Journal of Cancer Therapy
Vol.07 No.10(2016), Article ID:70922,10 pages
10.4236/jct.2016.710071
LKB1, a New Biomarker in breast cancer
Hanine Lattouf1,2,3,4, Coralie Poulard5, Isabelle Treilleux6, Nader Hussein4, Mona Diab-Assaf4, Muriel Le Romancer1,2,3*
1Inserm U1052, Centre de Recherche en Cancérologie de Lyon, Lyon, France
2CNRS UMR5286, Centre de Recherche en Cancérologie de Lyon, Lyon, France
3Université Lyon 1, Lyon, France
4Laboratory of Molecular and Cellular Biology, Doctoral School of Sciences and Technologies, Lebanese University, Beirut, Lebanon
5Department of Biochemistry and Molecular Biology, Comprehensive Cancer Center, University of Southern California, Los Angeles, CA, USA
6Centre Léon Bérard, Pathology Department, Lyon, France

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: June 13, 2016; Accepted: September 25, 2016; Published: September 28, 2016
ABSTRACT
Breast cancer is the most common cancer in women worldwide. Estrogen signaling pathways have been identified as efficient targets of breast cancer therapy, given their key role in promoting breast tumor growth. Agents blocking estrogen-mediated pathways are routinely used in clinical applications in patients displaying estrogen-sensitive breast cancer subtypes; however intrinsic or acquired resistance to treatment often occurs or develops, thus limiting their efficacy. This limitation has highlighted an imperative need to identify new predictive biomarkers. Recent findings have highlighted a role for the Liver Kinase B1 (LKB1) in breast cancer tumorigenesis. LKB1 is a serine/threonine kinase mutated in Peutz-Jeghers syndrome (PJS), implicated in many cellular processes including energy metabolism, cell polarization and cell cycle arrest and has also been shown to play an essential role as a tumor suppressor gene by negatively regulating the mTOR pathway. This review provides an overview of previous findings and ongoing research on LKB1, and substantiates the use of this kinase as a potential prognostic and predictive biomarker of breast cancer.
Keywords:
LKB1, biomarker, breast Cancer, estrogen Signaling

1. Introduction
Breast cancer (BC) is the most common cancer in women worldwide. In 2012, breast cancer was reported to cause the death of more than 522,000 women by the International Agency for Research on Cancer. Siegel et al. found that BC accounts for 29% of all new cancers in women in 2015 [1] . Estrogens stimulate the proliferation and the differentiation of breast epithelial cells through the activation of downstream mitogenic signaling pathways, namely via the estrogen receptors ERα and ERβ. These receptors function as ligand-dependent transcription factors [2] - [4] , acting either through the transcriptional regulation of genes (genomic pathway) or through membrane and cytoplasmic signaling cascades (non-genomic pathway). Current BC treatment options exclusively target the genomic signaling pathway. Even though ERα is expressed at low levels in normal mammary cells [5] , 70% of BC express high levels of ERα and are either treated by agents that suppress the function of the receptor (anti-estrogen) or the estrogen synthesis (aromatase inhibitors) [6] .
Despite the fact that ERα is a standard predictive marker used to prescribe endocrine therapy, about 40% of these patients initially display or eventually become resistant to treatment [7] [8] . This represents a burden on public healthcare and a serious issue to be considered when designing the treatment plan of the patients, highlighting an urgent need to identify new predictive markers of therapeutic response, to ensure a better survival rate for the patients. Therefore, the identification of new actors of estrogen signaling pathways should improve treatment specificity and thus overcome cancer drug resistance. For instance, it has been extensively shown that the PI3K/Akt/mTOR pathway is frequently activated in BC, leading to the use of the mammalian target of rapamycin (mTOR) inhibitors, such as everolimus, in clinical trials [9] [10] . Although the latter has become a standard treatment in hormone-resistant BC, its use is accompanied with many side effects (stomatitis, rash, diarrhea, arthralgia/myalgia and anemia) and only 50% of patients respond to the treatment. LKB1 is a known regulator of both mTOR [11] [12] and estrogen signaling pathways [13] [14] , and given its role in BC tumorigenesis [15] , it may emerge as an attractive novel cancer biomarker.
2. Breast cancer Subtypes and Therapies
Breast cancers can be classified into several subtypes based on their epidemiological risk factors, molecular markers and response to systemic and local therapies. Norum et al. presented an overview of the intrinsic molecular subtypes of breast cancer, as well as their prognostic and therapeutic implications [16] summarized in the Table 1. The most common breast cancer subtype is the luminal A, with the best prognosis, a low grade and an increased survival rate compared to other subtypes. Luminal A tumors are characterized by an estrogen receptor-positive (ERα+), a progesterone receptor-positive (PR+) and a human epidermal growth factor receptor 2-negative (HER2−) profile. The luminal B cancer subtype shares some similarities with the luminal A subtype. It is also ERα- and/or PR-positive, HER2-negative and has a high level of Ki67 expression, reflecting a high number of proliferating cancer cells. Endocrine therapy is usually the recommended treatment for these subtypes. It entails either targeting the estrogen signaling pathway, using ERα-blocking drugs in premonopausal women, primarily the selective estrogen receptor modulator tamoxifen, or inhibiting estrogen synthesis, using aromatase inhibitors in postmenopausal women, such as exemestane and anastrozole,

Table 1.Breast cancer subtypes: molecular marks, clinical features, general treatment and prognosis.
in order to reduce estrogen levels [17] .
The third subtype is known as the HER2-enriched subtype. It is generally negative for ERα/PR and positive for the HER2, with the amplification of the HER2 gene leading to the protein over expression and, thereby, to an uncontrolled mammary cell growth and division. It is associated with a poor prognosis and HER2-enriched BC patients frequently develop metastases. These tumors are targeted by monoclonal antibodies, such as trastuzumab [18] , and tyrosine kinase activity inhibitors, like lapatinib [19] . However, a more recent therapeutic strategy has been adopted, in which the specificity of monoclonal antibodies is combined with the efficacy of cytotoxic agents, such as the microtubule polymerization inhibitor called emtansine (DM1) [20] . Another monoclonal antibody targeting HER2, namely pertuzumab, has entered clinical trials. When combined to trastuzumab, this latter molecule significantly increases progression-free survival (PFS) and overall survival (OS) of HER2-positive metastatic/advanced breast cancer patients [21] [22] .
The final subtype contains tumors with a negative profile for ERα, PR and HER-2, also known as triple negative tumors, and constitutes the most aggressive cancer subtype, with the worst patient prognosis. So far, no specific targeted therapy has been established to cure these patients, but many drugs have been tested and the results were compared to define an optimal therapy [23] . The authors showed that some new drugs are emerging as potential treatments, such as anti-androgens in patients with androgen receptor positive cancers, Src family kinase inhibitors in patients having mesenchymal-like cancer subtypes, PARP inhibitors and platinum agents in patients who have BRCA mutations [23] .
Tinoco et al. reported that over the past two decades, breast cancer mortality rates have declined by approximately 30%, with corresponding improvements in the 5-year OS rate of patients of up to 90% [24] . However, all breast cancer therapies are eventually associated with treatment resistance, coupled with the activation of estrogen- independent growth and of survival signaling pathways. In addition, metastatic breast cancer remains incurable with an estimated 5-year OS rate of only 23% [24] [25] . Altogether these data present the current significant challenges encountered by researchers/clinicians and emphasize the need to identify new predictive biomarkers of the response of patients to treatment.
3. Estrogen Signaling in Breast Cancer
Estrogen is a member of the steroid hormones, which play important roles in cell proliferation and differentiation in many target tissues [26] . Estrogen exerts its effects by binding to its receptors ERα and ERβ, which belong to the nuclear receptor superfamily, leading to their activation. This, in turn, sequentially results in their conformational change, dimerization and their binding to specific estrogen response elements (EREs), which are located in the targeted gene promoter regions [27] . There, ERs recruit co- regulator complexes to modulate the transcriptional activity. This process is known as the estrogen genomic signaling pathway.
