 Advances in Materials Physics and Chemistry, 2013, 3, 178-184 http://dx.doi.org/10.4236/ampc.2013.32025 Published Online June 2013 (http://www.scirp.org/journal/ampc) Ab Initio Study of the Electronic and Vibrational Pr operties of 1-nm-Diameter Single-Walled Nanotubes Jesús Marquina1, Chrystian Power2, Jesús González2,3, Jean-Marc Broto4 1Centro de Estudios Avanzado en Óptica, Facultad de Ciencias, Universidad de los Andes, Mérida, Venezuela 2Centro de Estudios de Semiconductores, Facultad de Ciencias, Universidad de los Andes, Mérida, Venezuela 3Malta Consolider Team, CITIMAC, Facultad de Ciencias, Universidad de Cantabria, Santander, España 4LNCMI, Paul Sabatier University, Toulouse, France Email: castella@ula.ve, marquinajesus@gmail.com Received February 19, 2013; revised April 25, 2013; accepted May 10, 2013 Copyright © 2013 Jesús Marquina et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT The electronic structure, band gap, density of states of the (8,8), (14,0) and (12,3) single-walled carbon nanotubes by the SIESTA (Spanish Initiative for Electronic Simulations with Thousands of Atoms) method in the framework den- sity-functional theory (DFT) with the generalized gradients approximation (GGA) were studied. Also, we studied the vibrational properties of the (8,8) and (14,0) nanotubes. Only the calculated relaxed geometry for (12,3) nanotube show significant deviations from the ideal rolled graphene sheet configuration. The electronic transition energies of van Hove singularities were studied and compared with previous results. The calculated band structures, density of states and dis- persion curves for all tubes were in good agreement with theoretical and experimental results. Keywords: Carbon Nanotubes; Electronic Structure; Dispersion Curve 1. Introduction Carbon nanotubes can be regarded as graphitic cylinders generated just by rolling one sheet of graphene with a finite width [1]. Also it is known that a nanotube can be metallic or semiconducting depending on their diameter and chirality [2-4]. In recent years, it has attracted great attention in many fields of science and technology be- cause of its novel and unique structural and electronic properties [5]. Nanotubes are promising for applications, such as probes and sensors; tips for atomic force mi- croscopy, gas sensors, biosensors and physical sensors [6]. There has been much theorical and experimental work on the vibrational and electronic properties of carbon nanotubes. The first theorical calculation of SWCNT’s electronic structure was carried out using zone folding scheme [7,8]. Within this framework, all the armchair nanotubes (n,n) are expected to be metallic, while ap- proximately one third of zigzag (n,0) and chiral (n,m) tubes should be metallic, with remainder being semicon- ducting [3,4]. The zone-folding scheme in general works reasonably well and has proven immense success in pro- viding physically relevant information on the nature of electronic and vibrational interactions in carbon nano- tubes [9,10]. However, previous studies show the effects of the σ-π hybridization in small diameter SWCNT’s on their electronic properties [11,12]. An example of par- ticular importance is the (5,0) nanotube which has been demonstrated by ab initio (using local density approxi- mation in the framework of the density-functional theory) and symmetry-adapted non-orthogonal tight-binding cal- culations to be a metal [12-14] but not a semiconductor with a moderate gap as reported in the previous π-band tight-binding work [2,3]. The phonon dispersion curves over the full Brillouin zone are useful in such fields as the inelastic neutron scattering [15], Second-order Raman spectroscopy [16], double-resonance processes [17-20] and the electron- phonon interactions [21-24]. The calculations of the phonon dispersions have been made using both empirical methods and the accuracy ab initio calculations. Initial work on this problem using zone folding and force con- stant models [9,25-27] shows that the zone-folding me- thod has two shortcomings: the structural relaxation ef- fect has