 Journal of Water Resource and Protection, 2013, 5, 40-48 http://dx.doi.org/10.4236/jwarp.2013.54A007 Published Online April 2013 (http://www.scirp.org/journal/jwarp) Advances in Water Quality Monitoring of Inorganics: Current Trends Florence Bullough1, Cecilia Fenech2, Helen Bridle3 1Department of Earth Science and Engineering, Royal School of Mines, Imperial College London, London, UK 2School of Biotechnology, Dublin City University, Dublin, UK 3Institute for Biological Chemistry, Biophysics and Bioengineering, Heriot-Watt University, Edinburgh, UK Email: f.bullough10@imperial.ac.uk, cecilia.fenech@dcu.ie, h.l.bridle@hw.ac.uk Received February 8, 2013; revised March 9, 2013; accepted March 22, 2013 Copyright © 2013 Florence Bullough et al. This is an open access article distributed under the Creative Commons Attribution Li- cense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT New methods of analysis for water quality monitoring to detect inorganic substances are required to meet the demands of determining concentration, particularly at low detection limits, analysing speciation and even identifying the pollu- tion source. Such information is essential to inform public health decisions and to comply with more stringent legisla- tion. This paper concentrates on two case studies, reviewing the development in monitoring methods, and predicting future trends. Arsenic and nitrates detection was selected as these pollutants are particularly problematic from a human health perspective. Additionally, the challenges faced in developing monitoring methods for these chemicals are rele- vant to a wide range of other inorganics. The current state of the art in detection approaches for these chemicals are discussed along with recommendations for future research to further improve the methods. Keywords: Water Quality; Monitoring; Detection Methods; Arsenic; Nitrates; Speciation; Source Tracking 1. Introduction In water quality monitoring simply determining the pre- sence of pollutants is often insufficient. Accurate deter- mination of concentrations, speciation or sources can all be critical information to determine, for example, drink- ing water safety or identify the origin of pollution. Pol- lutants include pathogens, organics and inorganics. Pathogen monitoring has been a recent subject of fo- cus with a large EU grant recently awarded to develop new microbial methods of detection. This has been driven by the onset of molecular methods and the grow- ing realisation that faecal indicator monitoring is insuffi- ciently well-correlated with the presence of certain patho- gens [1]. At the same time concern regarding emerging pollutants, many of which are trace level organics have been mounting worldwide [2,3]. These chemicals are extremely challenging to detect at environmentally rele- vant ng/L, and extraction and concentration methods are a key part of addressing this problem. However, inorganics are another major class of water- borne pollutants, many of which have long-term chronic impacts upon human health [4]. The aim of this paper is to review the challenges facing monitoring for inorganic compounds in water by focusing on two key case stud- ies. Arsenic has been selected due to the widespread na- ture, and huge scale, of the issue. This example describes the challenges of meeting low detection limits, especially for field instruments, and the issues relating to speciation. This is a common problem for the detection, and analysis, of inorganic waterborne contaminants. Speciation can impact upon the fate and transport of an inorganic in the environment as well as be a key determinant in the health risk. Nitrates have been selected as this chemical repre- sents one the few short-term acute exposure risks. This example also highlights the challenges of environment forensics in identifying the source of pollution to meet ever-increasing legislation. 2. The Problems of Arsenic Concentration and Speciation Detection Arsenic contamination of drinking water, which leads to chronic poisoning, affects more than 140 million people across 70 countries in all six continents and is considered as the most challenging water pollutant on a global scale [5]. Arsenic in groundwater is a widespread contaminant in South East Asia affecting the quality of drinking water in Bangladesh, India [6] and Cambodia [7] and is also present to a lesser degree in the USA [8] and in parts of C opyright © 2013 SciRes. JWARP 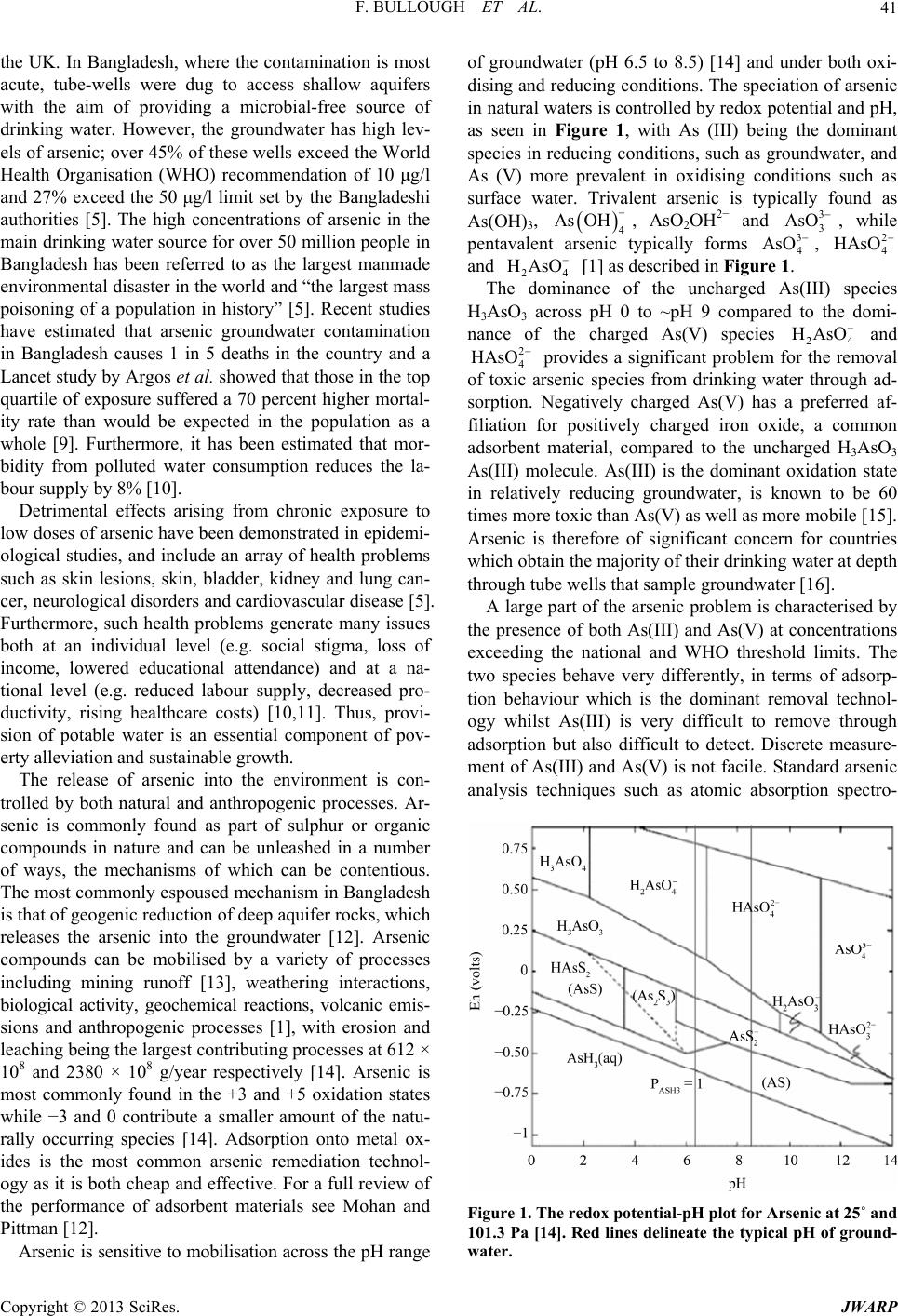 F. BULLOUGH ET AL. 41 the UK. In Bangladesh, where the contamination is most acute, tube-wells were dug to access shallow aquifers with the aim of providing a microbial-free source of drinking water. However, the groundwater has high lev- els of arsenic; over 45% of these wells exceed the World Health Organisation (WHO) recommendation of 10 μg/l and 27% exceed the 50 μg/l limit set by the Bangladeshi authorities [5]. The high concentrations of arsenic in the main drinking water source for over 50 million people in Bangladesh has been referred to as the largest manmade environmental disaster in the world and “the largest mass poisoning of a population in history” [5]. Recent studies have estimated that arsenic groundwater contamination in Bangladesh causes 1 in 5 deaths in the country and a Lancet study by Argos et al. showed that those in the top quartile of exposure suffered a 70 percent higher mortal- ity rate than would be expected in the population as a whole [9]. Furthermore, it has been estimated that mor- bidity from polluted water consumption reduces the la- bour supply by 8% [10]. Detrimental effects arising from chronic exposure to low doses of arsenic have been demonstrated in epidemi- ological studies, and include an array of health problems such as