However, estrogen also exerts non-genomic activities in the cytoplasm, where it binds to cytoplasmic ERα, leading to its dimerization and subsequent interaction with phosphatidylinositol 3-kinase (PI3K) and with the tyrosine kinase Src close to the plasma membrane (Figure 1). The formation of the complex results in the activation of downstream protein kinase cascades, particularly the Src/Ras/MAPK and the PI3K/Akt pathways [28] [29] . Le Romancer’s group also showed that ERα is regulated by the protein arginine methyltransferase 1 (PRMT1), through the methylation of Arg260 within the DNA binding domain [28] . Interestingly, they confirmed that this methylation is a prerequisite for the formation of the ERα non-genomic complex containing ERα/Src/ PI3K (Figure 1). In a cohort of 175 patients with invasive breast tumors, it was shown that the interaction between ERα and PI3K/Src was correlated with ERα methylation and the downstream activation of Akt. Of note, the ERα/Src/PI3K complex is highly expressed in aggressive breast tumors, independently of ERα expression, and may, therefore, constitute an efficient target for the treatment of BC [30] .
4. The Tumor Suppressor LKB1
4.1. History and Physiological Role
The lkb1 gene, also called STK11, codes for a highly conserved serine/threonine kinase,
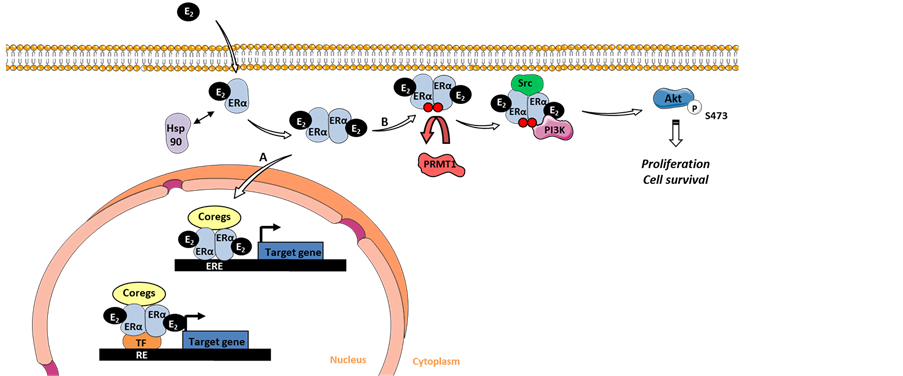
Figure 1. Estrogen signaling pathways. (A) Estrogen binds to its receptor ERα in the cytoplasm leading to its dimerization and its translocation into the nucleus to act through a genomic mechanism, where it binds directly on estrogen responsive elements (EREs) or indirectly via other transcription factors on responsive elements (RE), and initiate the transcription of target genes after recruitment of coregulator complexes. (B) Estrogen participates in the non-genomic pathway, in which the dimerized receptors stay in the cytoplasm where they are methylated by PRMT1 allowing it to form a complex with Src and PI3K, thus activating downstream Akt-de- pendent proliferation and cell survival pathways. E2: 17 β-oestradiol; Hsp90: Heat shock protein 90; ERα: Estrogen receptor α; PRMT1: Protein arginine methyltransferase 1; Coregs: Coregulators; TF: Transcription factor; ERE: Estrogen response elements; RE: Response element; PI3K: Phosphatidylinositide 3-kinase.
which is mutated in patients suffering from Peutz-Jeghers syndrome (PJS), a dominant autosomal inherited syndrome. PJS is characterized by mucocutaneous pigmentations on the lips, finger tips and oral mucosa, in addition to the appearance of multiple gastro-intestinal hamartomatous polyps and an increased risk of developing various types of cancers, including BC (32% - 54%) [31] [32] .
The human LKB1 consists of 433 amino acids, encompassing a nuclear localization signal (NLS) (38 - 43 aa) and a kinase domain (49 - 309 aa), which enables LKB1 to phosphorylate downstream kinases, such as the AMP-activated protein kinases AMPKs (Figure 2). LKB1 is ubiquitously expressed in fetal and adult tissues. During the developmental process, several organs display a predominant expression of LKB1 at the protein level, such as the heart, the lungs, the pancreas, the kidneys and gastrointestinal tract, while in adults, the highest levels of LKB1 protein expression are mostly found in epithelial tissues, as well as in the testes [33] [34] . Although a nuclear subcellular localization of LKB1 has principally been reported, LKB1 was also shown to have a cytoplasmic localization [31] [32] [35] . Since LKB1 lacks a nuclear export signal (NES), it interacts with two partners, the pseudokinase STE20-related adaptor (STRADα), lacking several crucial residues for the intrinsic catalytic activity, and the mouse protein 25 (MO25). This allows LKB1 to translocate into the cytoplasm and phosphorylate its substrates [36] [37] . Indeed, the adaptor protein MO25 binds to STRADα, thus forming a high-affinity ternary complex with LKB1 and driving the relocalization of nuclear LKB1

Figure 2. LKB1 post-translational modifications. Circles in red refer to autophosphorylation sites, blue circles reflect phosphorylation by other kinases and green circles symbolize prenylation sites. The dark blue square represents acetylation/deacetylation site.
into the cytoplasm. This event induces a 10-fold increase in the catalytic activity of this kinase toward its substrates [38] .
Furthermore, the activation of LKB1 in unpolarized epithelial cells was demonstrated to trigger cell repolarization, confirming its crucial role in regulating cell polarity, and the concomitant remodeling of the act in cytoskeleton to gap junctions [39] - [42] . In addition, LKB1 is involved in metabolic pathways through the phosphorylation of AMPK, a key regulator of cellular metabolic/energy homeostasis. The latter process results in the inactivation of the mammalian target of rapamycin (mTOR), a crucial protein involved in the initiation of protein synthesis, cell proliferation and survival mechanisms, thus highlighting the role of LKB1 as a tumor suppressor [11] [12] [41] [43] . Loss of LKB1 leads to an increase in the mTOR signaling pathways, a mechanism modulated by an increased phosphorylation of its effectors, such as the translational regulator S6K1 and the eukaryote translation initiation factor 4E-binding protein 1 (4E-BP1).
LKB1 is also involved in DNA damage checkpoint regulation, in which cells lacking LKB1 accumulate more DNA-damage than control cells [44] . Induced expression of wild type LKB1 in G361 melanoma cells results in cell growth inhibition, reflecting its implication in cell growth arrest [45] . LKB1 is also involved in other signaling pathways, e.g. the TGF-β pathway, by interacting with the liver-enriched inhibitory protein (LIP-1), a novel leucine-rich repeat containing protein, in the cytoplasm [46] . Indeed, LKB1 forms a complex with LIP-1 and the transcription factor Smad-4, inhibiting the translocation of Smad-4 to the nucleus and thus regulating the TGF-β pathway.
4.2. Regulation by Post-Translational Modifications (PTMs)
LKB1 is the target of several PTMs, such as phosphorylation, acetylation and prenylati- on, which serve to regulate its kinase activity, as well as its localization (Figure 2, Table 2).
LKB1 is autophosphorylated on Serine (Ser) and Threonine (Thr) residues, and is also phosphorylated by other kinases, regulating both its localization and/or enzymatic activity. Here, we will focus on the sites known to be associated with functional consequences.
Ser307:
In response to several stimuli, such as peroxynitrite (ONOO), metformin (an antidiabetic drug known as Glucophage) or AICAR (an analog of adenosine monophosphate), the Ser307 residue is phosphorylated by PKC-ζ, inducing the nucleocytoplasmic transport of LKB1 and the subsequent activation of AMPK, the suppression of angiogenesis and the regulation of cell cycle progression and proliferation [47] . This phosphorylation is also responsible for the activation of AMPK by LKB1 in the presence of berberine, a drug used to treat gut infections, diarrhea and diabetes [48] .

Table 2.Post-translational modification sites of LKB1. This table shows sites of phosphorylation, acetylation and prenylation within LKB1 and the effects of their PTM. The enzymes with an italic font have been reported on mouse LKB1 residues.