been completely ignored which makes the trans- ferability of the phonon spectra between the graphene and the SWCNT’s a serious question and that the vibra- tional eigenvectors are also different from the graphene to the SWCNT’s. The ab initio phonon dispersion is C opyright © 2013 SciRes. AMPC  J. MARQUINA ET AL. 179 advantageous because such calculations do not depend on any predefined parameter. There are different authors that have reported ab initio phonon dispersion of SWCNT’s, all using the supercell approach [11,28,29]. One way to study the structural and mechanical prop- erties of carbon nanotubes is by applying hydrostatic pressure. A recent study shows that in DWCNT’s the outer tube acts as a protection shield for the inner tube (at least up 11 GPa) [30,31]. This means that the inner tube retains its properties as single-walled nanotube. In Fig- ures 5 and 6 of Ref. [30], the Raman spectrums of the radial breathing modes (RBM) of the inner and the outer shells of pristine and filled DWCNT’s have been observed, respectively. In both in pristine or Te-filled DWCNT’s tubes with diameters around 1 nm for the inner tubes of DWCNT’s are presented. In this paper, we present first-principles calculations of the structural, electronic, and vibrational properties of SWCNT’s about 1 nm in diameter to address the point mentioned above. We will discuss the relaxed geometry of the (8,8), (14,0) and (12,3) nanotubes comparing it to the ideal cylindrical configuration. We also examine the importance of the detailed structure in discussing elec- tronic properties of nanotubes. Finally, we discuss about phonon dispersion curves for the (8,8) nanotube. 2. Computational Method Our first-principles calculations are based on density functional theory (DFT) employ a numerical-atomic- orbital basis set. We used the package SIESTA ab initio simulation package [32]. We worked within the gradient generalized approximation (GGA) as parametrized by Perdew-Burke-Ernzerhof (PBE) [33]. The core electrons were replaced by pseudopotentials of the type Troullier- Martins [34], the valence electrons were described by localized pseudo-atomic orbitals [35]. The valence elec- trons were described by a double-ζ polarized basis set with a cutoff radius of 4.99 a.u. for the s and 6.25 for the p and the polarizing d orbitals, as is determined from an energy shift of 50 meV by localization. These values have been tested by M. Machón et al. [12]. They found that an increasing of the cutting of the orbitals only pro- duces changes in the total energies less than 0.1 meV/ atom and none significant effect neither on the structure nor on the energies of electronic states was observed. The dynamical matrix was found by a finite-difference approach, this is, calculating the Hellman-Feynmann forces for displaced atoms. Calculations were performed for the following tubes: (8,8), (14,0) and (12,3). They were all considered as iso- lated. For that purpose, we used periodic-boundary con- ditions on supercell geometry with sufficient lateral separation among neighboring tubes. For the purpose of sampling the Brillouin zone in the direction of the tube axis, as well as for the computation of the force-constant matrix for phonon calculations, we used supercells con- sisted of five unit cells for the armchair (8,8) tube with 160 atoms and three unit cells for zigzag (14,0) tube with 162 atoms. The supercell length (about 1 nm) in the tube- axis direction is not too different from one another. We are aware that the supercell should be large enough so that the effect of the image displacement is negligible [11]. 