skin lesions, skin, bladder, kidney and lung can- cer, neurological disorders and cardiovascular disease [5]. Furthermore, such health problems generate many issues both at an individual level (e.g. social stigma, loss of income, lowered educational attendance) and at a na- tional level (e.g. reduced labour supply, decreased pro- ductivity, rising healthcare costs) [10,11]. Thus, provi- sion of potable water is an essential component of pov- erty alleviation and sustainable growth. The release of arsenic into the environment is con- trolled by both natural and anthropogenic processes. Ar- senic is commonly found as part of sulphur or organic compounds in nature and can be unleashed in a number of ways, the mechanisms of which can be contentious. The most commonly espoused mechanism in Bangladesh is that of geogenic reduction of deep aquifer rocks, which releases the arsenic into the groundwater [12]. Arsenic compounds can be mobilised by a variety of processes including mining runoff [13], weathering interactions, biological activity, geochemical reactions, volcanic emis- sions and anthropogenic processes [1], with erosion and leaching being the largest contributing processes at 612 × 108 and 2380 × 108 g/year respectively [14]. Arsenic is most commonly found in the +3 and +5 oxidation states while −3 and 0 contribute a smaller amount of the natu- rally occurring species [14]. Adsorption onto metal ox- ides is the most common arsenic remediation technol- ogy as it is both cheap and effective. For a full review of the performance of adsorbent materials see Mohan and Pittman [12]. Arsenic is sensitive to mobilisation across the pH range of groundwater (pH 6.5 to 8.5) [14] and under both oxi- dising and reducing conditions. The speciation of arsenic in natural waters is controlled by redox potential and pH, as seen in Figure 1, with As (III) being the dominant species in reducing conditions, such as groundwater, and As (V) more prevalent in oxidising conditions such as surface water. Trivalent arsenic is typically found as As(OH)3, 4 As OH , AsO2OH2− and 3 3 AsO , while pentavalent arsenic typically forms 4, 3 AsO 2 4 HAsO and 24 HAsO [1] as described in Figure 1. The dominance of the uncharged As(III) species H3AsO3 across pH 0 to ~pH 9 compared to the domi- nance of the charged As(V) species 24 HAsO and 4 2 HAsO provides a significant problem for the removal of toxic arsenic species from drinking water through ad- sorption. Negatively charged As(V) has a preferred af- filiation for positively charged iron oxide, a common adsorbent material, compared to the uncharged H3AsO3 As(III) molecule. As(III) is the dominant oxidation state in relatively reducing groundwater, is known to be 60 times more toxic than As(V) as well as more mobile [15]. Arsenic is therefore of significant concern for countries which obtain the majority of their drinking water at depth through tube wells that sample groundwater [16]. A large part of the arsenic problem is characterised by the presence of both As(III) and As(V) at concentrations exceeding the national and WHO threshold limits. The two species behave very differently, in terms of adsorp- tion behaviour which is the dominant removal technol- ogy whilst As(III) is very difficult to remove through adsorption but also difficult to detect. Discrete measure- ment of As(III) and As(V) is not facile. Standard arsenic analysis techniques such as atomic absorption spectro- Figure 1. The redox potential-pH plot for Arsenic at 25˚ and 101.3 Pa [14]. Red lines delineate the typical pH of ground- water. Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 42 metry (AAS); atomic fluorescence spectrometry (AFS); atomic emission spectrometry (AES); differential pulse polarography (DPP); electrothermal atomic absorption spectrometry (ETAAS) and mass spectrometry (ICP-MS) [17] do not measure individual species and much be used with front-end separation techniques, such as high pres- sure liquid chromatography (HPLC), to separate the As(III) and As(V) oxidation states. Following the recent reduction in threshold limit from 50 µg/L to 10 µg/L, the need for cheap and accurate spe- cies determination of arsenic has become intensified. The presence of As(III) at toxic levels in parts of Bangladesh [18] has created a greater need for discrete analysis as opposed to total arsenic. Many of the existing technolo- gies, including Gutzeit method and the aforementioned method are either unable to provide this distinction or complex and expensive. Due to the nature of much of arsenic contamination being in areas of the world with poor infrastructure and funds for remediation technology, arsenic analysis techniques need to be cheap, reliable, portable as much as possible and with a very low detec- tion limit, below 10 µg/L. One method that has been proposed to deliver field- tests of the bioavailable arsenic is that of genetically modified whole-cell biosensors [19,20]. In this approach bacteria are engineered such that a reporter gene, which generates a signal, is paired with a contaminant sensing component. In the presence of arsenic, the biosensors emit a signal, which could be emission of visible light or a pH change of the water sample. pH changes can also be easily visualised using an indicator dye [21]. Modified cells can be grown and then freeze dried to facilitate transportation. Reconstitution in the field, followed by over-night incubation (at ambient temperature), revives the cells and activates the biosensor. Such sensors are cheap and easy to use; however, there are challenges in achieving low detection limits as well as the issue of permissions for field use of genetically modified organ- isms. Alternatively, low detection limits can be achieved by the voltammetric analytical