Sapkota et al. showed that LKB1 is phosphorylated on the Ser325 residue, independently of its catalytic activity [49] . Zheng et al. identified this residue as a direct phosphorylation target of ERK in cells transformed by oncogenic B-RAF mutations, and revealed that this modification suppresses the ability of LKB1 to bind and, thereby, to activate AMPK [50] . Furthermore, they showed that this modification is important for cell proliferation and anchorage-independent growth, leading to their transformation. Indeed, cells that expressed the phosphorylation-deficient amino acid mutant proliferate significantly less than cells expressing the wild type LKB1 and form fewer colonies on soft agar than the control group.
Ser334:
The serine 334 residue is phosphorylated by the survival kinase Akt. This phosphorylation enables LKB1 to interact with the 14-3-3 protein, resulting in its increased nuclear retention, preventing the formation of the LKB1/STRADα complex and abolishing its tumor suppressor activity in vitro in breast cancer cell cultures and in vivo in mammary carcinoma in nude mice [51] .
Thr336:
Upon activation of LKB1 by STRADα, LKB1 is autophosphorylated on the Thr336 residue [49] . Bai et al. demonstrated that P-Thr336 is recognized by the 14-3-3 protein, fostering its binding to LKB1 and the inhibition of the LKB1 kinase activity without affecting the level of its autophosphorylation [52] .
Thr363:
Sapkota and colleagues reported that the Thr366 residue is phosphorylated in mouse LKB1 (corresponding to Thr363 in human LKB1) [49] . This phosphorylation does not regulate the activity of LKB1 or its nuclear localization in HeLa cell lines. Rather, exposure of cells to ionizing radiation results in the phosphorylation of Thr363 in vitro and in vivo by the ataxia telangiectasia mutated kinase (ATM) and in vitro by the DNA- dependent protein kinase DNA-PK [53] . As a result, this modification might be implicated in enabling LKB1-mediated cell growth suppression. In addition, Baas et al. demonstrated that Thr363 is also an autophosphorylation site [36] .
Ser428:
Following stimulation with EGF/12-O-tetradecanoylphorbol-13-acetate (TPA) or with the adenylate cyclase activator, forskolin, LKB1 is phosphorylated on the Ser431 residue (corresponding to Ser428 in human LKB1) by p90 ribosomal S6 kinase (P90RSK) and cAMP-dependent protein kinase (PKA), respectively [45] . This modification mediates the ability of LKB1 to suppress cell growth. Indeed, the transfection of G361 cells with a LKB1 protein containing a mutation on this site led to a 7-fold increase in the number of colonies formed in comparison with cells transfected with the wild-type LKB1. Shen et al. demonstrated that the phosphorylation of this residue by PKA regulates the asymmetric localization of LKB1 in Schwann cells and the initiation of myelination in vitro [54] . Moreover, upon activation by metformin, Xie et al. showed that PKC-ζ is also able to phosphorylate LKB1 on Ser428 and that this phosphorylation is required to enhance the activation of AMPK, through an increase in its phosphorylation on Thr172 [50] [55] . Indeed, this modification on Ser428 leads to the nuclear export of LKB1 and the subsequent phosphorylation of its target AMPK on Thr172 in endothelial cells [55] .
LKB1 acetylation has been the subject of fewer studies than its phosphorylation. By mass spectrometry, Lan et al. showed that LKB1 is acetylated on at least nine lysine residues over its three domains, although most studies have focused on the Lys48 residue on the N-terminal part, which interacts with SIRT1, a NAD+-dependent histone/pro- tein deacetylase [56] . Even though Lys48 deacetylation is known to be catalyzed by SIRT1, the identity of the acetylase involved remains unknown. Acetylation prevents STRADα from interacting with LKB1 and leads to an increased nuclear localization of LKB1, thereby, inhibiting its kinase activity. In contrast, the deacetylation of this lysine by SIRT1 causes a 2-fold increase in the binding of LKB1 to STRADα, in its cytoplasmic localization and in its activity.
In line with these findings, Zu et al. revealed that the levels of LKB1 and SIRT1 were inversely correlated during the progression of senescence in endothelial cells with an increase in the level of LKB1 and a decrease in that of SIRT1 [57] . In addition, these authors showed that the deacetylation of LKB1 reduces cell senescence by decreasing the activation of AMPK, and mediates the downregulation of LKB1, through ubiquitination and proteasome-mediated degradation. Recently, Bai et al. showed that the acetylation of LKB1 on the Lys64 residue triggers the formation a protein complex with SIRT1 leading to subsequent LKB1 proteasomal degradation [58] .
LKB1 contains a CAAX motif in its C-terminal domain, usually required for protein prenylation. Indeed, this modification takes place on the cysteine residue Cys430 of LKB1. Two studies demonstrated that the substitution of Cys430 with a serine residue reduced the level of LKB1 associated with the plasma membrane, thus confirming the role of prenylation in targeting LKB1 to the membrane [59] [60] . Based on in vivo studies, Houde et al. reported that knock-in mice, in which Cys433 (corresponding to Cys430 in human) was replaced by Ser433, displayed a lower level of LKB1 associated with the membrane, as well as a significantly reduced AMPK activity, suggesting that LKB1 localization is a prerequisite for its kinase activity. Upon activation by AICAR, an AMPK activator, these mice showed muscle contraction along with a significant decrease in lipid synthesis in their liver [60] .
Altogether, the identification of different PTMs of LKB1 shows that its localization appears to be closely linked to its kinase activity and this may affect its anti-tumoral properties.
4.3. Role of LKB1 in Cancer: Focus on breast Cancer in mouse Models
Patients with a predisposition to PJS, suffering from germline mutations of lkb1, presenta higher risk of developing cancer at a young age, such as lung, gastrointestinal, gynecological and breast cancers [32] [61] [62] . In addition, BC patients are reported to have frequent loss of heterozygosity in a chromosome locus, in which lkb1 is located [63] . These results are consistent with several reports showing that LKB1 possesses anti-tumoral properties.
Indeed, LKB1 was found to be overexpressed in breast cancer cells upon exposure to adiponectin [64] . As a result, LKB1 overexpression mediates the activation of the AMPK signaling pathway, thus inhibiting the activation of mTOR, leading to a decrease in the adhesion, invasion and migration abilities of cancerous cells. Finally, this anti-tumoral effect was reversed by the knockdown of LKB1.
Another study revealed that LKB1 limits the migration and the invasive capacity of breast cancer cell lines by decreasing levels of secreted matrix metalloproteinases and downregulating the expression of angiogenesis factors, such as VEGF, both in vitro and in vivo [65] . These authors demonstrated that LKB1 acts as a negative regulator of human BC, since LKB1-transfected breast cancer cells grew at a slower rate than control cells in nude mice, and in addition, LKB1-transfected xenografts displayed a significant regression of lung metastasis.
Conditional deletion of lkb1 in primary mouse mammary epithelial 3D cultures leads to the disruption of both epithelial architecture and basement membrane structure [66] . In vivo, this deletion disrupts the epithelial integrity and causes the appearance of a discontinuous basement membrane around lkb1-deficient mammary epithelial ducts. Loss of lkb1 is also significantly associated with the activation of c-Myc, which induces mammary tumorigenesis [66] . Indeed, the combination of Myc overexpression and loss of lkb1 intensely accelerated the formation of mammary tumors and shortened the mean latency of tumor formation in comparison to the wild type group.
McCarthy et al. generated transgenic mice carrying the lkb1 allele with provisional deletion of the exons coding for the LKB1 kinase domain, exclusively in the mammary gland [15] . Mammary gland tumors were developed, sharing some histological patterns with breast cancers developed in PJS patients. This mouse model offers a precious tool to test new therapeutic strategies and to understand the biological impact of the loss-of-function of lkb1in the development and the progression of mammary gland cancer.
Another breast cancer mouse model was developed in 2013, combining the loss of lkb1 with an activating mutation of the Her2 oncogene [67] . Here, the loss of LKB1 activity reduced the latency of tumor formation and significantly stimulated tumor growth. In parallel, breast tumor formation was strongly correlated with the hyperactivation of the mTOR signaling pathway and the alteration of cell metabolism, leading to tumors with a high metabolic activity. This model exposes the loss of lkb1 in HER2-positive tumors as a potential new marker for mTOR hyperactivation, thereby encouraging new therapeutic agents to target the LKB1-mTOR signaling pathway in BC.