3. Results and Discussion 3.1. Dispersion Curve of Graphene In Figure 1 we show the phonon frequencies of graphene calculated from first principles. This will serve as a test of accuracy of the calculation method. In general, a good agreement between our calculations and experiment (see Ref. [36]) for the TO, LO and LA branches is found. This shows the accuracy of the basis set used in this work. We optimized the bond length of a graphene sheet in a supercell with the interlayer distance being 10 Å, which can be considered enough to neglect the interlayer inter- action. The bond length of graphite obtained by this op- timization is 1.437 Å, which is 1.5% higher than the ex- perimental value (1.416 Å) [37]. The small splitting of the LO and LA phonons at the M point equal to 33 cm−1 coincides with the experimental value reported by inelas- tic x-ray scattering [36]. While, our calculated splitting between TO and LA modes at the K point 118 cm−1 is different from experimental value 71 cm−1. Other theo- retical values have been reported; Pavone et al. [38] ob- tained a splitting of 120 cm−1 and a value of 140 cm−1 by using LDA was also reported [28]. The zone center pho- non value 1575 cm−1 is slightly below the experimental values for graphite and grapheme (1582 cm−1) measured recently [39]. These differences can be explained by tak- ing into account that other authors have estimated uncer- tainties of 10 cm−1 for the frequencies higher than 1300 cm−1 [11,40]. Figure 1. Ab initio calculations of the phonon dispersion of graphene. We used a supercell with 98 atoms. The mesh cutoff was 350 Ry, with 3321 k points and a 50 meV PAO energy shift. Copyright © 2013 SciRes. AMPC 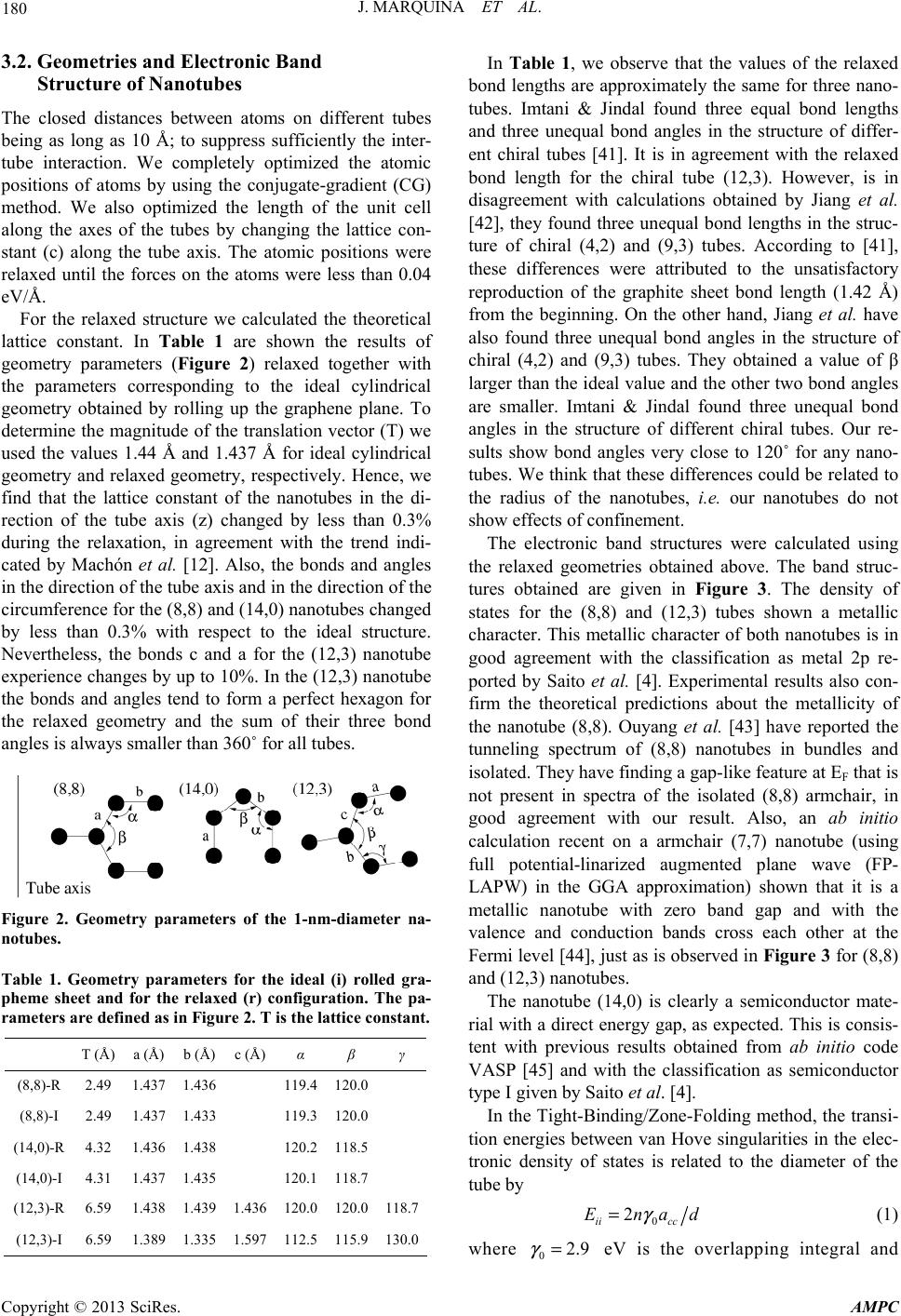 J. MARQUINA ET AL. 180 3.2. Geometries and Electronic Band Structure of Nanotubes The closed distances between atoms on different tubes being as long as 10 Å; to suppress sufficiently the inter- tube interaction. We completely optimized the atomic positions of atoms by using the conjugate-gradient (CG) method. We also optimized the length of the unit cell along the axes of the tubes by changing the lattice con- stant (c) along the tube axis. The atomic positions were relaxed until the forces on the atoms were less than 0.04 eV/Å. For the relaxed structure we calculated the theoretical lattice constant. In Table 1 are shown the results of geometry parameters (Figure 2) relaxed together with the parameters corresponding to the ideal cylindrical geometry obtained by rolling up the graphene plane. To determine the magnitude of the translation vector (T) we used the values 1.44 Å and 1.437 Å for ideal cylindrical geometry and relaxed geometry, respectively. Hence, we find that the lattice constant of the nanotubes in the di- rection of the tube axis (z) changed by less than 0.3% during the relaxation, in agreement with the trend indi- cated by Machón et al. [12]. Also, the bonds and angles in the direction of the tube axis and in the direction of the circumference for the (8,8) and (14,0) nanotubes changed by less than 0.3% with respect to the ideal structure. Nevertheless, the bonds c and a for the (12,3) nanotube experience changes by up to 10%. In the (12,3) nanotube the bonds and angles tend to form a perfect hexagon for the relaxed geometry and the sum of their three bond angles is always smaller than 360˚ for all tubes. Figure 2. Geometry parameters of the 1-nm-diameter na- notubes. Table 1. Geometry parameters for the ideal (i) rolled gra- pheme sheet and for the relaxed (r) configuration. The pa- rameters are defined as in Figure 2. T is the lattice constant. T (Å) a (Å) b (Å) c (Å) α β γ (8,8)-R 2.49 1.437 1.436 119.4 120.0 (8,8)-I 2.49 1.437 1.433 119.3 120.0 (14,0)-R 4.32 1.436 1.438 120.2 118.5 (14,0)-I 4.31 1.437 1.435 120.1 118.7 (12,3)-R 6.59 1.438 1.439 1.436 120.0 120.0118.7 (12,3)-I 6.59 1.389 1.335 1.597 112.5 115.9 130.0 In Table 1, we observe that the values of the relaxed bond lengths are approximately the same for three nano- tubes. Imtani & Jindal found three equal bond lengths and three unequal bond angles in the structure of differ- ent chiral tubes [41]. It is in agreement with the relaxed bond length for the chiral tube (12,3). However, is in disagreement with calculations obtained by Jiang et al. [42], they found three unequal bond lengths in the struc- ture of chiral (4,2) and (9,3) tubes. According to [41], these differences were attributed to the unsatisfactory reproduction of the graphite sheet bond length (1.42 Å) from the beginning. On the other hand, Jiang et al. have also found three unequal