning with the ischemic cascade is leading to a considerable development of phar- macological tools, suggesting that each step of this cascade might be a target for cytoprotection. Thus, many neuro- protective drugs, such as calcium channel blockers, anti- oxidants or free radical scavengers, GABA agonists, glu- tamate antagonists, growth factors, NO inhibitors, phos- phatidylcholine precursors and so on, have been studied in experimental stroke models (for review, see [3]). However, very few have shown efficacy in clinical trials. Among them, caspase inhibitors to reduce apoptosis and estrogen or its derivatives, phytoestrogens, have been proposed as future neuroprotecti ve treatm ent [4,5] . In vivo and in vitro studies have demonstrated powerful neuroprotective effects of estrogens against a variety of insults. In rodents, estrogen reduces injury caused by focal cerebral ischemia [6] and global ischemia [7]. In vitro,  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 51 similar neuroprotective effects have been demonstrated in primary cortical [8-10], hippocampal [11-13] and mesen- cephalic cultures [14]. Furthermore, these neuroprotective effects appear to utilize multiple mechanisms depending on the injury and cell type [5,15] . It is increasingly clear that physiological doses of isoflavones, which can behave as phytoestrogens, can mimic some of the neuroprotective effects of estrogens. [16-18]. In vitro some soy isoflavones can protect pri- mary neurons from glutamate toxicity [19], thapsigar- gin-induced apoptosis [20], and -amyloid toxicity [21]. Several studies have indicated that genistein admini- stration prevents delayed neuronal death after transient global cerebral ischemia [22], inhibits the lipid peroxi- dation induced by pro-oxidant agents in cultured corti- cal neurons [23] and attenuates the oxidative stress and neuronal damage following cerebral ischemia in rat hippocampus [24]. However, the mechanisms underly- ing protection from ischemic injury remain unclear. Recently, Schreihofera and Redmond, [17] have shown that pre-treatment with dietary levels of soy phytoes- trogens can mimic neuroprotective effects observed with estrogen. Nevertheless, in all these work s estrogen and phytoestrogens have been studied as neuroprotec- tive agents but it has not been studied their possible effects on neurorepair in post-ischemic period, in spite that it is in this time when protection is necessary after an ischemical event. The present study was performed to test the ability of the phytoestrogen genistein to in- hibit neuronal death in the post-ischemic period and to obtain further evidence about the molecular mecha- nisms by which phytoestrogens exercise these potential neuroprotective and neurorepairing effects. The study has been performed by using cortical neurons in pri- mary cultures subjected to oxygen-glucose deprivation (OGD), since this has been proved to be a good model to test the mechanisms of neuroprotective agents in brain ischemia. Our results suggest that genistein may protect neurons from OGD in the post-ischemic period by attenuating oxidative stress, lipid peroxidation, and necrotic and autophagic cell death. 2. Material and Methods 2.1. Materials Tetramethylrhodamine methyl ester (TMRM) and 2,7 dichlorodihydro-fluorescein diacetate (2,7 DCF-DA) and bis-[1,3-diethyl-thio-barbiturate]-trimethine-oxonol (bisoxonol) were purchased b y Molecular Probes ( E u ge n e , OR). Minimum Essential Eagle’s Medium (EMEM) was from Bio-Whittaker, and Foetal Calf (FCS) was from Sera-Lab (Sussex, England). Other chemicals were reactive grade products from Merck (Darmstadt, Germany). Anti-rabit microtubule associated protein light chain 3 (LC3B) antibody was from Cell Signal- lyng Technology Inc. (Danvers, MA). 2.2. Cell Isolation and Culture Foetal rat brains of 19 days gestation were used. Brain neurons were obtained as described in [25]. Isolated neurons were suspended in EMEM medium containing 0.3 g/l glutamine, 3 g/l glucose, 10% FCS, 100 U/ml penicillin and 100 µg/ml streptomycin. Cells, at density of 106 cells/cm/well, were placed on plastic Petri dishes, treated with 10 µg/ml poly-D-lysine, so that the cells could attach themselves to these plates. Incubations were made in a humidified incubator with 5% CO2/95% air at 37˚C. After 72 hours, the incubation medium was replaced by fresh medium to which 10 µM of cytosine arabinoside was added to prevent overgrowth of con- taminating glial cells. Cells were used after 7 days of culture. Cell purity was checked by both, cells staining with cresyl violet to identify neurons, and immunocy- tochemistry with the specific anti-GFAP antibody [25] to identify glial cells. Under these conditions, the glial cell number in the cultures was of 8.3 ± 3.6% of the total cell population (neurons + glial cells). 