Journal of Cancer Therapy
Vol.4 No.1A(2013), Article ID:26381,10 pages DOI:10.4236/jct.2013.41A003
The False Paradigm of RUNX3 Function as Tumor Suppressor in Gastric Cancer
![]()
Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel.
Email: *yoram.groner@weizmann.ac.il
Received November 14th, 2012; revised December 16th, 2012; accepted December 25th, 2012
Keywords: Gastric Cancer; RUNX3 Expression; Tumor Suppressor Genes; Promoter Methylation; Therapeutic Targets
ABSTRACT
Gastric cancer (GC) is a major cause of cancer mortality. GC studies that aim to identify relevant oncogenes and tumor suppressor genes (TSGs) are essential for devising effective new therapies. A decade ago, RUNX3, a gene that resides on human chromosome 1p36.1, was claimed to be a major TSG in GC. Since then, hundreds of studies involving thousands of GC patients have attempted to verify and extend the RUNX3 TSG paradigm. However, RUNX3 is not recognized as TSG and not listed in the “Cancer Gene Census” website. To be a TSG that protects normal cells against malignancy, the gene must be expressed in the normal tissue from which the cancer arose and its loss or inactivation should contribute to cancer development. This review summarizes compelling body of evidence challenging the RUNX3-TSG paradigm. Studies show unequivocally that RUNX3 is not expressed in normal gastric epithelium and that it fails to fulfill all other premises of a TSG. RUNX3 mutations and 1p36 deletions are not frequent in GC and RUNX3 is not associated with familial GC or with increased risk of GC. Accordingly, Runx3−/− mice do not develop tumors. RUNX3 promoter methylation, which has been reported to be a frequent event in GC, is not relevant to its alleged TSG function, since the gene is already silent in normal gastric epithelium. In sharp contrast, overexpression of RUNX3 was found in several types of human cancers, including GC, and the 1p36.1 region is amplified in B-cell lymphoma. Thus, it is possible that RUNX3 actually promotes cancer development rather than being a TSG. The true targets for GC therapy are discussed below. Those are genes frequently lost or amplified in GC and are well known for their tumor suppressive or oncogenic activity, respectively.
1. Introduction
GC is highly prevalent in China and other Far East countries, constituting a major cause of worldwide cancer mortality [1,2]. Etiological contribution of dietary factors, Helicobacter pylori [3] and EBV [4] infections and the resulting gastric inflammation to GC development are well established but the downstream crucial events leading to malignancy are complex and involve numerous genetic [5-8] and epigenetic [9] alterations. Many well established oncogenes and TSGs have been proposed as contributing factors in GC development. A decade ago, Li et al. suggested that the transcription factor (TF) RUNX3 functions as a novel TSG that plays a major role in GC [10]. This suggestion was based on their finding of high expression level of Runx3 in normal mouse gastrointestinal tract (GIT), gastric hyperplasia in Runx3−/− newborn mice, detection of a RUNX3 point mutation in a single GC patient, loss of RUNX3 heterozygosity (LOH) in 30% of GC patients, RUNX3 promoter DNA hypermethylation in GC versus normal gastric tissue and reduced RUNX3 mRNA expression by in situ hybridization in GC versus normal gastric tissues in 60% of GC patients.
Since then, hundreds of studies involving thousands of GC patients have attempted to verify and extend the RUNX3 TSG paradigm suggested by Li et al. in GC and/or in other GIT cancers [11]. Many of these studies (references included in Table S1 of [12]) focused on the issue of RUNX3 promoter DNA hypermethylation, but have not tested whether RUNX3 was indeed expressed in normal GIT and what are the consequences of this hypermethylation on RUNX3 expression in GIT. To qualify as a cell autonomous TSG in a given cell type requires that the gene be expressed in that cell type and that its loss by mutation, deletion, degradation or silencing would promote malignancy. However, compelling evidence from our laboratory and several other laboratories that demonstrated the lack of RUNX3 expression in normal and GC tissues and refuting the other specific requirements from a gene to be recognized as a genuine TSG, challenged the claim that RUNX3 is a TSG in GC. The large body of data disproving the possibility that RUNX3 is a TSG in GC is the subject of this review. Of note, chasing the wrong gene is counter-productive and interferes with efforts to discover the correct gene.