technique which has been found to accurately detect As(III) and As(V) discretely at sub 10 µg/L concentrations [22]. Voltammetry, as a form of elemental analysis, has been known to chemists for over 50 years [16]. It has re- ceived more attention in recent years, particularly in the analysis of arsenic, due to its sensitivity, low cost, reli- ability, relatively short analysis time [17] and its unique sensitivity for As(III) [16]. It has become particularly useful in the area of water contamination and remedia- tion because of its portability, which allows sample analysis to take place at, or close to the sampling point [16]. This avoids the issue of sample preservation be- tween sample taking and measurement in a laboratory. In addition to its selectivity for arsenic oxidation states, the development of voltammetry for stand-alone field use is of particular interest for the detection of arsenic in groundwater [16]. This is highly significant for measur- ing samples in situ without issues of preservation or al- teration of the arsenic in the sample vessel. Stripping Voltammetry exploits the electrochemically active nature of certain metal species to determine con- centrations. It involves the electrochemical deposition, on application of a current, through reduction of the ele- ment of interest on an electrode, for a given deposition time. The element is then oxidised back into the solution by a reverse potential scan [16]. During the deposition time the arsenic is pre-concentrated at the electrode which accounts for the sensitivity of the method [23]. The oxidation current which causes the stripping of the arsenic from the electrode back into solution is recorded and plotted against scan potential [16] to give an analyti- cal signal. The analysis of As(III) and As(V) is controlled through the potential at which the solution is held prior to stripping, where the potential is low enough to reduce the analyte and deposit it at the electrode [16]. In our system the As(III) determination is held at a potential −0.2 V and total arsenic (As(III) + As(V)) determination is held at −1.2 V. In both cases the arsenic is reduced to As(0) when it is deposited at the electrode at ~−0.1 V. The dif- ferential pulse anodic stripping voltammetric system (DPASV), used in the work by Alves et al. and also Saluan et al. [24] uses a three electrode system, a work- ing electrode, on to which the element of interest is de- posited, a reference electrode and an auxiliary electrode all of which vary depending on the type of determination being carried out [16]. The system is calibrated through standard addition [18] whereby once the sample has been analysed, an aliquot of a known stock solution of known concentration is added to the voltammetric cell: the re- sultant concentration is measured twice and then the process is repeated again. The benefit of the standard addition calibration is that each sample is calibrated indi- vidually at the time of analysis as opposed to a calibra- tion line which can incur more drift. This means that some of the differences in relative conditioning of the electrode should be removed through the standard addi- tion method. Electrode conditioning is a potential issue with the re- producibility of voltammetric determinations. When not being used, the reproducibility of data using solid elec- trodes can be impeded due to the formation of a surface oxide on the electrode surface, which limits sensitivity [25]. This can cause sensitivity issues both within runs and over the long term [16]. Electrodes are conditioned by either immersing the electrode in an appropriate acid or base over a period of time or electrochemically by Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 43 running voltammetric cycles in a pre-determined solution. No standard electrode pre-treatment has been established with many researcher using acids or bases with variations in the strength of the conditioning solution [16]. All voltammetric techniques require efficient mass transfer of ions in the cell so that the working electrode can ef- fectively pre-concentrate the analyte of interest. This is primarily achieved by a magnetic stirrer or a rotating electrode. This local motion enhances metal deposition [25] and can also minimise the H2 bubble formation at the working electrode, resulting in reduced noise and increased signal [26]. Much research has gone into optimising many parts of the process including the material the electrode is made from. Most commonly an Au electrode is used due to its stability and sensitivity. In recent publications, particu- larly from Salaun et al. they have tested an alternative Au microwire electrode. These studies found that As(III) could be determined in freshwaters and seawaters at any pH, thus excluding the addition of corrosive acids as electrolytes [27]. Optimum conditions included a deposi- tion potential of −1.0 kv for As(III) and As(V) and 30 s deposition time. They reported the use of 0.01 M HCl as an electrolyte with limits of detection of 14.98 ng·L−1 for As(III) and 22.47 ng/L for As(V) [27]. Later work de- veloped the use of cathodic stripping voltammetry (CSV) at a vibrating gold microwire electrode, where arsenite determination was possible with no pre-treatment of the sample at its original pH and open to ambient air [22]. This means that this method is suitable for on-site analy- sis. Total arsenic is then determined through the addition of acid to pH 1. The data compared well (within 10%) with the ASV method. To highlight the issue of sample preservation, a study in West Bengal waters showed that if analysed immediately then As(III) was the dominant species, however, upon storage there was significant ox- idation to As(V) and adsorption on particulate matter in the solution [22]. Immediate analysis of the arsenic sam- ples without the need for pre-treatment or expensive lab techniques for countries such as Bangladesh would be an important contribution to the ongoing effort to provide safe drinking water. 