2.3. Oxygen-glucose Deprivation (OGD) After 7 days of culture, the medium was removed and replaced by a glucose-free balanced salt Locke medium (composition (mM) NaCl 140, KCl 4.7, KH2PO4 1.2, CaCl2 2.5, MgSO4 1.2 and HEPES 15, pH 7.5). Multi- wells were then placed in a gassed chamber equipped with gas entry and outflow devices, the chamber was then sealed and placed in the gassed incubator in an atmosphere of 95% N2/5% CO2 at 37˚C for 1 or 2 h. OGD was terminated by removing the cultures from the chamber. These cells were considered as cells treated with OGD. In some multiwells, after this treatment, the OGD cultured medium was replaced by normal medium (EMEM containing 3 g/l glucose) and the multiwells were returned to the incubator under normoxic condi- tions (95% air/5% CO2). This treatment was considered as reperfusion. The time of reperfusion of OGD-neurons was of 24 h. Control experiments were performed al- ways in parallel, so, after 7 days of culture, the medium was removed and replaced by fresh normal medium EMEM containing 3 g/l of glucose. Control cells were kept at incubator under this condition during 1 or 2 h. Then, OGD-neurons were processed as indicated in each case. 2.4. Assessment of Cell Viability Cell viability was determined by the XTT test, based on the ability of living metabolically active cells to reduce  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 52 the yellow tetrazolium salt (XTT) to form an orange formazan dye, which quantity directly correlates to the number of living cells. Measurements were preformed as described [25]. Briefly, control and treated neurones (2 × 105) were incubated with the XTT solution at 0.3 mg/ml final concentration and the orange formazan dye formed was spectrophotometrically quantified using an ELISA plate reader , at 450 nm (reference 650 nm). Results were expressed as percentages with respect to the control cells. 2.5. Measurement of the ROS Formation To ass ay the ROS for mat i on, 2,7-dic hlorodihydro fluorescein diacetate (H2DCF-DA), a non-fluorescent lipophilic re- agent, was used. HDCF-DA en ters in to the cells, wh ere it is transformed into 2,7-dichloro dihydrofluorescein (H2DCF) by the action of intracellular estearases. H2DCF is oxi- dized to fluorescent DCF by hydrogen peroxide. H2DCF -DA (5 M) was added to the cells, before subjectting them to the different treatments and then the incubation medium was removed, the cells were washed twice with PBS and the fluorescence was measured in a FL600-Bio Tek spectrofluorimeter with filters of 485/20 nm exc and 530/25 nm em. Results were expressed as arbitrary fluo- rencence units (AFU) ratios. 2.6. Lactate Dehydrogenase (LDH) LDH activity was measured as the rate of decrease of the absorbance at 340 nm, resulting from the oxidation of NADH to NAD+ as described [25]. Briefly, the culture medium was collected and the neurones were lysed with 0.1 M Tris-HCl (pH 7.4) containing 0.1% Triton X-100, cell suspension was centrifuged at 13,000 g during 5 minutes and LDH activity measur ed in both prep arations, culture medium and cells supernatant. Activity of the LDH release is given as percentage of release of LDH with respect to the total LDH conten t (LDH in the cultu re medium plus LDH inside the cells). 2.7. Caspase-3 Activity Measurement Control and treated cortical neurones were washed rap- idly with PBS and lysed by cell lysis buffer (10 mM Tris-HCl, 10 mM NaH2PO4/Na2HPO4, pH 7.5, 130 mM NaCl, 0.5% Triton X-100, 10 mM Na4P2O7). Lysates were centrifuged at 13,000 g for 5 minutes and cas- pase-3 activity was measured in the supernatants. Su- pernatants with at least 20 g of protein were incubated in caspase-3 assay buffer (20 mM HEPES, pH 7.5, 10% glycerol, 2 mM DTT) , con tain ing 20 M Ac. DEVD-AMC. [N-acetyl-Asp-Glu-Val-Asp-(7-amino-4-methylcoumarin) at 37˚C during 2-4 h. The fluorogenic AMC liberated from Ac-DEVD-AMC was monitored using a spectro fluori- meter (Bio-Tek FL 600) with an excitation wavelength of 360/20 nm and an emission wavelength range 460/20 nm. Under these conditions, the caspase-3 activity was linear during 6 h. Enzymatic activity is expressed as arbitrary fluorescence units, after 3 hours, per g protein (AFU/3 h/g protein). The cell protein contents were monitored by the Brad- ford technique [26]. 2.8. Mitochondrial Membrane Potential Assay Mitochondrial membrane potential was measured ac- cording to Lalitha Tenneti et al. [27], with few modifica- tions. Control and treated cells were washed with PBS and incubated for 30 min with 500 nM of TMRM dis- solved in Locke medium (140 mM NaCl, 4.4 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 4 mM NaHCO3, 5.5 mM glucose and 10 mM HEPES, adjusted to pH 7.5). Then, the cells were washed with PBS, and the fluorescence was measured with a FL600-BioTek spectrofluorimeter using filters of 530/25 nm exc. and 590/35 nm em. 2.9. Plasma Membrane Potential Measurement Changes in the membrane potential (PMP) of neurons were monitored with the fluorescent dye bisoxonol (bis- [1,3-diethyl-thio-barbiturate]-trimethineoxono l), which is a lipophylic anion whose distribution across the mem- brane is dependent upon the membrane potential. Thus, an increase in bisoxonol fluorescence indicates that the membrane has been depolarised, allowing more of this negatively charged dye to enter the cells [28]. The con- trol neurons, and neurons after treatment were washed and incubated with 0.2 M bisoxonol for 30 min. After that, bisoxonol was removed, the cells were washed with PBS and suspended in PBS and fluorescence was meas- ured at wavelengths of 540 nm excitation and 565 nm emission, and monitored with a FL600-BioTek spectro- fluorimeter. Fluorescence intensity was reported in arbi- trary fluorescence units (AFU). 