However, several studies reported that LKB1 could also have oncogenic properties. In several breast cancer cell lines, Nath-Sain et al. showed that, in response to estrogen, LKB1 acts as a coactivator of ERα in the nucleus and thus interferes with the estrogen genomic signaling pathway [13] [33] . This results in an enhanced ERα-mediated transactivation of targeted genes, such as cathepsin D, c-myc and cyclin D1, thereby increasing cell proliferation and highlighting a signaling pathway, in which LKB1 acts as a tumor enhancer. Furthermore, LKB1 was shown to drive the proliferation of UVB-in- duced murine basal cell carcinoma, by leading to a constitutive activation of the Wnt/β-catenin signaling pathway [68] . In addition, LKB1 can play an important role in the phosphorylation of pro-apoptotic proteins by Akt, such as Bad, FoxO1 and others, which suppresses cell death in tumor cells with a constitutively active Akt [69] .
4.4. LKB1, A potentially Novel Biomarker of BC
PJS patients develop breast cancers that display either a loss or a reduced expression of LKB1 protein, the sporadic mutations being rarely detected [32] [35] . Indeed, in a cohort of 62 patients with primary BC and 17 established BC cell lines, Bignel et al. screened the full coding sequence and splice junctions of LKB1 for somatic mutations, to no avail [70] . A whole-genome sequence analysis was conducted on 560 BC samples to improve our understanding of the origins and the effects of somatic mutations in BC [71] . The authors identified 916 probable mutations as driver mutations in BC, including the mutation of the lkb1 gene, thereby giving rise to a protein mutated on the Cys151 residue.
Shen et al. studied the expression of LKB1 in a cohort of 116 invasive breast cancers and assessed the correlation with established clinicopathological prognostic factors [72] . They found that a low level of the protein LKB1 is associated with poor prognostic factors, such as high histological grade, increased tumor size, the presence of lymph node metastasis, in addition to a shorter RFS and poorer OS. In this study, LKB1 expression was analyzed in tumors by Western blot, an approach which does not take into consideration the heterogeneity of the tumors or the subcellular localization of LKB1.
More recently, Xiao et al. conducted a meta-analysis of 14 studies that encompassed 1915 patients and included 7 different solid tumors, such as BC and lung cancer [73] . They demonstrated that the decreased expression of LKB1 messenger RNA was associated with a poor OS, as well as a larger tumor size, lymph node metastasis and higher TNM stage, and they suggested that the low level of LKB1 could be used as a potential predictive marker for poor prognosis in patients with solid tumors.
Bouchekioua-Bouzaghou et al. described, for the first time, a dual localization of LKB1 both in the nucleus and in the cytoplasm in breast tumors. Moreover, they identified cytoplasmic LKB1 as a partner of methylated ERα in the estrogen non-genomic complex [14] . This interaction takes place not only in breast cell lines but also in primary breast tumors specifically expressing the methylated form of ERα. The authors showed that LKB1 does not alter the integrity or the stability of the complex, but that it acts as an auxiliary partner. Interestingly, they also studied the expression of LKB1 at the protein level in 154 BC samples [14] and revealed that a nuclear LKB1 expression was inversely correlated with its cytoplasmic expression. Furthermore, it was shown that high levels of cytoplasmic LKB1 are an independent poor prognostic factor of OS and disease-free survival (DFS) and are associated with bad prognostic factors, such as high SBR grade and negative ERα status, while the nuclear form of LKB1 is associated with good prognostic factors. The dual localization of LKB1 was confirmed in the MCF7 (cytoplasmic localization) and ZR75-1 (nuclear localization) BC cell lines. The expression of LKB1 in the cytoplasm was also associated with the expression of ERα/Src/PI3K, corroborating in vitro results showing that LKB1 is a partner of the non-genomic complex. Bouchekioua-Bouzaghou et al. speculated that LKB1 was trapped inside the nongenomic complex, thus inhibiting LKB1 from exerting its anti- proliferative role (Figure 3). Indeed, the level of phosphorylated 4E-BP1, a direct target of mTOR, was positively correlated with the expression of cytoplasmic LKB1 in the tested cohort. Accordingly, the presence of LKB1 within the estrogen non-genomic complex could impair the ability of LKB1 to phosphorylate its substrates, thus altering its anti-tumoral activity.
More recently, Azim et al. conducted a study on 35 Egyptian luminal breast tumors to establish correlations between the PI3K/AKT/m-TOR pathway effectors, mainly LKB1, and the known clinicopathological factors of the patients studied [74] . This study confirmed the dual localization of LKB1 and the authors demonstrated that the high level of nuclear LKB1 protein expression was predictive of a better DFS. They also found that it is associated with parameters of good prognosis, such as smaller tumor size, more ERα and PR positivity, a predominant HER2 negativity and a decreased risk of recurrence or death, thereby confirming the results obtained earlier by Bouchekioua- Bouzaghou et al. [14] .
In 2012, Bachelot et al. conducted the first randomized phase II trial combining eve- rolimus, an inhibitor of mTOR, with the endocrine therapy agent, tamoxifen, on 111
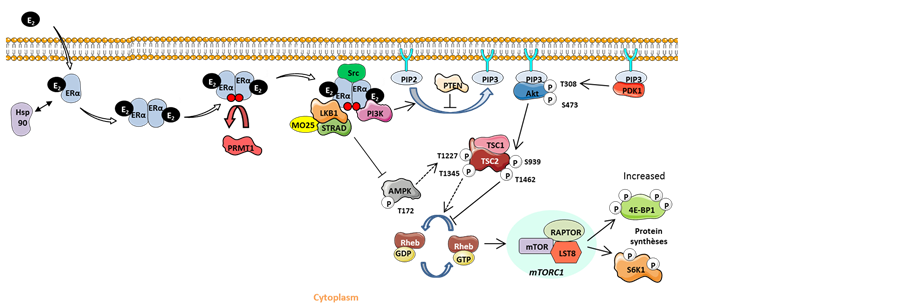
Figure 3. Model of the relationship between cytoplasmic LKB1 and the non-genomic ERα/Src/ PI3K complex. The association of LKB1 with the estrogen non-genomic complex prevents its ability to phosphorylate its substrates as AMPK, thus preventing the inhibition of the mTOR signaling pathway and leading to the increase of 4E-BP1 activation. E2: 17 β-oestradiol; Hsp90: Heat shock protein90; ERα: Estrogen receptor α; PRMT1: Protein arginine methyltransferase 1; MO25: Mouse protein 25; STRAD: STE-related adaptor; PI3K: Phosphatidylinositide 3-kinase; PIP2: Phosphatidylinositol-4, 5-bisphosphate; PIP3: Phosphatidylinositol-3, 4, 5-trisphosphate; PTEN: Phosphatase and tensin homolog; AMPK: AMP-activated protein kinase; GDP: Guanosine diphosphate; GTP: Guanosine triphosphate; TSC1/2: Tuberous sclerosis 1/2; mTOR: Mammalian target of rapamycin; PDK1: S6K1: S6 kinase 1.
postmenopausal women, suffering from metastatic BC that became resistant to the aromatase inhibitors treatment [75] . The trial aimed to evaluate the efficacy of the combination treatment on enhancing the prognosis of endocrine therapy-resistant patients. After a follow up of 6 months, this study, entitled TAMRAD, reported that the clinical benefit rate, the time to progression of cancer and the OS of patients increased for patients receiving the combination therapy compared to the tamoxifen monotherapy group. These results were confirmed by Baselga et al. who conducted a phase III randomized trial on 724 patients with hormone receptor-positive advanced BC [76] and found that the combination of everolimus with the aromatase inhibitors also enhanced the PFS of patients. From the TAMRAD trial, Treilleux et al. conducted a retrospective evaluation on 55 primary tumor samples to identify potential predictive biomarkers of the response to everolimus [77] . Focusing exclusively on the mTOR pathway, they found that low levels of cytoplasmic LKB1 expression was associated with a high level of P-4EBP1, low cytoplasmic P-Akt and low PI3K levels. The patients showing these expression profiles appeared to benefit more from the treatment with tamoxifen and everolimus. This evaluation highlighted the fact that LKB1 could be a predictive marker of the efficacy of administering everolimus in advanced BC.