3. The Challenge of Nitrate Source Determination Nitrate (3) occurs naturally within the environment. However, concern regarding its ever-increasing entry into the natural environment as a result of various an- thropogenic sources, such as inorganic fertilisers and effluents from wastewater treatment plants, have led to it being considered a contaminant of concern. This is largely as it has been linked to various environmental and health concerns. High nitrate concentrations within water bodies have been linked to such occurrences as eutro- phication [28,29]. High nitrate concentrations within drink- ing water have also been linked to methemoglobinemia in children (blue-baby disease) [30] and cancer [31] amongst other diseases. However, the presence of a di- rect link is still a factor for debate [32]. These factors have led to increasing interest in the development of en- vironmental forensics techniques for nitrate source de- termination, largely in relation to legislative requirements related to the Water Framework (2000/60/EC) and Ni- trates Directives (91/676/EEC). To date, various approaches have been adopted in an effort to distinguish between different sources of nitrate. The use of nitrate stable isotope compositions has been one of the most successful in this regard [33]. The dual isotope approach is the most successful approach for identifying the various sources of nitrate contamination, where isotopic fractionation for both the nitrogen (δ15N) and oxygen (δ18O) atoms within the nitrate ion is consid- ered (Figure 2) e.g. [33-37]. In particular, the dual isotope approach has been useful for the identification of hydrologic pathways [38-41]. The isotopic composition of a particular water body does not only reflect the composition of the original source or of mixed sources having different compositions but can also be influenced by isotopic fractionation during the transport and chemical transformation of the compounds [42,43]. Therefore, it allows for the source of contamina- tion and the pathways undertaken to be identified. How- ever, this method is not suitable for differentiating closely related sources of contamination, such as sewage and manure [44]. This is because both sewage and ma- nure undergo similar isotopic fractionation processes leading to overlapping isotopic compositions (as seen in Figure 2 where these elements cannot be distinguished) [44]. A range of approaches has been utilised for specifi- cally achieving faecal source tracking. The use of faecal indicator bacteria (FIB) represents the most commonly NO Figure 2. A general depiction of the normal range of δ18O and δ15N values for the dominant sources of nitrate [45]. Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 44 adopted faecal contamination markers of water bodies. However, whilst it is useful for the detection of faecal contamination, it is currently not possible to distinguish between microbial pathogens arising from human (sew- age) or animal (manure) sources on this basis. This is because FIB such as Escherichia coli and enterococci, which represent the commonly used FIB, do not dis- criminate between human and animal faecal matter sources [46]. The ratio of faecal coliforms (FC) to faecal streptococci (FS) was also proposed as a way to differen- tiate sewage and manure [47]. However, as a result of variable survival rates of the bacterial species and the differences in FC-FS ratios within different animals, the use of these ratios is no longer considered to be suitable [46]. For this reason, other tracers must be used to achieve this differentiation. The use of molecular techniques for Microbial Source Tracking (MST) through a variety of library dependent and library independent methods has also been investi- gated [48]. These include antibiotic resistance, bio- chemical fingerprinting, DNA fingerprinting, bacterio- phage occurrence and the use of genetic markers. How- ever, the application of MST for achieving faecal source tracking has met a number of challenges. Host specificity is one of the major challenges in the development of MST techniques. This is because, whilst it is commonly the case that differential distribution of the particular source identifier is present within the various sources, such that it is found at a higher frequency or density within certain hosts [49], it is known that a significant level of cosmopolitan strains (strain sharing) is present, such that incorrect source attribution might result [50, 51]. Furthermore, particular molecular source identifiers often also vary on temporal and spatial scales. Differ- ences in dietary regimes are amongst the major contribu- tors to this variability. This is because different dietary regimes would include the presence and levels of bacte- rial groups within the intestinal tract [49]. Hence, source identifiers that would be relevant within a specific tem- poral period and geographical area might not be relevant in a different scenario [49,50]. The environmental per- sistence of the various source identifiers selected is an- other consideration. This is because the clonal composi- tion of the species commonly differs between the envi- ronmental samples and the host populations [49,52]. Lastly, practical considerations, in particular related to the method’s transferability and applications must be taken into account. These include factors such as the technique’s availability and complexity, the cost of analysis and the level of expertise required for successful data interpretation [46]. Therefore, whilst the use of molecular techniques for MST allows for highly specific information on the pres- ence of faecal indicators, a number of challenges have been identified. In fact, although multiple LDMs and LIMs are currently available, many have not yet been fully tested and validated to the stage of application in field studies [48,49] and no specific method has been shown to be superior enough to be adopted as a standard [46]. In fact, a number of studies carried out by the US Geological Survey project to assess available techniques concluded that none of the methods investigated were ready for field application [51,53]. An additional consid- eration of using molecular techniques, is that they can only function in the identification of the host from which the source of nitrate (or faecal) contamination is initiated. Therefore, using such techniques it would not be possible to differentiate between raw and treated sources of con- tamination. A review of recent literature has identified the use of a suite of chemical markers, namely pharmaceuticals and related compounds such as caffeine, as providing the greatest potential in this regards [45]. The use of phar- maceuticals and related compounds, such as food addi- tives, as chemical markers of co-occurring discriminators of sewage and manure is believed to provide the greatest potential in this regards. They are ideal for such an ap- plication as they are generally relatively water soluble and non-volatile, and their natural background levels are low. The adoption of such an approach also renders in- creased temporal and spatial stability of the source iden- tifiers, as opposed to the use of molecular markers, since consumption of pharmaceuticals and related compounds such as food additives is largely stable, at least within the developed world. Furthermore, due to the wide variety of such com- pounds available, through an understanding of the che- mical marker’s environmental persistence, biodegrada- tion and environmental fate, it would be possible to se- lect the most appropriate suite of chemical markers for achieving identification of the required input. To date, most such environmental forensics studies have focussed on a single tracer approach. Caffeine has been one of the most studied chemical tracers of sewage to date [54,55]. However, it is only through the use of a suite of chemical markers, that it would be possible to achieve further characterisation of the sewage or manure input. For ex- ample, indicating the effectivity and the level of treat- ment being undergone within the DWWTS. The use of immunoassays for chemical marker detec- tion is an emerging technique for this purpose. Immuno- assays have been widely applied in other areas of science, in particular clinical analyses [56]. However, despite the first studies on using immunoassays to detect pharma- ceutical in surface waters showing up around 10 years ago [57] for the detection on Diclofenac, they have re- ceived limited further attention [58-60]. This may be due Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 45 to the limited availability of antibodies showing reactiv- ity to e.g. pharmaceuticals, as well as the skills set of environmental scientists. However, the use of immunoassays renders a number of advantages. Since the antibody-antigen complexes form through relatively weak interactions, which func- tion over short distances, a close antibody-antigen fit is required for complex formation [61]. This confers a high degree of specificity to antibody-antigen binding. There- fore, they have the capability of measuring antigens within complex matrices, with limited or no pre-treat- ment, extraction, purification or concentration, due to the potential for low detection limits being achieved [56]. Furthermore, they have the potential for high-throughput analysis [56]. This is particularly related to the use of 96 (and less commonly 384) well-plates for analysis and multi-channel pipettes, which greatly facilitate reagent and sample handling [62]. Combined with this are mul- tichannel-spectrophotometers, which allow for the com- plete sample plates to be read within a few seconds [62]. As with all analytical techniques, the use of immuno- assay techniques has a number of limitations, which need to be considered. One of the main limitations is the po- tential for cross-reactivity or interference within immu- noassay analyses. Therefore, factors such as the unique- ness of the epitope used are critical, as they determine antibody-antigen selectivity. Furthermore, the level of confirmatory detail on the presence of a particular ana- lyte within a sample is reduced as compared to that ob- tained through mass spectrometric analyses. This is es- pecially true when considering the potential variability in surface water matrices. However, the use of immunoas- says has wide potential as a fast-screening method for sample analysis, allowing for samples requiring chroma- tographic analysis to be decreased. This is particularly true, with the advent of multiplex screening which is a recent development and the potential for incorporation in lab-on-a-chip systems [63] augurs for high potential for such screening techniques. 