2.10. ATP Determination ATP analysis was performed by using the luciferase re- action. Cells were lysed with 0.4 M perchloric acid and the lysates neutralized with 1 M KOH under ice. The cellular extract was centrifuged and supernatant were kept at 0˚C. Aliquot samples were assayed immediately with firefly luciferase/D-luciferin as indicated in the test protocol (ATP Bio-Orbit Kit) and 2 mM of EDTA in 0.1 M Tris-acetate buffer, pH 7.5. The increase in chemilu- miniscence was recorded in a Bio Orbit 125 luminometer. For calibrating the light signals, 10 pmol of ATP dis- solved in Tris-acetic buffer was injected into each sample, and luminescence signal was determined. ATP cellular content was expressed as arbitrary fluorescence units (AFU). Intracellular ATP content in basal conditions was 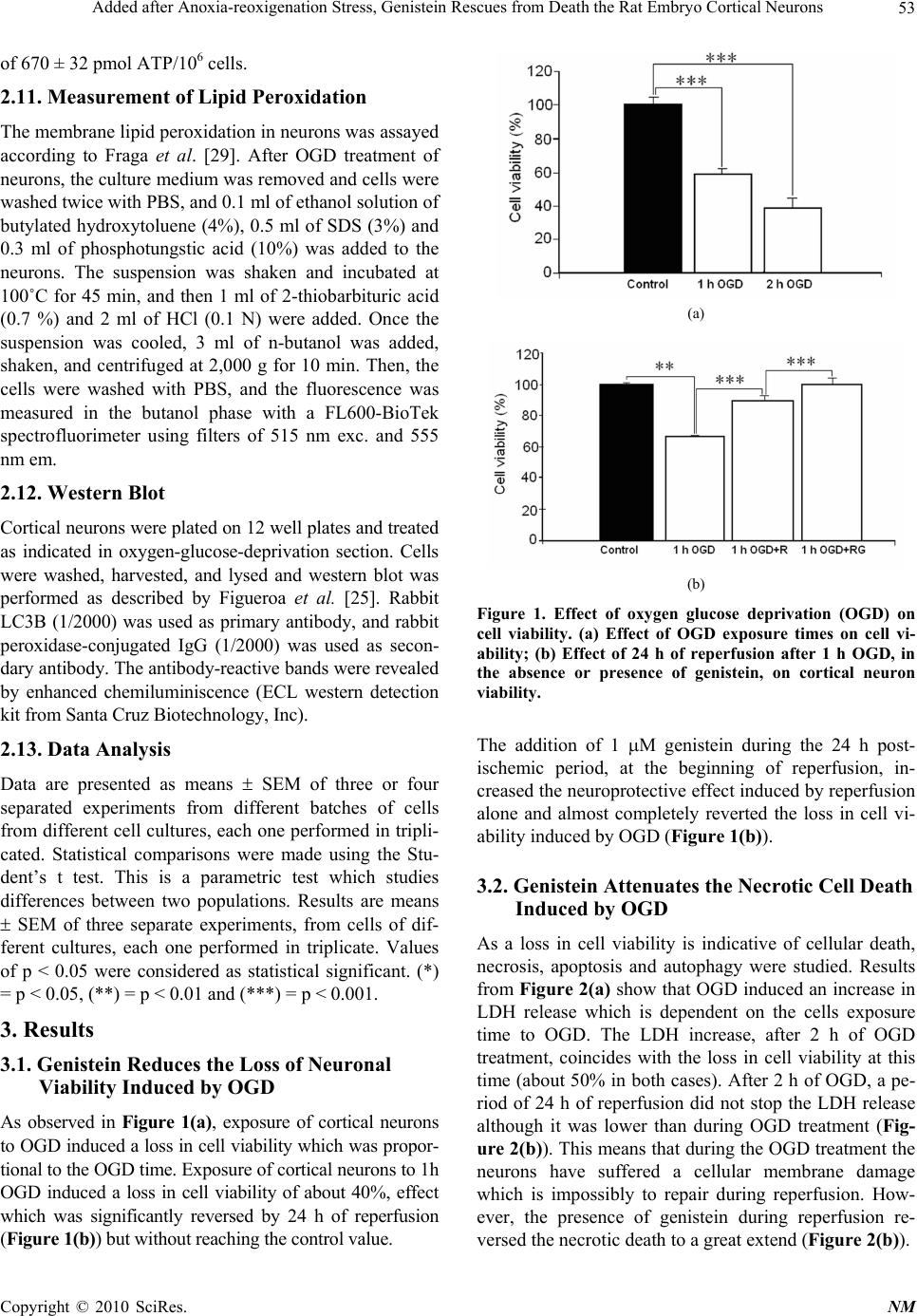 Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 53 of 670 ± 32 pmol ATP/106 cells. 2.11. Measurement of Lipid Peroxidation The membrane lipid peroxidation in neurons was assayed according to Fraga et al. [29]. After OGD treatment of neurons, the culture medium was removed and cells were washed twice with PBS, and 0.1 ml of ethanol solution of butylated hydroxytoluene (4%), 0.5 ml of SDS (3%) and 0.3 ml of phosphotungstic acid (10%) was added to the neurons. The suspension was shaken and incubated at 100˚C for 45 min, and then 1 ml of 2-thiobarbituric acid (0.7 %) and 2 ml of HCl (0.1 N) were added. Once the suspension was cooled, 3 ml of n-butanol was added, shaken, and centrifuged at 2,000 g for 10 min. Then, the cells were washed with PBS, and the fluorescence was measured in the butanol phase with a FL600-BioTek spectrofluorimeter using filters of 515 nm exc. and 555 nm em. 2.12. Western Blot Cortical neurons were plated on 12 well plates and treated as indicated in oxygen-glucose-deprivation section. Cells were washed, harvested, and lysed and western blot was performed as described by Figueroa et al. [25]. Rabbit LC3B (1/2000) was used as primary antibody, and rabbit peroxidase-conjugated IgG (1/2000) was used as secon- dary antibody. The an tibody-reactive bands were revealed by enhanced chemiluminiscence (ECL western detection kit from Sant a Cruz Bi ot echn ology , I nc). 2.13. Data Analysis Data are presented as means SEM of three or four separated experiments from different batches of cells from different cell cultures, each one performed in tripli- cated. Statistical comparisons were made using the Stu- dent’s t test. This is a parametric test which studies differences between two populations. Results are means SEM of three separate experiments, from cells of dif- ferent cultures, each one performed in triplicate. Values of p < 0.05 were considered as statistical significant. (*) = p < 0.05, (**) = p < 0.01 and (***) = p < 0.001. 3. Results 3.1. Genistein Reduces the Loss of Neuronal Viability Induced by OGD As observed in Figure 1(a), exposure of cortical neurons to OGD induced a loss in cell viability which was propor- tional to the OGD time. Exposure of cortical neurons to 1h OGD induced a loss in cell viability of about 40%, effect which was significantly reversed by 24 h of reperfusion (Figure 1(b)) but without reaching t he control val ue. (a) (b) Figure 1. Effect of oxygen glucose deprivation (OGD) on cell viability. (a) Effect of OGD exposure times on cell vi- ability; (b) Effect of 24 h of reperfusion after 1 h OGD, in the absence or presence of genistein, on cortical neuron viability. The addition of 1 M genistein during the 24 h post- ischemic period, at the beginning of reperfusion, in- creased the neuroprotective effect induced by reperfusion alone and almost completely reverted the loss in cell vi- ability induced by OGD (Figure 1(b)). 3.2. Genistein Attenuates the Necrotic Cell Death Induced by OGD As a loss in cell viability is indicative of cellular death, necrosis, apoptosis and autophagy were studied. Results from Figure 2(a) show that OGD induced an increase in LDH release which is dependent on the cells exposure time to OGD. The LDH increase, after 2 h of OGD treatment, coincides with the loss in cell viability at this time (about 50% in both cases). After 2 h of OGD, a pe- riod of 24 h of reperfusion did not stop the LDH release although it was lower than during OGD treatment (Fig- ure 2(b)). This means that during the OGD treatment the neurons have suffered a cellular membrane damage which is impossibly to repair during reperfusion. How- ever, the presence of genistein during reperfusion re- versed the necrotic death to a great extend (Fi gure 2(b)).  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 54 (a) (b) Figure 2. Effect of OGD on LDH release. (a) Effect of OGD time on LDH release; (b) Effect of 24 h of reperfusion after 2 h OGD, in the absence and presence of 1 M genistein on LDH secretion. 3.3. Genistein Protects Againts ATP Lost Since it is known that necrosis is associated with a loss in intracellular ATP, the intracellular levels of this parame- ter were studied. Results from Figure 3 show that after 2 h of OGD there was a great reduction in the intracellular ATP levels which is in accordance with the cellular death produced by necrosis (see Figure 2(a)). During reperfu- sion the ATP levels continuously decay although at lower levels than in the case of the OGD condition alone (Fig- ure 3). However, when reperfusion was performed in the presence of 1 M genistein, a light but significant in- crease in the ATP content was observed with respect to that in the absence of genistein (Figure 3). 