Of note, it is hard to associate the expression profile of LKB1 with a specific BC subtype, since the cohorts studied give rise to contradictory results. This could be due to
the difference in the cohorts’ size, the methods used to detect LKB1 (western blot, immunohistochemistry, RT-PCR…) and the fact that LKB1 localization is not always taken into account [14] [72] [78] .
5. Conclusion
At present, ERα is the only factor routinely used to predict and prescribe endocrine therapy. Based on recent findings, it seems that combining targeted therapy to hormonotherapy could significantly increase the survival rate of patients with BC. However, the occurrence of side effects and the cost of targeted therapies disable the clinicians from targeting the right patients, giving thereby rise to an urgent need for novel and efficient predictive markers. LKB1, as a regulator of the mTORC1 pathway, could be a potential predictive marker of the efficacy of everolimus in advanced breast cancer. Based on recent results, it seems that the dual localization of LKB1 within tumor cells should be taken in account to determine its prognostic value, since it can act either as a tumor suppressor or as an oncogene, according to its localization. It would be of interest to determine which PTM regulates LKB1 shuttling in BC.
Acknowledgements
We thank the Lebanese University, Fondation de France, Fondation Arc Cancer and The National Institutes of Health for their support to this project.
Cite this paper
Lattouf, H., Poulard, C., Treilleux, I., Hussein, N., Diab- Assaf, M. and Le Romancer, M. (2016) LKB1, A New Biomarker in Breast Cancer. Journal of Cancer Therapy, 7, 680-699. http://dx.doi.org/10.4236/jct.2016.710071
References
- 1. Siegel, R.L., Miller, K.D. and Jemal, A. (2015) Cancer Statistics, 2015. CA: A Cancer Journal for Clinicians, 65, 5-29.
http://dx.doi.org/10.3322/caac.21254 - 2. Bocchinfuso, W.P. and Korach, K.S. (1997) Mammary Gland Development and Tumorigenesis in Estrogen Receptor Knockout Mice. Journal of Mammary Gland Biology and Neoplasia, 2, 323–334.
http://dx.doi.org/10.1023/A:1026339111278 - 3. Colditz, G.A. (1998) Relationship between Estrogen Levels, Use of Hormone Replacement Therapy, and Breast Cancer. Journal of the National Cancer Institute, 90, 814-823.
http://dx.doi.org/10.1093/jnci/90.11.814 - 4. Renoir, J.M., Marsaud, V. and Lazennec, G. (2013) Estrogen Receptor Signaling as a Target for Novel Breast Cancer Therapeutics. Biochemical Pharmacology, 85, 449–465.
http://dx.doi.org/10.1016/j.bcp.2012.10.018 - 5. Crandall, D.L., Busler, D.E., Novak, T.J., Weber, R.V. and Kral, J.G. (1998) Identification of Estrogen Receptor Beta RNA in Human Breast and Abdominal Subcutaneous Adipose Tissue. Biochemical and Biophysical Research Communications, 248, 523-526.
http://dx.doi.org/10.1006/bbrc.1998.8997 - 6. Harvey, J.M., Clark, G.M., Osborne, C.K. and Allred, D.C. (1999) Estrogen Receptor Status by Immunohistochemistry Is Superior to the Ligand-Binding Assay for Predicting Response to Adjuvant Endocrine Therapy in Breast Cancer. Journal of Clinical Oncology, 17, 1474-1481.
- 7. Ali, S. and Coombes, R.C. (2002) Endocrine-Responsive Breast Cancer and Strategies for Combating Resistance. Nature Reviews Cancer, 2, 101-112.
http://dx.doi.org/10.1038/nrc721 - 8. Musgrove, E.A. and Sutherland, R.L. (2009) Biological Determinants of Endocrine Resistance in Breast Cancer. Nature Reviews Cancer, 9, 631-643.
http://dx.doi.org/10.1038/nrc2713 - 9. Burstein, H.J. (2011) Novel Agents and Future Directions for Refractory Breast Cancer. Seminars in Oncology, 38, 17-24.
http://dx.doi.org/10.1053/j.seminoncol.2011.04.002 - 10. Yardley, D.A., Bosserman, L.D., O’Shaughnessy, J.A., Harwin, W.N., Morgan, S.K., Priego, V.M., Peacock, N.W., Bass, J.D., Burris Iii, H.A. and Hainsworth, J.D. (2015) Paclitaxel, Bevacizumab, and Everolimus/Placebo as First-Line Treatment for Patients with Metastatic HER2-Negative Breast Cancer: A Randomized Placebo-Controlled Phase II Trial of the Sarah Cannon Research Institute. Breast Cancer Research and Treatment, 154, 89-97.
http://dx.doi.org/10.1007/s10549-015-3599-5 - 11. Shaw, R.J., Kosmatka, M., Bardeesy, N., Hurley, R.L., Witters, L.A., DePinho, R.A. and Cantley, L.C. (2004) The Tumor Suppressor LKB1 Kinase Directly Activates AMP-Activated Kinase and Regulates Apoptosis in Response to Energy Stress. Proceedings of the National Academy of Sciences U.S.A., 101, 3329-3335.
- 12. Dong, L., Sun, L., Zhang, X., Pan, L., Lian, L., Chen, Z. and Zhong, D. (2013) Negative Regulation of mTOR Activity by LKB1-AMPK Signaling in Non-Small Cell Lung Cancer Cells. Acta Pharmacologica Sinica, 34, 314-318.
http://dx.doi.org/10.1038/aps.2012.143 - 13. Nath-Sain, S. and Marignani, P.A. (2009) LKB1 Catalytic Activity Contributes to Estrogen Receptor α Signaling. Molecular Biology of the Cell, 20, 2785-2795.
http://dx.doi.org/10.1091/mbc.E08-11-1138 - 14. Bouchekioua-Bouzaghou, K., Poulard, C., Rambaud, J., Lavergne, E., Hussein, N., Billaud, M., Bachelot, T., Chabaud, S., Mader, S., Dayan, G., Treilleux, I., Corbo, L. and Le Romancer, M. (2014) LKB1 When Associated with Methylated ERα Is a Marker of Bad Prognosis in Breast Cancer. International Journal of Cancer, 135, 1307-1318.
- 15. McCarthy, A., Lord, C.J., Savage, K., Grigoriadis, A., Smith, D.P., Weigelt, B., Reis-Filho, J.S. and Ashworth, A. (2009) Conditional Deletion of the Lkb1 Gene in the Mouse Mammary Gland Induces Tumour Formation. Journal of Pathology, 219, 306-316.
http://dx.doi.org/10.1002/path.2599 - 16. Norum, J.H. andersen, K. and Sorlie, T. (2014) Lessons Learned from the Intrinsic Subtypes of Breast Cancer in the Quest for Precision Therapy. British Journal of Surgery, 101, 925-938.
http://dx.doi.org/10.1002/bjs.9562 - 17. Lumachi, F., Santeufemia, D.A. and Basso, S.M. (2015) Current Medical Treatment of Estrogen Receptor-Positive Breast Cancer. World Journal of Biological Chemistry, 6, 231-239.
http://dx.doi.org/10.4331/wjbc.v6.i3.231 - 18. Goldhirsch, A., Wood, W.C., Coates, A.S., Gelber, R.D., Thürlimann, B., Senn, H.J. and Panel Members (2011) Strategies for Subtypes—Dealing with the Diversity of Breast Cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Annals of Oncology, 22, 1736-1747.
http://dx.doi.org/10.1093/annonc/mdr304 - 19. Geyer, C.E., Forster, J., Lindquist, D., Chan, S., Romieu, C.G., Pienkowski, T., Jagiello-Gruszfeld, A., Crown, J., Chan, A., Kaufman, B., Skarlos, M., Campone, N., Davidson, M., Berger, C., Oliva, S.D., Rubin, S., Stein, D. and Cameron, D. (2006) Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. New England Journal of Medicine, 355, 2733-2743.
http://dx.doi.org/10.1056/NEJMoa064320 - 20. Michel, L.L., Bermejo, J.L., Gondos, A., Marmé, F. and Schneeweiss, A. (2015) T-DM1 as a New Treatment Option for Patients with Metastatic HER2-positive Breast Cancer in Clinical Practice. Anticancer Research, 35, 5085-5090.