2. Runx3 Expression Pattern in Normal Healthy Mouse Tissues
RUNX3 is one of the three mammalian Runt domain TFs (RUNX1, RUNX2 and RUNX3) that comprise the highly conserved and structurally similar RUNX gene family. RUNX TFs are key gene expression regulators in several important developmental processes including hematopoiesis, osteogenesis and neurogenesis [13]. Runx3 was originally cloned based on its similarity to Runx1 [14] and subsequently localized on human and mouse chromosomes 1 and 4, respectively [14,15]. Tissue-specific Runx3 expression is transcriptionally regulated by two alternative control regions, designated the distal (P1) and proximal (P2) promoters [13,16]. We have previously published a detailed survey of the spatio-temporal expression of Runx3 and Runx1 during embryonic development, using immunohistochemistry (IHC) and knock-in (KI) b-galactosidase activity (LacZ staining) [17]. Runx3 and Runx1 were readily detected in different compartments of the hematopoietic system as well as in dorsal root ganglia (DRG), epidermal appendages and developing skeletal elements [17]. However, regarding epithelia an interesting distinction was noted in the expression pattern of Runx1 and Runx3. While Runx1 was expressed in various epithelia including mucosa of the esophagus and stomach, the salivary glands ducts and the olfactory and respiratory mucosa, Runx3 expression in these epithelia was undetectable [17].
Subsequently, using a different b-gal gene KI mouse strain, Li et al. reported that Runx3 was highly expressed in GIT epithelium of E14.5 embryos and adult mice [10]. Clearly, this report contradicted our earlier results that showed no Runx3 expression in the GIT, which was particularly intriguing since all other LacZ-expressing sites shown in Li et al., including cartilage, epidermal appendages and DRG corresponded with those reported previously by our own group [17,18]. It is interesting to note that using Runx3-specific quantitative reverse transcriptase polymerase chain reaction (RT-qPCR) assay, Ito et al. reported in a later study that normal mouse gastric epithelial cells expressed 10-fold less Runx3 compared to blood leukocytes [19], which contradicts their own earlier finding [10] of a very strong Runx3 expression in mouse gastric epithelium. In order to resolve these contradicting results we have reanalyzed Runx3 expression using a variety of biochemical techniques, including 8 different anti-Runx3 antibodies (Abs), qRT-PCR as well as 35S-RNA in situ hybridization and have created new RUNX3 reporter mouse strains, Runx3P1-AFP/+, Runx3P2-EGFP/+, R26-LacZ/Runx3Cre and R26-tdTomato/ Runx3Cre [12]. The Cre recombinase reporter strains display amplified expression of LacZ or tdTomato following Cre-mediated excision of loxP-flanked (Floxed) transcriptional “stop” sequences [20,21]. Upon crossing the Runx3Cre with the Cre recombinase reporter strains, the reporter (i.e. LacZ or tdTomato) is switched-on in all cells that express Runx3 and from then on constitutively expressed within the R26 locus. This occurrence generates a permanent genetic mark which is transmitted to all progeny cells allowing to trace not only constant but also transient expression of Runx3 even in rare cell populations.