4. Conclusions and Future Outlook As our two case studies illustrate, new methods of analy- sis for inorganics in water samples are required. While existing techniques can successfully detect the presence of compounds, new approaches are needed to meet the demands of determining concentration, particularly at low detection limits, analysing speciation and even iden- tifying the pollution source. This information is essential to inform public health decisions and to comply with more stringent legislation. Our first case study, arsenic detection, illustrated the first two challenges of low de- tection limit analysis in addition to the need to obtain species information. Our second case study, nitrate source determination, discussed how chemical markers can be applied to identify the origin of pollution. In addi- tion to these factors, new detection approaches also have to be cheap, reliable and ideally portable. Portability en- ables detection in the field negating the challenges of sample processing and transportation. This paper concentrated on two case studies and re- viewed the challenges as well as the existing state-of- the-art in detection technologies. For arsenic we have seen that detection at low concentration along with spe- ciation were key challenges, essential to appropriate risk assessments and public health interventions. From a re- view of the literature we found that voltammetry is one of the most promising potential solutions to this chal- lenge as it offers many advantages including sensitivity, ability to speciate, low-cost and short analysis times. Whole-cell biosensors are also interesting from the per- spective of field testing instrument. This paper summarised many of the recent develop- ments to optimise the voltammetric detection approach for arsenic. The current state of this technology still re- quires significant research and testing to move towards analysis with less pre-treatment of samples so as to im- prove detection in real sea-water and terrestrial water samples. In order to deliver field-ready instrumentation, the stability of the samples and equipment in field sce- narios needs to be tested as well as the reproducibility of concentrations in complex real water samples. For nitrate, we have seen that new environmental fo- rensics methods are required to meet recent legislation and enable tracing of sources of pollution. Our literature review revealed that various methods are available in- cluding isotope approaches, molecular methods and de- tection of a range of chemical markers. Overall, it is ex- pected that no single method would allow for complete source characterisation. The most appropriate approach largely depends upon the specific scenario and the con- text of the study. The use of chemical markers is a rela- tively recent development. Its use for such studies is par- ticularly promising as it allows for the entry pathway to be identified, such as the differentiation of raw and treated sewage inputs. At present this approach has been applied to a number of small scale studies and would benefit from further research into field testing and vali- dation of alternative analytical techniques, such as im- munoassay analyses, to replace costly and time-intensive chromatographic and mass spectrometric analysis, which would facilitate catchment scale studies. In conclusion, it is clear from this review of water- borne inorganics detection that current approaches are moving towards more detailed analysis and this demand for improved information raises significant challenges for detection technologies. However, for all the case studies discussed here, progress is being made towards this goal. Further research developing, characterising and Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 46 validating such new techniques, for these case studies as well as other inorganic substances, should be a priority. 5. Acknowledgements All authors would like to acknowledge the European Science Foundation and thank them for the organization and invitation to the Junior Summit on Water: Unite and Divide. FB would like to acknowledge the Department of Earth Science and Engineering for financial support of her PhD. CF would like to acknowledge the financial support of the Marie Curie Initial Training Network funded by the EU FP7 People Programme, ATWARM (Advanced Technologies for Water Resource Manage- ment, ITN No. 238273) HB would like to acknowledge the Royal Academy of Engineering/EPSRC for her re- search fellowship. REFERENCES [1] I. Brettar and M. G. Höfle, “Molecular Assessment of Bacterial Pathogens—A Contribution to Drinking Water Safety,” Current Opinion in Biotechnology, Vol. 19, No. 3, 2008, pp. 274-280. doi:10.1016/j.copbio.2008.04.004 [2] M. Schriks, et al., “Toxicological Relevance of Emerging Contaminants for Drinking Water Quality,” Water Re- search, Vol. 44, No. 2, 2010, pp. 461-476. doi:10.1016/j.watres.2009.08.023 [3] R. M. Sharp and N. E. Skakkebaek, “Male Reproductive Disorders and the Role of Endocrine Disruption: Ad- vances in Understanding and Identification of Areas for Future Research,” Pure and Applied Chemistry, Vol. 75, No. 11-12, 2003, pp. 2023-2038. doi:10.1351/pac200375112023 [4] World Health Organisation, “Guidelines for Drinking- Water Quality,” 2011. [5] A. H. Smith, E. O. Lingas and M. Rahman, “Contamina- tion of Drinking-Water by Arsenic in Bangladesh: A Pub- lic Health Emergency,” Bulletin of the World Health Or- ganisation, Vol. 78, No. 9. 