3.4. Genistein Protects Cortical Neurons against ROS Generation Induced by OGD As the ROS formation could be an inductor of death by necrosis, this parameter was also measured. Results from Figure 4(a) indicate that the ROS generation is depend- ent on time of cells exposure to OGD. During the 24 h of reperfusion, after 2 h of OGD, ROS levels were signifi- cantly reduced in about a 30% with respect to the ROS induced for OGD. The treatment with 1 M genistein, during the 24 h reperfusion period after OGD, induced a total reversion of the ROS levels induced by the OGD treatment (Figure 4(b)). Figure 3. Effects of OGD and reperfusion, in absence and presence of 1 µM genistein, on intracellular levels of ATP. (a) (b) Figure 4. Effect of OGD on ROS formation. (a ) Effect of OGD exposure times o n ROS formatio n; (b) Effect of 24 h reperfusion, in absence or pre sence of genis tein, after 2 h of OGD. 3.5. Genistein Protects Neurons against Lipid Peroxidation Induced by OGD It is known that ROS may produce lipid peroxidation, being this process the responsible for the necrotic cell death. We measured this parameter under the OGD con- ditions and the results showed an increase in lipid per- oxidation after 2 h of OGD (Figure 5). During the 24 h of reperfusion, after 2 h of OGD, lipid peroxidation lev- els were significantly reduced in about a 65% with re- spect to that induced by OGD, although they were higher  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 55 than in the control. The treatment with 1 M genistein, during the 24 h reperfusion period after 2 h of OGD treat- ment, produced a total reversion of the increase in lipid peroxidation induced by the OGD treatment (Figure 5). 3.6. Genistein Doest not Protect Neurons against Apoptotic Cell Death Induced by OGD Knowing that in this OGD model neurons die by necrosis, the possibility of an apoptotic cell death was also checked and caspase-3 activity was measured as indicator of this process. In our OGD model the cells were cultured in absence of serum, then, as the serum deprivation induces apoptosis in cortical neurons [25] a positive control of apoptosis like the absenc e of serum (trophic factors) was used. Results from Figure 6(a), indicate that the only serum deprivation was able to induce an increase in cas- pase-3 activation, being this effect directly proportional to the time the cells were cultured without serum. On the other hand, when these cells were subjected to OGD for different times, the caspase-3 activity was also signifi- cantly increased with respect to the treatment times. These effects were higher than those found for neurons cultured in absence of serum. Thus, the OGD treatment “perse” was also able to induce additional increases in caspase-3 activation (Figure 6(a)), although, at the low OGD treatment (1 to 5 h), the increase in caspase-3 ac- tivity with respect to the cells cultured in absence of se- rum was lower than those produced at the longer OGD conditions (24 h). The increase in caspase-3 activity in- duced by 2 h O GD was significan tly reduced during 24 h of reperfusion and, in this case, 1 M genisteine had not any additional effect (Figure 6(b)). 3.7. Genistein Protects Mitochondrial Membrane Potential without Affecting Plasma Membrane Potential One of the mechanisms of apoptosis cell death induction is mitochondria malfunction. In order to check the in Figure 5. Effects of OGD and reperfusion on lipid peroxidation, in the absence and presence of 1 M g enist ein . (a) (b) Figure 6. Caspase-3 activation in cortical neurons exposed to OGD. (a) (-OGD) = action of time serum deprivation on cortical neurons in culture. (+OGD) = Effect of different OGD exposure times on caspase-3 activity. The activity of caspase-3 at 0 time (control) was 5.9 ± 0.4. (b) Effects of 24 h of reperfusion after 2 h of OGD exposure in absence and presence of 1 µM genistein. volvement of mitochondria in apoptotic neuronal death induced by OGD, the possible variations in mitochondrial membr ane pot entia l (MMP) ind uced by the OGD trea tment were checked. Results from Figure 7(a) show that 1 h of OGD produ ces a sign ific ant in crea se in MMP. On the other hand, 24 h of reperfusion in absence of genistein, after 1 h of OGD, decr eased the TMR sign al wi th r espect to the con- trol. However, when the reperfusion is performed in pres- ence of 1 M genistein the MMP reach the values of the control cells. By contrast with plasma membrane potential (PMP), 1 h of OGD tr eatment did not aff ect th e potent ial of the cellular membrane, in this case genistein had not any effect on this parameter (Figure 7(b)). 