bond angles in the structure of chiral (4,2) and (9,3) tubes. They obtained a value of β larger than the ideal value and the other two bond angles are smaller. Imtani & Jindal found three unequal bond angles in the structure of different chiral tubes. Our re- sults show bond angles very close to 120˚ for any nano- tubes. We think that these differences could be related to the radius of the nanotubes, i.e. our nanotubes do not show effects of confinement. The electronic band structures were calculated using the relaxed geometries obtained above. The band struc- tures obtained are given in Figure 3. The density of states for the (8,8) and (12,3) tubes shown a metallic character. This metallic character of both nanotubes is in good agreement with the classification as metal 2p re- ported by Saito et al. [4]. Experimental results also con- firm the theoretical predictions about the metallicity of the nanotube (8,8). Ouyang et al. [43] have reported the tunneling spectrum of (8,8) nanotubes in bundles and isolated. They have finding a gap-like feature at EF that is not present in spectra of the isolated (8,8) armchair, in good agreement with our result. Also, an ab initio calculation recent on a armchair (7,7) nanotube (using full potential-linarized augmented plane wave (FP- LAPW) in the GGA approximation) shown that it is a metallic nanotube with zero band gap and with the valence and conduction bands cross each other at the Fermi level [44], just as is observed in Figure 3 for (8,8) and (12,3) nanotubes. The nanotube (14,0) is clearly a semiconductor mate- rial with a direct energy gap, as expected. This is consis- tent with previous results obtained from ab initio code VASP [45] and with the classification as semiconductor type I given by Saito et al. [4]. In the Tight-Binding/Zone-Folding method, the transi- tion energies between van Hove singularities in the elec- tronic density of states is related to the diameter of the tube by 0 2 ii cc Enad= 02.9 (1) = eV is the overlapping integral and where Copyright © 2013 SciRes. AMPC  J. MARQUINA ET AL. AMPC 181 Figure 3. Calculated electronic band structures and DOS of the relaxed SWCNT’s. The Fermi level is at 0 eV. One thousand k points for (8,8) nanotube and five thousand k points for (14,0) and (12,3) nanotubes were employed and the mesh cutoff was at 1600 Ry for (8,8) nanotube and at 1200 Ry for (14,0) and (12,3) nanotubes. 1.437a= () 112 224 cc Å (Table 1) is the carbon-carbon distance mean of the nanotubes. The integer n has the values 1, 2, 4, 5 ... for the transitions E11, E22, E33, E44, of the semi- conducting tubes, respectively, and 3, 6, ... for the tran- sitions E11, E22, of the metallic tubes, respectively [46]. In Table 2 are shown electronic transition energies (Eii). In this table is observed that the calculated values for the gap energy (Eg) are in good agreement with the previous report. On the contrary, the values for energies E11 and E22 differ from values reported usign different techniques. The energies Eii obtained from Zone-Folding (Z-F) and Resonance Raman Spectroscopy (RRS) are based in Tight-Binding approximation [46,47]. Thus, the differences between our results and those obtained by Z-F or RSS are due to the type of approximation used for calculations of the energies Eii, i.e. Tight-Binding or Density Functional Theory. On the other hand, they find that the experimental values 1.45 and 2.73 eV for the (8,8) nanotube shown in [43] are below our values. Although, even the approach used is better than those obtained by other methods, it does not reproduce well the valence bands in the electronic band structure. This is a problem inherent in all DFT-based methods. Figure 4. Phonon dispersion curves. We have used used a supercell with 160 atoms for the (8,8) nanotube and 162 atoms for the (14,0) nanotube. Seventy- five k points for (8,8) nanotube and fifty k points for (14,0) and (12,3) nanotubes were employed and the mesh cutoff was at 350 Ry for all nanotubes. Table 2. Energy-gap values and electronic transitions ener- gies values. The Figure 4 shows phonon dispersion