2000, p. 1093. [6] M. A. Hossain, et al., “Ineffectiveness and Poor Reliabil- ity of Arsenic Removal Plants in West Bengal, India,” Environmental Science & Technology, Vol. 39, No. 11, 2005, pp. 4300-4306. doi:10.1021/es048703u [7] H. A. L. Rowland, et al., “Geochemistry of Aquifer Sediments and Arsenic-Rich Groundwater from Kandal Province, Cambodia,” Applied Geochemistry, Vol. 23, No. 11, 2008, pp. 3029-3046. doi:10.1016/j.apgeochem.2008.06.011 [8] T. L. Root, et al., “Arsenic Geochemistry and Hydros- tratigraphy in Midwestern US Glacial Deposits,” Ground Water, Vol. 48, No. 6, 2010, pp. 903-912. doi:10.1111/j.1745-6584.2009.00637.x [9] M. Argos, et al., “Arsenic Exposure from Drinking Water, and All-Cause and Chronic-Disease Mortalities in Bang- ladesh (HEALS): A Prospective Cohort Study,” The Lan- cet, Vol. 376, No. 9737, 2010, pp. 252-258. doi:10.1016/S0140-6736(10)60481-3 [10] R. T. Carson, P. Koundouri and C. L. Nauges, “Arsenic Mitigation in Bangladesh: A Household Labor Market Approach,” American Journal of Agricultural Economics, Vol. 93, No. 2, 2011, pp. 407-414. [11] A. Rahman, “Towards an Arsenic Safe Environment in Bangladesh,” BCAS, 2010. [12] K. Gupta and U. C. Ghosh, “Arsenic Removal Using Hydrous Nanostructure Iron(III)-titanium(IV) Binary Mixed Oxide from Aqueous Solution,” Journal of Hazardous Materials, Vol. 161, No. 2-3, 2009, pp. 884-892. doi:10.1016/j.jhazmat.2008.04.034 [13] J. S. Yamani, et al., “Enhanced Arsenic Removal Using Mixed Metal Oxide Impregnated Chitosan Beads,” Water Research, Vol. 46, No. 14, 2012, pp. 4427-4434. doi:10.1016/j.watres.2012.06.004 [14] D. Mohan and C. U. Pittman, “Arsenic Removal from Water/Wastewater Using Adsorbents—A Critical Review,” Journal of Hazardous Materials, Vol. 142, No. 1-2, 2007, pp. 1-53. doi:10.1016/j.jhazmat.2007.01.006 [15] K. S. Liu, et al., “Preparation of Large-Pore Mesoporous Nanocrystalline TiO2 Thin Films with Tailored Pore Di- ameters,” Journal of Physical Chemistry B, Vol. 109, No. 40, 2005, pp. 18719-18722. doi:10.1021/jp054546p [16] D. E. Mays and A. Hussam, “Voltammetric Methods for Determination and Speciation of Inorganic Arsenic in the Environment—A Review,” Analytica Chimica Acta, Vol. 646, No. 1-2, 2009, pp. 6-16. doi:10.1016/j.aca.2009.05.006 [17] S. Yilmaz, et al., “Direct Quantitative Determination of Total Arsenic in Natural Hotwaters by Anodic Stripping Voltammetry at the Rotating Lateral Gold Electrode,” Current Analytical Chemistry, Vol. 5, No. 1, 2009, pp. 29-34. doi:10.2174/157341109787047934 [18] S. B. Rasul, et al., “Electrochemical Measurement and Speciation of Inorganic Arsenic in Groundwater of Bang- ladesh,” Talanta, Vol. 58, No. 1, 2002, pp. 33-43. doi:10.1016/S0039-9140(02)00254-0 [19] H. Strosnider, “Whole-Cell Bacterial Biosensors and the Detection of Bioavailable Arsenic,” US Environmental Protection Agency, 2003. [20] C. French, et al., “Development of Biosensors for the Detection of Arsenic in Drinking Water, in Arsenic in the Environment: The Metabolism of Arsenite,” CRC Press— Taylor and Francis Group, London, 2012. [21] K. De Mora, et al., “A pH-Based Sensor for Detection of Arsenic in Drinking Water,” Analytical and Bioanalytical Chemistry, Vol. 400, No. 4, 2011, pp. 1031-1039. doi:10.1007/s00216-011-4815-8 [22] K. Gibbon-Walsh, P. Salaun and C. M. G. van den Berg, “Arsenic Speciation in Natural Waters by Cathodic Strip- ping Voltammetry,” Analytica Chimica Acta, Vol. 662, No. 1, 2010, pp. 1-8. doi:10.1016/j.aca.2009.12.038 [23] F. G. Bodewig, P. Valenta and H. W. Nurnberg, “Trace Determination of As(III) and As(V) in Natural-Waters by Differential Pulse Anodic-Stripping Voltammetry,” Fre- senius Zeitschrift Fur Analytische Chemie, Vol. 311, No. 3, 1982, pp. 187-191. doi:10.1007/BF00476644 [24] G. M. S. Alves, et al., “Simultaneous Electrochemical Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. 47 Determination of Arsenic, Copper, Lead and Mercury in Unpolluted Fresh Waters Using a Vibrating Gold Mi- crowire Electrode,” Analytica Chimica Acta, Vol. 703, No. 1, 2011, pp. 1-7. doi:10.1016/j.aca.2011.07.022 [25] A. Profumo, D. Merli and M. Pesavento, “Voltammetric Determination of Inorganic As(III) and Total Inorganic as in Natural Waters,” Analytica Chimica Acta, Vol. 539, No. 1-2, 2005, pp. 245-250. doi:10.1016/j.aca.2005.02.062 [26] R. Prakash, R. C. Srivastava and P. K. Seth, “Direct Es- timation of Total Arsenic Using a Novel Metal Side Disk Rotating Electrode,” Electroanalysis, Vol. 15, No. 17, 2003, pp. 1410-1414. doi:10.1002/elan.200302658 [27] P. Salaun, B. Planer-Friedrich and C. M. G. van den Berg, “Inorganic Arsenic Speciation in Water and Seawater by Anodic Stripping Voltammetry with a Gold Microelec- trode,” Analytica Chimica Acta, Vol. 585, No. 2, 2007, pp. 312-322. doi:10.1016/j.aca.2006.12.048 [28] C. Kendall, “Tracing Nitrogen Sources and Cycling in Catchments,” In: M. J. Kendall, Ed., Isotope Tracers in Catchment Hydrology, Elsevier Science B.V., 1998, pp. 519-576. doi:10.1016/B978-0-444-81546-0.50023-9 [29] R. M. Monaghan, et al., “Linkages between Land Man- agement Activities and Water Quality in an Intensively Farmed Catchment in Southern New Zealand,” Agricul- ture Ecosystems & Environment, Vol. 118, No. 1-4, 2007, pp. 211-222. doi:10.1016/j.agee.2006.05.016 [30] N. E. Camp, “Methemoglobinemia,” Journal of Emer- gency Nursing, Vol. 33, No. 2, 2007, pp. 172-174. doi:10.1016/j.jen.2006.11.007 [31] World Health Organisation, “Rolling Revision of the WHO Guidelines for Drinking-Water Quality,” Nitrates and Ni- trites in Drinking Water, 2004. [32] D. S. Powlson, T. M. Addiscott and N. Benjamin, “When Does Nitrate Become a Risk for Humans?” Journal of Environmental Quality, Vol. 37, No. 2, 2008, pp. 291- 295. doi:10.2134/jeq2007.0177 [33] S. S. Kaushal, et al., “Tracking Nonpoint Source Nitrogen Pollution in Human-Impacted Watersheds,” Environmental Science Technology, Vol. 45, No. 19, 2011, pp. 8225- 8232. doi:10.1021/es200779e [34] L. I. Wassenaar, “Evaluation of the Origin and Fate of Nitrate in the Abbotsford Aquifer Using the Isotopes of 15N and 18O in 3 NO,” Applied Geochemistry, Vol. 10, No. 4, 1995, pp. 391-405. doi:10.1016/0883-2927(95)00013-A [35] B. Deutsch, et al., “Quantification of Diffuse Nitrate In- puts into a Small River System Using Stable Isotopes of Oxygen and Nitrogen in Nitrate,” Organic Geochemistry, Vol. 37, No. 10, 2006, pp. 1333-1342. doi:10.1016/j.orggeochem.2006.04.012 [36] G. Bordeleau, et al., “Determination of the Origin of Groundwater Nitrate at an Air Weapons Range Using the Dual Isotope Approach,” Journal of Contaminant Hy- drology, Vol. 