- 21. McCormack, P.L. (2015) Pertuzumab in HER2-Positive Breast Cancer: A Guide to Its Use. Drugs & Therapy Perspectives, 32, 35-41.
http://dx.doi.org/10.1007/s40267-015-0267-0 - 22. Rocca, A., Andreis, D., Fedeli, A., Maltoni, R., Sarti, S., Cecconetto, L., Pietri, E., Schirone, A., Bravaccini, S., Serra, P., Farolfi, A. and Amadori, D. (2015) Pharmacokinetics, Pharmacodynamics and Clinical Efficacy of Pertuzumab in Breast Cancer Therapy. Expert Opinion on Drug Metabolism & Toxicology, 11, 1647-1663.
http://dx.doi.org/10.1517/17425255.2015.1078311 - 23. Mirzania, M. (2016) Approach to the Triple Negative Breast Cancer in New Drugs Area. International Journal of Hematology-Oncology and Stem Cell Research, 10, 115-119.
- 24. Tinoco, G., Warsch, S., Glück, S., Avancha, K. and Montero, A.J. (2013) Treating Breast Cancer in the 21st Century: Emerging Biological Therapies. Journal of Cancer, 4, 117-132.
http://dx.doi.org/10.7150/jca.4925 - 25. Mohamed, A., Krajewski, K., Cakar, B. and Ma, C.X. (2013) Targeted Therapy for Breast Cancer. American Journal of Pathology, 183, 1096-1112.
http://dx.doi.org/10.1016/j.ajpath.2013.07.005 - 26. Bjornstrom, L. and Sjoberg, M. (2005) Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Molecular Endocrinology, 19, 833-842.
http://dx.doi.org/10.1210/me.2004-0486 - 27. Nilsson, S., Makela, S., Treuter, E., Tujague, M., Thomsen, J., Andersson, G., Enmark, E., Pettersson, K., Warner, M. and Gustafsson, J.A. (2001) Mechanisms of Estrogen Action. Physiological Reviews, 81, 1535-1565.
- 28. Le Romancer, M., Treilleux, I., Leconte, N., Robin-Lespinasse, Y., Sentis, S., Bouchekioua-Bouzaghou, K., Goddard, S., Gobert-Gosse, S. and Corbo, L. (2008) Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Molecular Cell, 31, 212-221.
http://dx.doi.org/10.1016/j.molcel.2008.05.025 - 29. Le Romancer, M., Poulard, C., Cohen, P., Sentis, S., Renoir, J.M. and Corbo, L. (2011) Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocrine Reviews, 32, 597-622.
http://dx.doi.org/10.1210/er.2010-0016 - 30. Poulard, C., Treilleux, I., Lavergne, E., Bouchekioua-Bouzaghou, K., Goddard-Léon, S., Chabaud, S., Trédan, O., Corbo, L. and Le Romancer, M. (2012) Activation of Rapid Oestrogen Signalling in Aggressive Human Breast Cancers. EMBO Molecular Medicine, 4, 1200-1213.
http://dx.doi.org/10.1002/emmm.201201615 - 31. Smith, D.P., Spicer, J., Smith, A., Swift, S. and Ashworth, A. (1999) The Mouse Peutz-Jeghers Syndrome Gene LKB1 Encodes a Nuclear Protein Kinase. Human Molecular Genetics, 8, 1479-1485.
http://dx.doi.org/10.1093/hmg/8.8.1479 - 32. Korsse, S.E., Peppelenbosch, M.P. and van Veelen, W. (2013) Targeting LKB1 Signaling in Cancer. Biochimica et Biophysica Acta, 1835, 194-210.
http://dx.doi.org/10.1016/j.bbcan.2012.12.006 - 33. Herrmann, J.L., Byekova, Y., Elmets, C.A. and Athar, M. (2011) Liver Kinase B1 (LKB1) in the Pathogenesis of epithelial Cancers. Cancer Letters, 306, 1-9.
http://dx.doi.org/10.1016/j.canlet.2011.01.014 - 34. Rowan, A., Churchman, M., Jefferey, R., Hanby, A., Poulsom, R. and Tomlinson, I. (2000) In Situ Analysis of LKB1/STK11 mRNA Expression in Human Normal Tissues and Tumours. Journal of Pathology, 192, 203-206.
http://dx.doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH686>3.0.CO;2-J - 35. Sanchez-Cespedes, M. (2007) A Role for LKB1 Gene in Human Cancer beyond the Peutz-Jeghers Syndrome. Oncogene, 26, 7825-7832.
http://dx.doi.org/10.1038/sj.onc.1210594 - 36. Baas, A.F., Boudeau, J., Sapkota, G.P., Smit, L., Medema, R., Morrice, N.A., Alessi, D.R. and Clevers, H.C. (2003) Activation of the Tumour Suppressor Kinase LKB1 by the STE20-Like Pseudokinase STRAD. EMBO Journal, 22, 3062-3072.
http://dx.doi.org/10.1093/emboj/cdg292 - 37. Boudeau, J., Baas, A.F., Deak, M., Morrice, N.A., Kieloch, A., Schutkowski, M., Prescott, A. R., Clevers, H.C. and Alessi, D.R. (2003) Mo25α/β Interact with STRADα/β Enhancing Their Ability to Bind, Activate and Localize LKB1 in the Cytoplasm. EMBO Journal, 22, 5102-5114.
http://dx.doi.org/10.1093/emboj/cdg490 - 38. Dorfman, J. and Macara, I.G. (2008) STRADα Regulates LKB1 Localization by Blocking Access to Importin-α, and by Association with Crm1 and Exportin-7. Molecular and Cellular Biology, 19, 1614-1626.
http://dx.doi.org/10.1091/mbc.E07-05-0454 - 39. Baas, A.F., Kuipers, J., van der Wel, N.N., Batlle, E., Koerten, H.K., Peters, P.J. and Clevers, H.C. (2004) Complete Polarization of Single Intestinal Epithelial Cells upon Activation of LKB1 by STRAD. Cell, 116, 457-466.
http://dx.doi.org/10.1016/S0092-8674(04)00114-X - 40. Alessi, D.R., Sakamoto, K. and Bayascas, J.R. (2006) LKB1-Dependent Signaling Pathways. Annual Review of Biochemistry, 75, 137-163.
http://dx.doi.org/10.1146/annurev.biochem.75.103004.142702 - 41. Shackelford, D.B. and Shaw, R.J. (2009) The LKB1-AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nature Reviews Cancer, 9, 563-575.
http://dx.doi.org/10.1038/nrc2676 - 42. Momcilovic, M. and Shackelford, D.B. (2015) Targeting LKB1 in Cancer—Exposing and Exploiting Vulnerabilities. British Journal of Cancer, 113, 574-584.
http://dx.doi.org/10.1038/bjc.2015.261 - 43. Gan, R.Y. and Li, H.B. (2014) Recent Progress on Liver Kinase B1 (LKB1): Expression, Regulation, Downstream Signaling and Cancer Suppressive Function. International Journal of Molecular Sciences, 15, 16698-16718.
http://dx.doi.org/10.3390/ijms150916698 - 44. Zhou, W., Zhang, J. and Marcus, A.I. (2014) LKB1 Tumor Suppressor: Therapeutic Opportunities Knock when LKB1 Is Inactivated. Genes & Diseases, 1, 64-74.