3.8. Genistein Attenuates the Autophagic Cell Death Induced by OGD and Reperfusion It is known that neurons can undergo more than one type of programmed cell death (PCD), not only apoptosis (or type I PCD) but also autophagy (or type II PCD). Auto- phagy was initially described as a process in which cyto- plasmic material is degraded in lysosomes, providing nutrients for survival. However, autophagic features have also been observed in cells exposed to pathological in- sults as well as in remodelling tissues [30,31]. Because  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 56 Figure 7. Effect of OGD and reperfusion in absence and presence of 1 µM genistein on mitochondrial (MMP) and cellular membrane (PMP) potential [A]: Action on MMP [B] Effect on PMP. several recent studies have shown that autophagy is in- volved in ischemic brain damage [32,33] and nutrient deprivation has been shown to lead cell to autophagia, we check in this paper the possibility that in the OGD experimental ischemia model used here, cortical neurons could drive to enter in an autophagic mechanism. To characterize autophagy, we carried out the assay of LC3B lipidation (increase in LC3B-II from LC3BI) measured by western blot. As it could be appreciated in Figure 8, OGD was able to induce a significant increase in LC3B-I lipidation to the autophagosomal LC3B-II in cortical neurons. A utophagic cell death induced by OGD was kept among 1 to 5 h of treatment, although the maximum autophagia levels were observed at 1 h of OGD treatment. At higher OGD exposure 2 and 5 h, the autophagia was lower than at 1 h exposure but the values were higher than in control cells. 24 h of reperfusion re- versed in part this parameter. However, the reperfusion in presence of 1 M of genistein completely reversed LC3B lipidation. 4. Discussion and Conclusions Epidemiologic data show that populations which con- sume diets rich in phytoestrogens have low incidence in coronary artery diseases [34,35]. Several studies have indicated that genistein administration prevents delayed Figure 8. Time-course effects of OGD and reperfusion, in absence and presence of 1 M genistein, on LC3B lipidation. Western blot analysis of LC3BI and LC3BII was performed in extracts of cortical neurons subjected to OGD exposure and 24 h reperfusion, in the absence or presence of 1 M genistein, at indicate times, using a LC3B antibody. (a) Representative LC3B western blot is shown. (b) Data are expressed as ratios over basal control and are mean ± SEM of two experiments each one performed in duplicate. neuronal death after transient global cerebral ischemia [22,36,37] and attenuates oxidative stress and neuronal damage following cerebral ischemia in rat hippocampus [24] and inhibits the lipid peroxidation induced by pro-oxidant agents in cultured cortical neurons [23]. However, in all these works phytoestrogens have been studied as neuroprotective agents when they are admin- istered in the period previous to ischemia and not in the postischemic period. Nevertheless it is in this time when protection is necessary after an ischemic event. Therefore, the present study was performed to test the ability of the phytoestrogen genistein to inhibit neuronal death in the post-ischemic period. In the OGD model described here we found that there was a loss in cell viability which was accompanied by necrotic and apoptotic cell death, and we demonstrated that both death processes are time-dependent. The exis- tence of a necrotic cortical neuron death, induced by the OGD model, is supported by the decrease in the intracel- lular ATP levels, the great ROS generation and the pro- duction of lipid peroxidation. This is in accordance with Traystman et al. [38] and Matsuo et al. [39] who found that a great ROS generation together with the lipid per- oxidation may be the possible cause of the necrosis. In our OGD model there is a time-dependent ROS forma- tion during the OGD treatment and lipid peroxidation is also induced. The ROS formation could look strange because this treatment was performed in absence of O2 (95% N2/5 % CO2). However, mitochondrial ROS gen- eration during hypoxia and anoxia has been shown by several authors [24,14,40-43] although the mechanism of this ROS generation, under this condition has not been  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 57 well resolved. During reperfusion the ROS generation, the loss in ATP content and the lipid peroxidation reduction are maintained below control values, which is in accor- dance with the necrotic death that is produced during this time, although this death is lower than during the OGD treatment. This could mean th at, after an ischemic process, and when the blood flow is restored, there is a group of neurons which have suffered a big damage and that they are unable to recover themselves, dying by necrosis. If this reperfusion is performed in presence of 1 M genistein, a total reversion of necrotic cell death, ROS formation and lipid peroxidation were found as well as a protection for increase in intracellular ATP content, in- dicating that genistein protects against death through reduction of both ROS and lipid peroxidation formation. This protection against lipid peroxidation may be the cause of a cellular membrane protection. Similar neuro- protection against ischemia mediated by flavonoids has been found by Zhang et al. [44] in gerbil’s brain sub- jected to ischemia/reperfusion and by Liang et al. [24] in brain hippocampus after global cerebral ischemia. In the OGD model used here, apoptotic and autophagic cell deaths were also found. Apoptotic death is probably due to the deprivation of trophic factors during the OGD, at least in the shortest treatments (1-5 h), because during these times the higher caspase-3 activity is found when cells are serum deprived and the increment due to the OGD treatment is too low, although it is statistically sig- nificant. However, at 24 h of OGD, additional mecha- nisms seem to be involved since caspase-3 activity in- duced by the OGD treatment is about a 33% higher than the caspase-3 activity found in cells