98, No. 3-4, 2008, pp. 97-105. doi:10.1016/j.jconhyd.2008.03.004 [37] D. Kaown, et al., “Identification of Nitrate and Sulfate Sources in Groundwater Using Dual Stable Isotope Ap- proaches for an Agricultural Area with Different Land Use (Chuncheon, Mid-Eastern Korea),” Agriculture, Eco- systems & Environment, Vol. 132, No. 3-4, 2009, pp. 223- 231. doi:10.1016/j.agee.2009.04.004 [38] J. Böttcher, et al., “Using Isotope Fractionation of Ni- trate-Nitrogen and Nitrate-Oxygen for Evaluation of Mi- cro-Bial Denitrification in a Sandy Aquifer,” Journal of Hydrology, Vol. 114, No. 3-4, 1990, pp. 413-424. doi:10.1016/0022-1694(90)90068-9 [39] R. Aravena and W. D. Robertson, “Use of Multiple Iso- tope Tracers to Evaluate Denitrification in Ground Water: Study of Nitrate from a Large-Flux Septic System Plume,” Ground Water, Vol. 36, No. 6, 1998, pp. 975-982. doi:10.1111/j.1745-6584.1998.tb02104.x [40] W. A. Battaglin, et al., “Chemical and Isotopic Evidence of Nitrogen Transformation in the Mississippi River 1997-1998,” Hydrological Processes, Vol. 15, No. 7, 2001, pp. 1285-1300. d oi:10.1002/hyp.214 [41] H. M. Baulch, et al., “Isotopic Character of Nitrous Oxide Emitted from Streams,” Environmental Science & Tech- nology, Vol. 45, No. 11, 2011, pp. 4682-4688. doi:10.1021/es104116a [42] C. A. I. Cravotta, “Use of Stable Isotopes of Carbon, Nitrogen, and Sulfur to Identify Sources of Nitrogen in Surface Waters in the Lower Susquehanna River Basin,” USGS, Pennsylvania, 1997. [43] A. Nestler, et al., “Isotopes for Improved Management of Nitrate Pollution in Aqueous Resources: Review of Sur- face Water Field Studies,” Environmental Science and Pollution Research, Vol. 18, No. 4, 2011, pp. 519-533. doi:10.1007/s11356-010-0422-z [44] D. Xue, et al., “Present Limitations and Future Prospects of Stable Isotope Methods for Nitrate Source Identification in Surface and Groundwater,” Water Research, Vol. 43, No. 5, 2009, pp. 1159-1170. doi:10.1016/j.watres.2008.12.048 [45] C. Fenech, et al., “The Potential for a Suite of Isotope and Chemical Markers to Differentiate Sources of Nitrate Con- tamination: A Review,” Water Research, Vol. 46, No. 7, 2012, pp. 2023-2041. doi:10.1016/j.watres.2012.01.044 [46] J. Stewart, J. W. Santo Domingo and T. J. Wade, “Fecal Pollution, Public Health, and Microbial Source Tracking,” In: S. M. Santo and J. W. Domingo, Eds., Microbial Source Tracking, ASM Press, Washington DC, 2007, pp. 1-32. [47] E. E. Geldreich and B. A. Kenner, “Concepts of Fecal Streptococci in Stream Pollution,” Water Pollution Con- trol Federation, Vol. 41, No. 8, 1969, pp. R336-R352. [48] K. G. Field and M. Samadpour, “Fecal Source Tracking, the Indicator Paradigm, and Managing Water Quality,” Water Research, Vol. 41, No. 6, 2007. pp. 3517-3538. doi:10.1016/j.watres.2007.06.056 [49] V. J. Harwood, H. Ryu and J. Santo Domingo, “Microbial Source Tracking,” In: M. J. Sadowsky and R. L. Whitman, Eds., The Fecal Bacteria, ASM Press, Washington DC, 2011, pp. 189-216. [50] P. Roslev and A. S. Bukh, “State of the Art Molecular Markers for Fecal Pollution Source Tracking in Water,” Applied Microbiology and Biotechnology, Vol. 89, No. 5, 2011, pp. 1341-1355. doi:10.1007/s00253-010-3080-7 [51] D. M. Stoeckel, et al., “Semi-Quantitative Evaluation of Copyright © 2013 SciRes. JWARP  F. BULLOUGH ET AL. Copyright © 2013 SciRes. JWARP 48 Fecal Contamination Potential by Human and Ruminant Sources Using Multiple Lines of Evidence,” Water Re- search, Vol. 45, No. 10, 2011, pp. 3225-3244. doi:10.1016/j.watres.2011.03.037 [52] D. M. Gordon, “Geographical Structure and Host Speci- ficity in Bacteria and the Implications for Tracing the Source of Coliform Contamination,” Microbiology, Vol. 147, No. 5, 2001, pp. 1079-1085. [53] C. Hagedorn, A. R. Blanch and V. J. Harwood, “Micro- bial Source Tracking: Methods, Applications, and Case Studies,” Springer, New York, 2011. doi:10.1007/978-1-4419-9386-1 [54] I. J. Buerge, et al., “Combined Sewer Overflows to Sur- face Waters Detected by the Anthropogenic Marker Caf- feine,” Environmental Science & Technology, Vol. 40, No. 13, 2006, pp. 4096-4102. doi:10.1021/es052553l [55] S. Sauvé, et al., “Fecal Coliforms, Caffeine and Car- bamazepine in Stormwater Collection Systems in a Large Urban Area,” Chemosphere, Vol. 86, No. 2, 2012, pp. 118- 123. doi:10.1016/j.chemosphere.2011.09.033 [56] R. Edwards, “Immunoassays: Essential Data,” Wiley: Published in Association with BIOS Scientific Publishers, Chichester, Oxford, 1996. [57] A. Deng, et al., “Residue Analysis of the Pharmaceutical Diclofenac in Different Water Types Using ELISA and GC-MS,” Environmental Science & Technology, Vol. 37, No. 15, 2003, pp. 3422-3429. doi:10.1021/es0341945 [58] A. Bahlmann, et al., “Monitoring Carbamazepine in Sur- face and Wastewaters by an Immunoassay Based on a Monoclonal Antibody,” Analytical and Bioanalytica l Chem - istry, Vol. 395, No. 6, 2009, pp. 1809-1820. doi:10.1007/s00216-009-2958-7 [59] V. Calisto, et al., “Application of an ELISA to the Quan- tification of Carbamazepine in Ground, Surface and Wastewaters and Validation with LC-MS/MS,” Chemos- phere, Vol. 84, No. 11, 2011, pp. 1708-1715. doi:10.1016/j.chemosphere.2011.04.072 [60] J. Carvalho, et al., “A Highly Sensitive Caffeine Immu- noassay Based on a Monoclonal Antibody,” Analytical and Bioanalytical Chemistry, Vol. 396, No. 7, 2010, pp. 2617-2628. doi:10.1007/s00216-010-3506-1 [61] J. Kuby, et al., “Immunology,” W. H. Freeman, New York, 2007. [62] J. R. Crowther and N. J. Totowa, “ELISA: Theory and Practice,” Humana Press, New York, 1995. [63] S. Rebe Raz and W. Haasnoot, “Multiplex Bioanalytical Methods for Food and Environmental Monitoring,” TRAC Trends in Analytical Chemistry, Vol. 30, No. 9, 2011, pp. 1526-1537. doi:10.1016/j.trac.2011.04.016
|