http://dx.doi.org/10.1016/j.gendis.2014.06.002 - 45. Sapkota, G.P., Kieloch, A., Lizcano, J.M., Lain, S., Arthur, J.S., Williams, M.R., Morrice, N., Deak, M. and Alessi, D.R. (2001) Phosphorylation of the Protein Kinase Mutated in Peutz-Jeghers Cancer Syndrome, LKB1/STK11, at Ser431 by p90RSK and cAMP-Dependent Protein Kinase, but Not Its Farnesylation at Cys433, Is Essential for LKB1 to Suppress Cell Growth. Journal of Biological Chemistry, 276, 19469-19482.
http://dx.doi.org/10.1074/jbc.M009953200 - 46. Nony, P., Gaude, H., Rossel, M., Fournier, L., Rouault, J.P. and Billaud, M. (2003) Stability of the Peutz-Jeghers Syndrome Kinase LKB1 Requires Its Binding to the Molecular Chaperones Hsp90/Cdc37. Oncogene, 22, 9165-9175.
http://dx.doi.org/10.1038/sj.onc.1207179 - 47. Xie, Z., Dong, Y., Zhang, J., Scholz, R., Neumann, D. and Zou, M.H. (2009) Identification of the Serine 307 of LKB1 as a Novel Phosphorylation Site Essential for Its Nucleocytoplasmic Transport and Endothelial Cell Angiogenesis. Molecular and Cellular Biology, 29, 3582-3596.
http://dx.doi.org/10.1128/MCB.01417-08 - 48. Han, Y., Wang, Q., Song, P., Zhu, Y. and Zou, M.H. (2010) Redox Regulation of the AMP—Activated Protein Kinase. PLoS ONE, 5, e15420.
http://dx.doi.org/10.1371/journal.pone.0015420 - 49. Sapkota, G.P., Boudeau, J., Deak, M., Kieloch, A., Morrice, N. and Alessi, D.R. (2002) Identification and Characterization of Four Novel Phosphorylation Sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the Protein Kinase Mutated in Peutz-Jeghers Cancer Syndrome. Biochemical Journal, 362, 481-490.
- 50. Zheng, B., Jeong, J.H., Asara, J.M., Yuan, Y.Y., Granter, S.R., Chin, L. and Cantley, L.C. (2009) Oncogenic B-RAF Negatively Regulates the Tumor Suppressor LKB1 to Promote Melanoma Cell Proliferation. Molecular Cell, 33, 237-247.
http://dx.doi.org/10.1016/j.molcel.2008.12.026 - 51. Liu, L., Siu, F.M., Che, C.M., Xu, A. and Wang, Y. (2012) Akt Blocks the Tumor Suppressor Activity of LKB1 by promoting Phosphorylation-Dependent Nuclear Retention through 14-3-3 Proteins. American Journal of Translational Research, 4, 175-186.
- 52. Bai, Y., Zhou, T., Fu, H., Sun, H. and Huang, B. (2012) 14-3-3 Interacts with LKB1 via Recognizing Phosphorylated Threonine 336 Residue and Suppresses LKB1 Kinase Function. FEBS Letters, 586, 1111-1119.
http://dx.doi.org/10.1016/j.febslet.2012.03.018 - 53. Sapkota, G.P., Deak, M., Kieloch, A., Morrice, N., Goodarzi, A.A., Smythe, C., Shiloh, Y., Lees-Miller, S.P. and Alessi, D.R. (2002) Ionizing Radiation Induces Ataxia Telangiectasia Mutated Kinase (ATM)-Mediated Phosphorylation of LKB1/STK11 at Thr-366. Biochemical Journal, 368, 507-516.
http://dx.doi.org/10.1042/bj20021284 - 54. Shen, Y.A.A., Chen, Y., Dao, D.Q., Mayoral, S.R., Wu, L., Meijer, D., Ullian, E.M., Chan, J.R. and Lu, Q.R. (2014) Phosphorylation of LKB1/Par-4 Establishes Schwann Cell Polarity to Initiate and Control Myelin Extent. Nature Communications, 5, Article Number: 4991.
http://dx.doi.org/10.1038/ncomms5991 - 55. Xie, Z., Dong, Y., Scholz, R., Neumann, D. and Zou, M.H. (2008) Phosphorylation of LKB1 at Serine 428 by Protein Kinase C-Zeta Is Required for Metformin-Enhanced Activation of the AMP-Activated Protein Kinase in Endothelial Cells. Circulation, 117, 952-962.
http://dx.doi.org/10.1161/CIRCULATIONAHA.107.744490 - 56. Lan, F., Cacicedo, J.M., Ruderman, N. and Ido, Y. (2008) SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1. Possible Role in AMP-Activated Protein Kinase Activation. Journal of Biological Chemistry, 283, 27628-27635.
http://dx.doi.org/10.1074/jbc.M805711200 - 57. Zu, Y., Liu, L., Lee, M.Y.K., Xu, C., Liang, Y., Man, R.Y., Vanhoutte, P.M. and Wang, Y. (2010) SIRT1 Promotes Proliferation and Prevents Senescence through Targeting LKB1 in Primary Porcine Aortic Endothelial Cells. Circulation Research, 106, 1384-1393.
http://dx.doi.org/10.1161/CIRCRESAHA.109.215483 - 58. Bai, B.A., Man, W.C., Yang, K., Guo, Y., Xu, C., Tse, H.F., Han, W., Bloksgaard, M., Mey, J. G.R.D., Vanhoutte, P.M., Xu, A. and Wang, Y. (2016) Endothelial SIRT1 Prevents Adverse Arterial Remodeling by Facilitating HERC2-Mediated Degradation of Acetylated LKB1. Oncotarget, 7, 39065-39081.
http://dx.doi.org/10.18632/oncotarget.9687 - 59. Collins, S.P., Reoma, J.L., Gamm, D.M. and Uhler, M.D. (2000) LKB1, A Novel Serine/ Threonine Protein Kinase and Potential Tumour Suppressor, Is Phosphorylated by cAMP-Dependent Protein Kinase (PKA) and Prenylated in Vivo. Biochemical Journal, 345, 673-680.
http://dx.doi.org/10.1042/bj3450673 - 60. Houde, V.P., Ritorto, M.S., Gourlay, R., Varghese, J., Davies, P., Shpiro, N., Sakamoto, K. and Alessi, D.R. (2014) Investigation of LKB1 Ser431 Phosphorylation and Cys433 Farnesylation Using Mouse Knockin Analysis Reveals an Unexpected Role of Prenylation in Regulating AMPK Activity. Biochemical Journal, 458, 41-56.
http://dx.doi.org/10.1042/BJ20131324 - 61. Lim, W., Hearle, N., Shah, B., Murday, V., Hodgson, S.V., Lucassen, A., Eccles, D., Talbot, I., Neale, K., Lim, A.G., O’Donohue, J., Donaldson, A., Macdonald, R.C., Young, I.D., Robinson, M.H., Lee, P.W.R., Stoodley, B.J., Tomlinson, I., Alderson, D., Holbrook, A.G., Vyas, S., Swarbrick, E.T., Lewis, A.M., Phillips, R.K.S. and Houlston, R.S. (2003) Further Observations on LKB1/STK11 Status and Cancer Risk in Peutz-Jeghers Syndrome. British Journal of Cancer, 89, 308-313.
http://dx.doi.org/10.1038/sj.bjc.6601030 - 62. Van Lier, M.G.F., Wagner, A., Mathus-Vliegen, E.M.H., Kuipers, E.J., Steyerberg, E.W. and van Leerdam, M.E. (2010) High Cancer Risk in Peutz-Jeghers Syndrome: A Systematic Review and Surveillance Recommendations. American Journal of Gastroenterology, 105, 1258-1264.
http://dx.doi.org/10.1038/ajg.2009.725 - 63. Ollila, S. and Makela, T.P. (2011) The Tumor Suppressor Kinase LKB1: Lessons from Mouse Models. Journal of Molecular Cell Biology, 3, 330-340.