only deprived of serum. During the reperfusion period, after 2 h of OGD, the apoptosis is practically abolished with respect to the cells treated with OGD. This is, perhaps, due to the presence of trophic factors during this time. Under this condition genistein had no additional neuroprotective effect, result which c ould be due to the fact that apoptosis is inhibited, under the reperfusion condition. The mecha- nism of this apoptotic death induced by OGD, and medi- ated by caspase-3 activation could be due to the intrinsic mitochondrial apoptotic pathway because we found a depolarization of the mitochondrial membrane induced by OGD, which may show a mitochondrial membrane alteration and, as a consequence, an influence in the cas- pase-3 activation. Mitochondrial membrane depolariza- tion with caspase-3 activation and cytochrome c release was found by Figueroa et al. [25] in cortical neurons treated with SNAP, a NO donor. However, an apoptotic cell death mediated by extrinsic pathway could not be discarded. Another point to comment is th e fact that dur- ing the 24 h of reperfusion, after 1 h of OGD treatment the mitochondrial membrane continue modified, although in this case an hyperpolarization instead of a depolariza- tion was observed. This mitochondrial membrane altera- tion could also be the responsible for the necrotic death during the reperfusion time in absence of genistein. The fact that when reperfusion is performed in presence of genistein there was a total reversion of necrosis could signify that this phytoestrogen could mediate mitochon- drial membrane reparation. This asseveration is sup- ported by its ability to revert the mitochondrial mem- brane potential to the control values. The cortical neurons subjected to OGD also present a process of autophagy because lipidation of protein LC3B is increased. The autop hagy was more evident in the first hour of OGD treatment, after, the autophagy decreased but with values higher than in control cells. Under these conditions, this autophagic process could represent a survival mechanism induced to keep the ATP levels as it is shown by several authors [45-47]. However, in our case, during OGD process there is a great ATP depletion which could indicate that in this case autophagy could not be a protective signal. On the other hand, during reperfusion in which cells have the enough nutrients and oxygen, the autophagy is decreased with respect to the OGD treatment but the values are higher than in control cells after 1 and 3 h of OGD. On the other hand, the ATP levels continuously decay and in this case, autophagy only could mean a mechanism of cell death. So it would be possible than during the reperfusion condition many cells die not only by necrosis and apoptosis but also by autophagia. However, when reperfusion is performed in presence of 1 M genistein, after 1 h of OGD treatment, a total reversion of autophagy was found, indicating that genistein also protects against this death type, although the molecular mechanism of this neuroprotective effect needs to be studied. The interaction among autophagy, apoptosis and necrosis in the hypoxia-ischemia is com- plex and still a matter of debate and further work is nec- essary to understand the relationships between these three death mechanisms in brain ischemia. In conclusion, from all these results we may infer that the OGD treatment (O2, trophic factors, nutrients and glucose deprivation) produces the following effects: 1) Necrotic cell death which could be induced by the ROS formation, lipid peroxidation increase and the ATP de- pletion. 2) Apoptotic cell death mediated by caspase-3 activation probably through an intrinsic pathway. 3) Autophagic cell death. Genistein, applied during the reperfusion period, pro- tects the cells against necrosis and autophagy, by means of a mechanism which could be mediated by their anti- oxidant action and by a protector effect on cellular and mitochondrial membrane. Thus, our protective post-stress data on neuroprotection, could serve as basis to plan a  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 58 clinical trial with genistein in stroke patients. 5. Acknowledgements This study was supported by the grants SAF2009-11219 and SAF2010-20337 (Ministry of Science and Tech- nology (MCYT), Spain), CCG07-UCM/SAL-3024 (Com- plutense University/Madrid Community) and RD06/0026/ 0012 (Instituto de Salud Carlos III). E. Sánchez-Mendoza has a contract from RD06/0026/0012. The au thors t ha n k t o Mikel Erdocia for his help in improving the manuscript. REFERENCES [1] M. P. Mattson and R. J. Mark. “Excitotoxicity and Exci- toprotection in Vitro,” Advances in Neurology, Vol.71, 1996, pp 1-30. [2] U. Dirnagl, C. Iadecola, and M. A. Moskowitz, “Pathobi- ology of Ischemic Stroke: An Integrated View,” Trends Neuroscience, Vol. 22, No. 9, 1999, pp 391-397. doi:10.1016/S0166-2236(99)01401-0 [3] L. A. Labiche and J. C. Grotta, “Clinical Trials for Cyto- protection in Stroke,” NeuroRx, Vol. 1, No. 1, 2004, pp. 46-70. doi:10.1602/neurorx.1.1.46 [4] M. A. de Leciñana and J. A. Egi do, “E stro gens as Ne uropr o- tectants against Ischemic Stroke,” Cerebrovascular Diseases , Vol. 21 No. 2, 20 06, pp. 48-53. doi:10.1159/000091703 [5] S. Suzuki, C. M. Brown and P. M. Wise, “Neuroprotec- tive Effects of Estrogens Following Ischemic Stroke,” Frontiers in Neuroendocrinology, Vol. 30, No. 