curves of (8,8) and (14,0) nanotubes. The phonon-dispersion curve for (10,10) nanotube reported by Ye et al. [29] is very simi- lar to the nanotube (8,8). And also, the dispersion curve of (14,0) zigzag tube is in good qualitative agreement with a dispersion curve for the (11,0) nanotube reported in [29]. According to group theory there are eight modes Raman active for an armchair carbon nanotube, with symmetries and number of optically active phonon modes given by [48]: Eg (eV) E11 (eV) E22 (eV) Our Rep.Our Rep. Our Rep. (8,8) 0.00 0.00c 1.84 2.28a 2.22b, 1.45d 3.084.56a 2.73d (14,0) 0.67 0.62e 0.67 0.76a 1.16 1.51a (12,3) 0.00 0.00c 1.69 2.31a 2.04b 2.084.62a aRef. [46]; bRef [47]; cRef. [4]; dRef. [43]; eRef. [45]. The A modes are non-degenerate and the E modes are doubly degenerate. gg RA EEΓ=⊗ ⊗ 1 3u , (2) () In Table 3 we have assigned the symmetry of the dif- ferent modes of vibration calculated to the (8,8) nanotube, REΓ=. (3) Copyright © 2013 SciRes.  J. MARQUINA ET AL. 182 Table 3. Vibrational modes in zone center for (8,8) nano- tubes. R and IR mean Raman-Active and infrar ed-active. Symmetry Our Ab inicio (GGA) Ref. [29] Ab initio (LDA) Ref. [54] Tight-Binding Ref. [49] Exp. E2g (R) 1603 1593 1698 1609 A1g (R) 1589 1580 1672 1593 E1u (IR) 1583 1574 1683 - E1g (R) 1568 1561 1668 1567 following the assignment given in [29] taking into account the Equations (2) and (3). In a recent paper presented earlier by Rao et al. Raman bands were observed for an isolated nanotube with chirality that are consistent with an ensemble of (8,8), (9,9), (10,10), and (11,11) tubes [49]. They observed seven Raman peaks and failed to detect the lowest frequency mode in the experiment because of the strong Rayleigh scattering back- ground. These values are used for comparison with our calculations. From Ta ble 3 is clear that in the high frequency region the calculations made with the GGA approximation agree much better with experiment than those using LDA or Tight-binding. It is known that, the LDA and Tight-binding approxima- tions tends to over estimate the high-energy phonons. In the high-frequency region the phonon dipersions is well reproduced in our calculations. For example, the modes for A1g symmetry are 1589 and 1593 cm−1 for (8,8) and (14,0) tubes, respectively. These values are just be- low the highest frequency mode. On the other hand, tak- ing into account that the strongest G band (assigned to A1g symmetry [50,51]) at about 1590 cm−1 does not de- pend on the tube diameter and that the G bands of the outer tubes (in pristine DWCNT’s) fall in the same spec- tral range as the G+ of an isolated semiconducting SWCNT (1589 - 1593 cm−1) [30,52,53]. We conclude that our results for the zone-center phonons in the high- frequency region are in very good agreement with the experimental values. 4. Conclusion We have made a study of DFT as implemented in the code SIESTA, using localized atomic wave functions as a basis set with the GGA, and applied it to the study of SWCNT’s about 1 nm in diameter. We found that the structure for (8,8) and (14,0) nanotubes does not show significant change compared with the rolled graphene sheet, while the (12,3) nanotube shows significant change. It has been observed a natural tendency to form regular hexagons that dominates in the later case. Our results predict that the electronic band structure for (8,8) and (12,3) nanotubes has metallic behavior, that is, behaves as metal at 0 K. Whereas the (14,0) nanotube is a semi- conducting. Also, it is found that the accuracy of the method for predicting the transition energies of Van Hove singularities is insufficient. This is a problem in- herent in DFT-based methods because it does not repro- duce well the valence bands in the electronic band struc- ture. Finally, we have shown that our results are compa- rable with the experimental values, this proves that the method used can be quite accurate for the high-energy vibrations because of their reliable description of the chemical bonds. 