http://dx.doi.org/10.1093/jmcb/mjr016 - 64. Taliaferro-Smith, L., Nagalingam, A., Zhong, D., Zhou, W., Saxena, N.K. and Sharma, D. (2009) LKB1 Is Required for Adiponectin-Mediated Modulation of AMPK-S6K Axis and Inhibition of Migration and Invasion of Breast Cancer Cells. Oncogene, 28, 2621-2633.
http://dx.doi.org/10.1038/onc.2009.129 - 65. Zhuang, Z.G., Di, G.H., Shen, Z.Z., Ding, J. and Shao, Z.M. (2006) Enhanced Expression of LKB1 in Breast Cancer Cells Attenuates Angiogenesis, Invasion, and Metastatic Potential. Molecular Cancer Research, 4, 843-849.
http://dx.doi.org/10.1158/1541-7786.MCR-06-0118 - 66. Partanen, J.I., Tervonen, T.A., Myllynen, M., Lind, E., Imai, M., Katajisto, P., Dijkgraaf, G. J.P., Kovanen, P.E., Makela, T.P., Werb, Z. and Klefstrom, J. (2012) Tumor Suppressor Function of Liver Kinase B1 (LKB1) Is Linked to Regulation of Epithelial Integrity. Proceedings of the National Academy of Sciences of the United States of America, 109, 388-397.
http://dx.doi.org/10.1073/pnas.1120421109 - 67. Andrade-Vieira, R., Xu, Z., Colp, P. and Marignani, P.A. (2013) Loss of LKB1 Expression Reduces the Latency of ErbB2-Mediated Mammary Gland Tumorigenesis, Promoting Changes in Metabolic Pathways. PLoS ONE, 8, e56567.
http://dx.doi.org/10.1371/journal.pone.0056567 - 68. Byekova, Y.A., Herrmann, J.L., Xu, J., Elmets, C.A. and Athar, M. (2011) Liver Kinase B1 (LKB1) in the Pathogenesis of UVB-Induced Murine Basal Cell Carcinoma. Archives of Biochemistry and Biophysics, 508, 204-211.
http://dx.doi.org/10.1016/j.abb.2011.01.006 - 69. Zhong, D., Liu, X., Khuri, F.R., Sun, S.Y., Vertino, P.M. and Zhou, W. (2008) LKB1 Is Necessary for Akt-Mediated Phosphorylation of Pro-Apoptotic Proteins. Cancer Research, 68, 7270-7277.
http://dx.doi.org/10.1158/0008-5472.CAN-08-1484 - 70. Bignell, G.R., Barfoot, R., Seal, S., Collins, N., Warren, W. and Stratton, M.R. (1998) Low Frequency of Somatic Mutations in the LKB1/Peutz-Jeghers Syndrome Gene in Sporadic Breast Cancer. Cancer Research, 58, 1384-1386.
- 71. Nik-Zainal, S., Davies, H., Staaf, J., Ramakrishna, M., Glodzik, D., Zou, X., Martincorena, I., Alexandrov, L.B., Martin, S., Wedge, D.C., Van Loo, P., Ju, Y.S., Smid, M., Brinkman, A.B., Morganella, S., Aure, M.R., Lingjaerde, O.C., Langerod, A., Ringnér, M., Ahn, S.M., Boyault, S., Brock, J.E., Broeks, A., Butler, A., Desmedt, C., Dirix, L., Dronov, S., Fatima, A., Foekens, J.A., Gerstung, M., Hooijer, G K.J., Jang, S.J., Jones, D.R., Kim, H.Y., King, T.A., Krishnamurthy, S., Lee, H.J., Lee, J.Y., Li, Y., McLaren, S., Menzies, A., Mustonen, V., O’Meara, S., Pauporté, I., Pivot, X., Purdie, C.A., Raine, K., Ramakrishnan, K., Rodríguez-González, F.G., Romieu, G., Sieuwerts, A.M., Simpson, P.T., Shepherd, R., Stebbings, L., Stefansson, O.A., Teague, J., Tommasi, S., Treilleux, I., Van den Eynden, G.G., Vermeulen, P., Vincent-Salomon, A., Yates, L., Caldas, C., van’t Veer, L., Tutt, A., Knappskog, S., Tan, B.K.T., Jonkers, J., Borg, A., Ueno, N.T., Sotiriou, C., Viari, A., Futreal, P.A., Campbell, P.J., Span, P.N., Van Laere, S., Lakhani, S.R., Eyfjord, J.E., Thompson, A. M., Birney, E., Stunnenberg, H.G., van de Vijver, M.J., Martens, J.W.M., Borresen-Dale, A.L., Richardson, A.L., Kong, G., Thomas, G. and Stratton, M.R. (2016) Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature, 534, 47-54.
http://dx.doi.org/10.1038/nature17676 - 72. Shen, Z., Wen, X.-F., Lan, F., Shen, Z.Z. and Shao, Z.M. (2002) The Tumor Suppressor Gene LKB1 Is Associated with Prognosis in Human Breast Carcinoma. Clinical Cancer Research, 8, 2085-2090.
- 73. Xiao, J., Zou, Y., Chen, X., Gao, Y., Xie, M., Lu, X., Li, W., He, B., He, S., You, S. and Chen, Q. (2016) The Prognostic Value of Decreased LKB1 in Solid Tumors: A Meta-Analysis. PLoS ONE, 11, e0152674.
http://dx.doi.org/10.1371/journal.pone.0152674 - 74. Azim, H.A., Kassem, L., Treilleux, I., Wang, Q., El Enein, M.A., Anis, S.E. and Bachelot, T. (2016) Analysis of PI3K/mTOR Pathway Biomarkers and Their Prognostic Value in Women with Hormone Receptor-Positive, HER2-Negative Early Breast Cancer. Translational Oncology, 9, 114-123.
http://dx.doi.org/10.1016/j.tranon.2016.01.001 - 75. Bachelot, T., Bourgier, C., Cropet, C., Ray-Coquard, I., Ferrero, J.M., Freyer, G., Abadie-Lacourtoisie, S., Eymard, J.C., Debled, M., Spaeth, D., Legouffe, E., Allouache, D., El Kouri, C. and Pujade-Lauraine, E. (2012) Randomized Phase II Trial of Everolimus in Combination with Tamoxifen in Patients with Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer with Prior Exposure to Aromatase Inhibitors: A GINECO Study. Journal of Clinical Oncology, 30, 2718-2724.
http://dx.doi.org/10.1200/JCO.2011.39.0708 - 76. Baselga, J., Campone, M., Piccart, M., Burris, H.A., Rugo, H.S., Sahmoud, T., Noguchi, S., Gnant, M., Pritchard, K.I., Lebrun, F., Beck, J.T., Ito, Y., Yardley, D., Deleu, I., Perez, A., Bachelot, T., Vittori, L., Xu, Z., Mukhopadhyay, P., Lebwohl, D. and Hortobagyi, G.N. (2012) Everolimus in Postmenopausal Hormone-Receptor-Positive Advanced Breast Cancer. New England Journal of Medicine, 366, 520-529.
http://dx.doi.org/10.1056/NEJMoa1109653 - 77. Treilleux, I., Arnedos, M., Cropet, C., Wang, Q., Ferrero, J.M., Abadie-Lacourtoisie, S., Levy, C., Legouffe, E., Lortholary, A., Pujade-Lauraine, E., Bourcier, A.V., Eymard, J.C., Spaeth, D. and Bachelot, T. (2015) Translational Studies within the TAMRAD Randomized GINECO Trial: Evidence for mTORC1 Activation Marker as a Predictive Factor for Everolimus Efficacy in Advanced Breast Cancer. Annals of Oncology, 26, 120-125.
http://dx.doi.org/10.1093/annonc/mdu497 - 78. Chen, C., Chang, Y.C., Lu, Y.S., Chung, K.P., Huang, C.S., Lu, T.P., Kuo, W.H., Wang, M.Y., Kuo, K.T., Wu, P.F., Hsueh, T.H., Shen, C.Y., Lin, C.H. and Cheng, A.L. (2016) Clinical Relevance of Liver KinaseB1 (LKB1) Protein and Gene Expression in Breast Cancer. Scientific Reports, 6, Article Number: 21374.