2, 2009, pp. 201-211. doi:10.1016/j.yfrne.2009.04.007 [6] C. L. Gibson, L. J. Gray, S. P. Murphy and P. M. Bath, “Estrogens and Experimental Ischemic Stroke: A Sys- tematic Review,” Journal of Cerebral Blood Flow & Metabolism, Vol. 26, No. 9, 2006, pp. 1103-1113. [7] T. Jover, H. Tanaka, A. Calderone, K. Oguro, M. V. Bennett, A. M. Etgen and R. S. Zukin, “Estrogen Protects against Global Ischemia-Induced Neuronal Death and Prevents Activation of Apoptotic Signaling Cascades in the Hippocampal Ca1,” Journal of Neuroscience, Vol. 22, No. 6, 2002, pp. 2115-2124. [8] C. A. Singer, X. A. Figueroa-Masot, R. H. Batchelor and D. M. Dorsa. “The Mitogen-Activated Protein Kinase P ath - way Mediates Estrogen Neuroprotection after Glutamate Toxicity in Primary Cortical Neurons,” Journal of Neu- roscience, Vol. 19, No. 7, 1999, pp. 2455-2463. [9] K. Honda, H. Sawada, T. Kihara, M. Urushitani, T. Na- kamizo, A. Akaike and S. Shimohama, “Phosphatidy- linositol 3-Kinase Mediates Neuroprotection by Estrogen in Cultured Cortical Neurons,” Journal of Neuroscience Research, Vol. 60, No. 3, 2000, pp. 321-327. doi:10.1002/(SICI)1097-4547(20000501)60:3<321::AID- JNR6>3.0.CO;2-T [10] E. A. Sribnick, S. K. Ray, M. W. Nowak, L. Li and N. L. Banik, “17 Beta-Estradiol Attenuates Glutamate-Induced Apop- tosis and Preserves Electrophysiologic Function in Primary Cortical Neurons,” Journal of Neuroscience Research, Vol. 76, No. 5, 2004, pp. 688-696. doi:10.1002/jnr.20124 [11] J. Nilsen, and R. D. Brinton, “Impact of Progestins on Estrogen-Induced Neuroprotection: Synergy by Proges- terone and 19-Norprogesterone and Antagonism by Me- droxyprogesterone Acetate,” Endocrinology, Vol. 13, No. 6, 2002, pp. 205-212. doi:10.1210/en.143.1.205 [12] H. C i m a r o s t i , I. R . S i q u e i r a , L . L . Z a mi n , M. Na s s i f , R . B a lk, R . Frozza, C. Dalmaz, C. A. Netto and C. Salbego, “ Neuroprotec- tion and Protein Damage Prevention by Estradio l Repla cement in Rat Hippocampal Slices Exposed to Oxygen-Glucose Dep- rivation,” Neurochemical Research, Vol. 30, 2005, pp. 583-589. doi:10.1007/s11064-005-2693-1 [13] S. Chen, J. Nilsen and R. D. Brinton, “Dose and Tempo- ral Pattern of Estrogen Exposure Determines Neuropro- tective Outcome in Hippocampal Neurons: Therapeutic Implications,” Endocrinology, Vol. 147, No. 11, 2006, pp. 5303-5313. doi:10.1210/en.2006-0495 [14] E. Candelario-Jalil, N. H. Mhadu, S. M. Al-Dalain, G. Martinez and O. S. León, “Time Course of Oxidative Damage in Differenr Brain Regions Following Transient Cerebral Ischemia in Gerbis,” Neurochemical Research, Vol. 41, No. 3, 2001, pp. 233-241. [15] D. Amantea, R. Russo, G. Bagetta and M. T.Corasaniti. “From Clinical Evidence to Molecular Mechanisms Un- derlying Neuroprotection Afforded by Estrogens,” Phar- macol Research, Vol. 52, No. 2, 2005, pp. 119-132. doi:10.1016/j.phrs.2005.03.002 [16] V. N. Trieu and F. M. Uckun, “Genistein is Neuroprotec- tive in Murine Models of Familial Amyotrophic Lateral Sclerosis and Stroke,” Biochemical and Biophysical Re- search Communications, Vol. 258, No. 3, 1999, pp 685- 688. doi:10.1006/bbrc.1999.0577 [17] D. A. Schreihofer and L. Redmond. “Soy Phytoestrogens are Neuroprotective against Stroke-Like Injury in Vitro,” Neuroscience, Vol. 158, No. 2, 2009, pp 602-609. doi:10.1016/j.neuroscience.2008.10.003 [18] M. C. Burguete, G. Torregrosa, F. J. Perez-Asensio, M. Castello-Ruiz, J. B. Salom, J. V. Gil and E. Alborch, “Die- tary Phytoestrogens Improve Stroke Outcome after Tran- sient Focal Cerebral Ischemia in Rats,” European Journal of Neuroscience, Vol. 23, No. 2, 2006, pp. 703-710. doi:10.1111/j.1460-9568.2006.04599.x [19] L. Zhao, Q. Chen and R. D. Brinton, “Neuroprotective and Neurotrophic Efficacy of Phytoestrogens in Cultured Hippocampal Neurons,” Experimental Biology and Medi- cine, Vol. 227, No. 7, 2002, pp. 509-519. [20] N. J. Linford and D. M Dorsa, “17 Beta-Estradiol and the Phytoestrogen Genistein Attenuate Neuronal Apoptosis In- duced by the Endoplasmic Reticulum Calcium-Atpase In- hibito r Thapsigargin,” Steroids, Vol. 6 7, No. 13- 14, 200 2, pp. 1029-1040. doi:10.1016/S0039-128X(02)00062-4 [21] H. Zeng, Q. Chen and B. Zhao, “Genistein Ameliorates Beta- Amyloid Peptide (25-35)-Induced Hippocampal Neuronal Apoptosis,” Free Radical Biology and Medicine, Vol. 36, No. 2, 2004, pp. 180-188. doi:10.1016/j.freeradbiomed.2003.10.018 [22] H. Inoue, H. Ohtaki, T. Nakamachi, S. Shioda and Y. Okada, “Anion Channel Blockers Attenuated Delayed  Added after Anoxia-reoxigenation Stress, Genistein Rescues from Death the Rat Embryo Cortical Neurons Copyright © 2010 SciRes. NM 59 Neuronal Cell Death Induced by Transient Forebrain Ischemia,” Journal of Neuroscience Research, Vol. 85, No. 7, 2007, pp. 1427-1435. doi:10.1002/jnr.21279 [23] K. P. Ho, L. Li, L. Zhao and Z. M. Qjan, “Genistein Protects Primary Cortical Neurons from Iron Induced Lipid Peroxida- tion,” Molecular and Cellular Biochemistry, Vol. 247, No. 1-2, 2003, pp. 2 19-22 2. doi:10.1023/A:1024142004575 [24] H. W. Liang, S. F. Qju, J. Shen, L. N. Sun, J. Y. Wang, I. C. Bruce and Q. Xia, “Genistein Attenuates Oxidative Stress and Neuronal Damage Following Transient Global Cerebral Ische- mia in Rat Hippocampus,” Neuroscience Letters, Vol. 438. No. 1, 2008, pp. 116-120. doi:10.1016/j.neulet.2008.04.058 [25] S. Figueroa, M. J. Oset-Gasque, C. Arce, C. Martínez- Honduvilla and