5. Acknowledgements The authors are grateful to the PCP Carbon Nanotubes (France)-MCT (Venezuela) for financial support. We also would like to express our gratitude to Nicolas RENON and the CALMIP (Calcul en Midi-Midi-Pyré- nées) team for giving us access to the supercomputer used for parallel computing. One of us (J. M.) would like to thank Dr. L. Rincón for his interest in this work and the useful suggestions. REFERENCES [1] S. Lijima, “Helical Microtubules of Graphitic Carbon,” Nature, Vol. 354, 1991, pp. 56-58. doi:10.1038/354056a0 [2] N. Hamada, S.-I. Sawada and A. Oshiyama, “New One- Dimensional Conductors: Graphitic Microtubules,” Phy- sical Review Letters, Vol. 68, No. 10, 1992, pp. 1579- 1581. doi:10.1103/PhysRevLett.68.1579 [3] R. Saito, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, “Electronic Structure of Chiral Graphene Tubules,” Ap- plied Physics Letters, Vol. 60, No. 18, 1992, pp. 2204- 2206. doi:10.1063/1.107080 [4] R. Saito, K. Sato, Y. Oyama, J. Jiang, G. G. Samsonidze, G. Dresselhaus and M. S. Dresselhaus, “Cutting Lines near the Fermi Energy of Single-Wall Carbon Nano- tubes,” Physical Review B, Vol. 72, No. 15, 2005, Article ID: 153413. doi:10.1103/PhysRevB.72.153413 [5] M. S. Dresselhaus, G. Dresselhaus and P. C. Ecklund, “Science of Fullerene and Carbon Nanotubes,” Academic Press, San Diego, 1996. [6] P. J. F. Harris, “Carbon Nanotube Science,” Cambridge University Press, London, 2009. doi:10.1017/CBO9780511609701 [7] R. Saito, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, “Electronic Structure of Graphene Tubules Based on C60,” Physical Review B, Vol. 46, No. 3, 1992, pp. 1804-1811. doi:10.1103/PhysRevB.46.1804 [8] J. W. Mintmire and B. I. Dunlap, C. T. White. “Are Fullerene Tubules Metallic?” Physical Review Letters, Vol. 68, No. 5, 1992, pp. 631-634. doi:10.1103/PhysRevLett.68.631 [9] R. Saito, T. Takeya, T. Kimura, G. Dresselhaus and M. S. Dresselhaus, “Raman Intensity of Single-Wall Carbon Nanotubes,” Physical Review B, Vol. 57, No. 7, 1998, pp. 4145-4153. doi:10.1103/PhysRevB.57.4145 Copyright © 2013 SciRes. AMPC  J. MARQUINA ET AL. 183 [10] R. A. Jishi, L. Venkataraman, M. S. Dresselhaus and G. Dresselhaus, “Symmetry Properties of Chiral Carbon Na- notubes,” Physical Review B, Vol. 51, No. 16, 1995, pp. 11176-11179. doi:10.1103/PhysRevB.51.11176 [11] D. Sánchez-Portal, E. Artacho, J. M. Soler, A. Rubio and P. Ordejón, “Ab Initio Structural, Elastic, and Vibrational Properties of Carbon Nanotubes,” Physical Review B, Vol. 59, No. 19, 1999, pp. 12678-12688. doi:10.1103/PhysRevB.59.12678 [12] M. Machón, S. Reich, C. Thomsen, D. Sánchez-Portal and P. Ordejon, “Ab Initio Calculations of the Optical Properties of 4-Å-Diameter Single-Walled Nanotubes,” Physical Review B, Vol. 66, No. 15, 2002, Article ID: 155410. doi:10.1103/PhysRevB.66.155410 [13] K. Kanamitsu and S. Saito, “Geometries, Electronic Pro- perties, and Energetics of Isolated Single Walled Carbon Nanotubes,” Journal of the Physical Society of Japan, Vol. 71, 2002, pp. 483-486. doi:10.1143/JPSJ.71.483 [14] V. N. Popov, “Curvature Effects on the Structural, Elec- tronic and Optical Properties of Isolated Single-Walled Carbon Nanotubes within a Symmetry-Adapted Non- Orthogonal Tight-Binding Model,” New Journal of Phys- ics, Vol. 6, 2004, pp. 1-17. doi:10.1088/1367-2630/6/1/017 [15] S. Rols, Z. Benes, E. Anglaret, J. L. Sauvajol, P. Papanek, J. E. Fischer, G. Coddens, H. Schober and A. J. Dianoux, “Phonon Density of States of Single-Wall Carbon Nano- tubes,” Physical Review Letters, Vol. 85, No. 24, 2000, pp. 5222-5225. doi:10.1103/PhysRevLett.85.5222 [16] C. Thomsen, “Second-Order Raman Spectra of Single and Multiwalled Carbon Nanotubes,” Physical Review B, Vol. 61, No. 7, 2000, pp. 4542-4544. doi:10.1103/PhysRevB.61.4542 [17] J. Maultzsch, S. Reich and C. Thomsen, “Chirality-Se- lective Raman Scattering of the D Mode in Carbon Nanotubes,” Physical Review B, Vol. 64, No. 12, 2001, Article ID: 121407. doi:10.1103/PhysRevB.64.121407 [18] J. Maultzsch, J. S. Reich and S. C. Thomsen, “Raman Scattering in Carbon Nanotubes Revisited,” Physical Re- view B, Vol. 65, No. 23, 2002, Article ID: 233402. doi:10.1103/PhysRevB.65.233402 [19] V. Zólyomi and J. Kiirti, “Calculating the Discrepancy between the Stokes and Anti-Stokes Raman D Band of Carbon Nanotubes Using Double Resonance Theory,” Physical Review B, Vol. 66, No. 7, 2002, Article ID: 073418. doi:10.1103/PhysRevB.66.073418 [20] G. G. Samsonidze, R. Saito, A. Jorio, A. G. S. Filho, A. Griineis, M. P. Pimenta, G. Dresselhaus and M. S. Dres- selhaus, “Phonon Trigonal Warping Effect in Graphite and Carbon Nanotubes,” Physical Review Letters, Vol. 90, No. 2, 2003, Article ID: 027403. doi:10.1103/PhysRevLett.90.027403 [21] G. L. Kane, E. J. Mele, R. S. Lee, J. E. Fischer, P. Petit, H. Dai, A. Thess, R. E. Smalley, A. R. M. Verschueren, S. J. Tans and G. Dekker, “Temperature-Dependent Resistivity of Single-Wall Carbon Nanotubes,” EPL (Europhysics Letters), Vol. 41, No. 6, 1998, pp. 683-689. doi:10.1209/epl/i1998-00214-6 [22] Z. Yao, G. L. Kane and G. Dekker, “High-Field Electrical Transport in Single-Wall Carbon Nanotubes,” Physical Review Letters, Vol. 84, No. 13, 2000, pp. 2941-2944. doi:10.1103/PhysRevLett.84.2941 [23] H. Suzuura and T. Ando, “Phonons and Electron-Phonon Scattering in Carbon Nanotubes,” Physical Review B, Vol. 65, No. 23, 2002, Article ID: 235412. doi:10.1103/PhysRevB.65.235412 [24] A. Sédéki, L. G. Caron and G. Bourbonnais, “Electron- Phonon Coupling and Peierls Transition in Metallic Car- bon Nanotubes,” Physical Review B, Vol. 62, No. 11, 2000, pp. 6975-6978. doi:10.1103/PhysRevB.62.6975 [25] Z. J. Q. Ding, Q. Jiang and D. Tian, “Vibrational Spec- trum of Carbon Nanotubes,” Fullerene Science and Te- chnology, Vol. 4, No. 1, 1996, pp. 31-37. doi:10.1080/10641229608001535 [26] A. Charlier, E. McRae, M.-F. Charlier, A. Spire and S. Forster, “Lattice Dynamics Study of Zigzag and Armchair Carbon Nanotubes,” Physical Review B, Vol. 57, No. 11, 1998, pp. 6689-6696. doi:10.1103/PhysRevB.57.6689 [27] R. A. Jishi, L. Venkataraman, M. S. Dresselhaus and G. Dresselhaus, “Phonon Modes in Carbon Nanotubules,” Chemical Physics Letters, Vol. 209, No. 1-2, 1993, pp. 77-82. doi:10.1016/0009-2614(93)87205-H [28] O. Dubay and G. Kresse, “Accurate Density Functional Calculations for the Phonon Dispersion Relations of Gra- phite Layer and Carbon Nanotubes,” Physical Review B, Vol. 67, No. 3, 2003, Article ID: 035401. doi:10.1103/PhysRevB.67.035401 [29] L. H. Ye, B. G. Liu, D. S. Wang and R. Han, “Ab Initio Phonon Dispersions of Single-Wall Carbon Nanotubes,” Physical Review B, Vol. 69, No. 23, 2004, Article ID: 235409. doi:10.1103/PhysRevB.69.235409 [30] E. Belandria, M. Millot, J. M. Broto, E. Flahaut, F. Rod- riguez, R. Valiente and J. González, “Pressure Depend- ence of Raman Modes in Double Wall Carbon Nanotubes Filled with 1D Tellurium,” Carbon, Vol. 48, No. 9, 2010, pp. 2566-2572. doi:10.1016/j.carbon.2010.03.036 [31] J. Marquina, E. Flahaut and J. González, “High-Pressure Optical Absorption Studies of Double-Walled Carbon Nanotubes,” Physica Status Solidi (RRL)—Rapid Research Letters, Vol. 6, No. 9-10, 2012, pp. 382-384. doi:10.1002/pssr.201206280 [32] D. Sánchez-Portal, P. Ordejón, E. Artachoa and J. M. Soler, “Density-Functional Method for Very Large Sys- tems with LCAO Basis Sets,” International Journal of Quantum Chemistry, Vol. 65, No. 5, 1997, pp. 453-461. doi:10.1002/(SICI)1097-461X(1997)65:5<453::AID-QU A9>3.0.CO;2-V [33] J. P. Perdew, K. Burke and M. Ernzerhof, “Generalized Gradient Approximation Made Simple,” Physical Review Letters, Vol. 77, No. 18, 1996, pp. 3865-3868. doi:10.1103/PhysRevLett.77.3865 [34] N. Troullier and J. L. Martins, “Efficient Pseudopotentials for Plane-Wave Calculations,” Physical Review B, Vol. 43, No. 3, 1991, pp. 1993-2006. doi:10.1103/PhysRevB.43.1993 [35] J. Junquera, O. Paz, D. Sánchez-Portal and E. Artacho, “Numerical Atomic Orbitals for Linear-Scaling Calcula- Copyright © 2013 SciRes. AMPC  J. MARQUINA ET AL. Copyright © 2013 SciRes. AMPC 184 tions,” Physical Review B, Vol. 64, No. 23, 2001, Article ID: 235111. doi:10.1103/PhysRevB.64.235111 [36] J. Maultzsch, S. Reich, C. Thomsen, H. Requardt and P. Ordejón, “Phonon Dispersion in Graphite,” Physical Re- view Letters, Vol. 92, No. 7, 