M. P. González, “Mitochondrial Involvement in Nitric Oxide-Induced Cellular Death in Cortical Neurons in Culture,” Journal of Neuroscience Research, Vol. 83, No. 3, 2006, pp. 441-449. doi:10.1002/jnr.20739 [26] M. Bradford, “A Rapid Sensitive Method for the Quantization of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding,” Analytica l Biochemistry, Vol. 72, 1976, pp. 248-254. doi:10.1016/0003-2697(76)90527-3 [27] L. Tenneti, D. M. D'Emilia, C. M. Troy and S.A Lipton, “Role of Caspases in N-Methyl-D-Aspartate-Induced Apop- tosis in Cerebrocortical Neurons,” Journal of Neurochemis- try, Vol. 71, No. 3, 1998, pp. 946-959. doi:10.1046/j.1471-4159.1998.71030946.x [28] A. S. Waggoner. “The Use of Cyanine Dyes for the De- termination of Membrane Potentials in Cells, Organelles, and Vesicles,” Methods in Enzymology, Vol. 55, 1979, pp 689-695. doi:10.1016/0076-6879(79)55077-0 [29] C. G. Fraga, P. I. Oteiza, M. S. Golup, M. E. Gershwin and C. L. Keen, “Effects of Aluminium on Brain Lipid Peroxidation,” Toxicology Letters, Vol. 51, No. 2, 1990, pp. 213-219. doi:10.1016/0378-4274(90)90212-5 [30] L. Xue, G. C. Fletcher and A. M. Tolkovsky, “Autophagy is Activated by Apoptotic Signalin g in S ympathetic Neu rons: An Alternative Mechanism of Death Execution,” Molecular and Cellular Neuroscience, Vol. 14, No. 3, 1999, pp. 180-198. doi:10.1006/mcne.1999.0780 [31] P. Codogno and A. J. Mejier, “Autophagy and Signalling: Their Role in Cell Survival and Cell Death,” Cell Death and Differentiation, Vol. 12 No. S2, 2005, pp. 1509-1518. doi:10.1038/sj.cdd.4401751 [32] F. Adhami, G. Liao, Y. M. Morozov, A. Schloemer, V. J. Schmithorst, J. N. Lorenz, R. S. Dunn, C. V. Vorhees, M. Wills-Karp, J. L. Degen, R. J. Davis, N. Mizushima, P. Rakic, B. J. Dardzinski, S. K. Holland, F. R. Sharp and C. Y. Kuan, “Cerebral Ischemia-Hypoxia Induces Intravas- cular Coagulation and Autophagy,” American Journal of Pathology, Vol. 169, No. 2, 2006, pp. 566-583. doi:10.2353/ajpath.2006.051066 [33] W. Balduini, S. Carloni and G. Buonocore, “Autophagy in Hypoxia-Ischemia Induced Brain Injury: Evidence and Speculations,” Autophagy, Vol. 5, No. 2, 2009, pp. 221- 223. doi:10.4161/auto.5.2.7363 [34] S. Barnes, “Evolution of the Health Benefits of Soy Isofla- vones,” Proceedings of the Society f or Expe rime nt al Biolo gy and Medicine , Vol. 21 7, No . 3, 1998, pp. 38 6-392. [35] D. Altavilla, A. Crisafulli, H. Marini, M. Esposito, R. D. Anna, F. Corrado, A. Bitto and F. Squadrito, “Cardiovascular Effects of th e Phytoest rogen Genist ein,” Cardiovascular and Hematologi cal Agents in Medi cinal Chem istry, Vol 2, No. 2, 2004, pp. 179-186. doi:10.2174/1568016043477297 [36] H. J. Wu and W. H. Chan, “Genistein Protects Methylgli- oxal-Induced Oxidative DNA Damage and Cell Injury in Human Mononuclear Cells,” Toxicology in Vitro, Vol. 21, No. 3, 2007, pp. 335-342. doi:10.1016/j.tiv.2006.09.002 [37] M. S. Kindy, “Inhibition of Tyrosine Phosphorylation Prevents Delayed Neuronal Death Following Cerebral Ischemia,” Journal of Cerebral Blood Flow & Metabo- lism, Vol. 13, No.3, 1993, pp. 372-377. [38] R. Traystman, J. R. Kirsch and C. Koehler, “Oxygen Radicals Mechanism of Brain Injuria Following Ischemia and Reperfusion,” Journal of Applied Physiology, Vol. 71, No. 4, 1991, pp. 1185-1195. [39] Y. Matsuo, T. Kihara, M. Ikeda, M. Ninomiya, H. Ono- dera and K. Kogure, “Role of Platelet-Activating Factor and Thromboxane A2 in Radical Production during Ischemia and Reperfusion of the Rat Brain,” Brain Re- search, Vol. 709, No. 2, 1996, pp. 296-302. doi:10.1016/0006-8993(95)01324-5 [40] T. Kimova and N. S. Chandel, “Mitochondrial Complex Iii Regulates Hypoxic Activation of HIF,” Cell Death and Differentiation, Vol. 15, No. 4, 2008, pp. 660-666. doi:10.1038/sj.cdd.4402307 [41] P. H. Chan, “Role of Oxidants in Ischemic Brain Dam- age,” Stroke, Vol. 27, No. 6, 1996, pp. 1124-1129. [42] H. Friberg, T. Wieloch and R. F. Castilho, “Mitochon- drial Oxidative Stress after Global Brain Ischemia in Rats,” Neuroscience Letters, Vol. 334, No. 2, 2002, pp. 111-114. doi:10.1016/S0304-3940(02)01116-3 [43] M. A. Moro , A. Almeida, J. P. Bola ños and I. Lizasoain, “Mito- chondrial Respiratory Chain and Free Radical Generation in Stroke,” Free Radical Biology & Medicine, Vol. 39, No. 10, 2005, pp. 1291-1304. doi:10.1016/j.freeradbiomed.2005.07.010 [44] D. L. Zhang, Y. T. Zhang, J. J. Yin and B. L. Zhao, “Oral Administration of Crataegus Flavonoids Protects against Ischemia/Reperfusion Brain Damage in Gerbils,” Journal of Neurochemistry, Vol. 90, No. 1, 2004, pp. 211-219. doi:10.1111/j.1471-4159.2004.02480.x [45] V. E. Dosenko, V. S. Nagibin, L. V. Tumanovska and A. A. Moibenko, “Protective Effect of Autophagy in An- oxia-Reoxigenation of Isolated Cardiomyocyte?” Auto- phagy, Vol. 2, No. 4, 2006, pp. 305-306. [46] M. Komatsu, S. Waguri, T. Chiba, S. Murata, J. Iwata, I. Ta- nida, T. Ueno, M. Koike, Y. Uchiyama, E. Komonani and K. Tanaka, “Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice,” Nature, Vol. 441, No. 7095, 2006, pp. 880-884. doi:10.1038/nature04723 [47] T. Hara, K. Nakamura, M. Mat sui, A. Yamamoto, Y. Naka- hara, R. Suzuki-Migishima, M. Yokoyama, K. Mishima, I. Saito, H. Okano and N. Mizushima, “Supression of Basal Autophagy in Neuronal Cells Causes Neurodegenerative Disease in Mice,” Nature, Vol. 441, No. 7095, 2006, pp. 885-889. doi:10.1038/nature04724 |