2004, Article ID: 075501. doi:10.1103/PhysRevLett.92.075501 [37] M. Cardona, “In Electrons and Photons in Solids,” Scuola Normale, Pisa, 2001. [38] P. Pavone, R. Bauer, K. Karch, O. Schtt, S. Vent, W. Windl, D. Strauch, S. Baroni and S. de Gironcoli, “Ab Initio Phonon Calculations in Solids,” Physica B: Con- densed Matter, Vol. 219-220, 1996, pp. 439-441. doi:10.1016/0921-4526(95)00771-7 [39] J. Marquina, Ch. Power and J. González, “Espectroscopia Raman del Grafeno Monocapa y el Grafito: Acopla- miento Electrón Fonón y Efectos no Adiabáticos,” Re- vista Tumbaga, Vol. 1, No. 5, 2010, pp. 183-194. [40] Y. Miyamoto, M. L. Cohen and S. G. Louie, “Ab Initio Calculation of Phonon Spectra for Graphite, BN, and BC2N Sheets,” Physical Review B, Vol. 52, No. 20, 1995, pp. 14971-14975. doi:10.1103/PhysRevB.52.14971 [41] A. N. Imtani and V. Jindal, “Structure of Chiral Single- Walled Carbon Nanotubes under Hydrostatic Pressure,” Computational Materials Science, Vol. 46, No. 2, 2009, pp. 297-302. doi:10.1016/j.commatsci.2009.03.002 [42] H. Jiang, P. Zhang, B. Liu, Y. Huang, P. H. Geubelle, H. Gao and K. C. Hwang, “The Effect of Nanotube Radius on the Constitutive Model for Carbon Nanotubes,” Com- putational Materials Science, Vol. 28, No. 3-4, 2003, pp. 429-442. doi:10.1016/j.commatsci.2003.08.004 [43] M. Ouyang, J. L. Huang, C. L. Cheung and C. M. Lieber, “Energy Gaps in ‘Metallic’ Single-Walled Carbon Nano- tubes,” Science, Vol. 292, No. 5517, 2001, pp. 702-705. doi:10.1126/science.1058853 [44] H. S. Khadigeh, “Ab Initio Study of Electronic Properties of a Armchair (7,7) Carbon Nanotube,” Advances in Materials Physics and Chemistry, Vol. 2, 2012, pp. 159- 162. doi:10.4236/ampc.2012.23024 [45] T. Ito, K. Nishidate, M. Baba and M. Hasegawa, “First Principles Calculations for Electronic Band Structure of Single-Walled Carbon Nanotube under Uniaxial Strain,” Surface Science, Vol. 514, No. 1-3, 2002, pp. 222-226. doi:10.1016/S0039-6028(02)01633-3 [46] M. Hulman, R. Pfeiffer and H. Kuzmany, “Raman Spec- troscopy of Small-Diameter Nanotubes,” New Journal of Physics, Vol. 6, 2004, pp. 1-17. doi:10.1088/1367-2630/6/1/001 [47] C. Fantini, A. Jorio, M. Souza, M. S. Strano, M. S. Dres- selhaus and M. A. Pimenta, “Optical Transition Energies for Carbon Nanotubes from Resonant Raman Spectros- copy: Environment and Temperature Effects,” Physical. Review Letters, Vol. 93, No. 14, 2004, Article ID: 147406. doi:10.1103/PhysRevLett.93.147406 [48] O. E. Alon, “Number of Raman- and Infrared-Active Vi- brations in Single-Walled Carbon Nanotubes,” Physical Review B, Vol. 63, No. 20, 2001, Article ID: 201403. doi:10.1103/PhysRevB.63.201403 [49] A. M. Rao, E. Richter, S. Bandow, B. Chase, P. C. Ek- lund, K. A. Williams, S. Fang, K. R. Subbaswamy, M. Menon, A. Thess, R. E. Smalley, G. Dresselhaus and M. S. Dresselhaus, “Diameter-Selective Raman Scattering from Vibrational Modes in Carbon Nanotubes,” Science, Vol. 275, No. 5297, 1997, pp. 187-191. doi:10.1126/science.275.5297.187 [50] H. D. Sun, Z. K. Tang, J. Chen and G. Li, “Polarized Raman Spectra of Single-Wall Carbon Nanotubes Mono- Dispersed in Channels of ALPO4-5 Single Crystals,” Solid State Communications, Vol. 109, No. 6, 1999, pp. 365-369. doi:10.1016/S0038-1098(98)00588-2 [51] C. Thomsen, “Second-Order Raman Spectra of Single and Multiwalled Carbon Nanotubes,” Physical Review B, Vol. 61, No. 7, 2000, pp. 4542-4544. doi:10.1103/PhysRevB.61.4542 [52] A. Jorio, A. G. Souza Filho, G. Dresselhaus, M. S. Dres- selhaus, A. K. Swan, M. S. Ünlü, B. B. Goldberg, M. A. Pimenta, J. H. Hafner, C. M. Lieber and R. Saito, “G- Band Resonant Raman Study of 62 Isolated Single-Wall Carbon Nanotubes,” Physical Review B, Vol. 65, No. 15, 2002, Article ID: 155412. doi:10.1103/PhysRevB.65.155412 [53] M. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, “Raman Spectroscopy of Carbon Nanotubes,” Physics Reports, Vol. 409, No. 2, 2005, pp. 47-99. doi:10.1016/j.physrep.2004.10.006 [54] D. Kahn and J. P. Lu, “Vibrational Modes of Carbon Nanotubes and Nanoropes,” Physical Review B, Vol. 60, No. 9, 1999, pp. 6535-6540. doi:10.1103/PhysRevB.60.6535
|