Using all these new biochemical and genetic techniques we have found that Runx3 was not detected at all in GIT epithelium, but was readily detected in all other organs known to express Runx3 [12]. Furthermore, using flow cytometry on R26-tdTomato/Runx3Cre E16.5 embryos we have found that while Runx3 expression was readily detected in the GIT intraepithelial leukocytes, no Runx3 was detected in GIT epithelial cells [12]. Runx3 was also undetectable in isolated adult GIT epithelial cells assayed by RT-qPCR [12]. Taken together these results demonstrate that Runx3 expression was undetectable in embryonic and adult GIT epithelium and that Runx3 was not even transiently expressed at any time point during epithelial lineage development. The results also preclude the possibility that Runx3 is expressed even in small progenitor compartments of GIT epithelial cells or the possibility that the detection of Runx3 expression in GIT epithelium by Li et al., but not by our laboratory, was due to different targeting vectors.
We have also analyzed the original Li et al. Runx3LacZ/LacZ mice, which were provided to us by Dr. Y. Ito, the PI and principal author of the Li et al. report [10]. LacZ staining of these Runx3LacZ/+ and Runx3LacZ/LacZ E14.5 embryos revealed Runx3-LacZ expression in DRG and skeletal elements [12] at intensities similar to those previously observed in these mice [10,22], yet no Runx3-LacZ was detected in GIT [12]. It is now also clear that using their Runx3LacZ/LacZ mice, Dr. Ito’s laboratory cannot replicate the key experimental finding of Runx3-LacZ staining in GIT [23] as reported in 2002 [10]. Thus, the inevitable conclusion is that normal GIT epithelium lacks Runx3 expression and the Li-2002 report, which indicated expression of Runx3 in this epithelium, is flawed. Accordingly, our failure to detect LacZ in Li-2002-LacZ mice and the current inability of Dr. Ito to reproduce his 2002 data [23] is almost certainly due to the fact that Runx3 was never expressed in GIT epithelium of the LiRunx3-LacZ mouse described in 2002. For additional details concerning the issue of Runx3 expression in normal GIT epithelium see http://www.weizmann.ac.il/molgen/Groner/uploads/runx 3.pdf
3. Specificity of Anti-Runx3 Abs Used for IHC
The issue of detecting Runx3 by IHC requires specific attention. As alluded to above, a detailed survey of the spatio-temporal expression of Runx3 and Runx1 proteins during embryonic development, using IHC, was originally published in 2001 [17]. By using 8 different Abs raised against different regions of Runx3, including 3 anti-Runx3 monoclonal Abs raised in Dr. Ito’s laboratory (R3-1E10, R3-8C9 and R3-3F12), it was more recently confirmed that Runx3 protein was not expressed in GIT epithelium in embryos and adult mice [12]. Most of these Abs specifically detected Runx3 in other cell types in the same or adjacent tissues, such as GIT-embedded leukocytes (in the adult GIT) and in embryonic DRG neurons [12]. These results clearly demonstrate an excellent correspondence between Runx3 mRNA and protein expression in different tissues. The R3-1E10 failed to comply with this criterion. Although this Ab has been reported to detect Runx3 in GIT epithelium [19,24], it did not detect Runx3 in leukocytes [12] nor in DRG [19]. Given that GIT epithelial cells are notorious for their high degree of nonspecific antibody binding, the Abs used in IHC experiments should be very carefully controlled for specificity. Such analysis clearly demonstrates that R3-1E10 is not suitable for detecting Runx3 by IHC, particularly when its reaction with GIT epithelium serves as the sole evidence for Runx3 expression in that tissue [24]. Therefore, it is unfortunate and scientifically unsound that of all available anti-Runx3 Abs, R3-1E10 was the one used for detecting Runx3 expression in GIT epithelium without proper disclosure of any information regarding its abnormal properties [24].
4. RUNX3 Protein in Human GC versus Normal Gastric Epithelium
In contrast to the reported expression of RUNX3 in normal human gastric epithelium [10,11,24,25], two independent reports that analyzed more than 100 patients did not detect RUNX3 mRNA and protein in normal gastric epithelium but detected nuclear RUNX3 protein in gastric tissue-embedded leukocytes in normal and H. pylori-induced gastritis patients [26,27]. Moreover, nuclear RUNX3 protein was detected in cancer cells of ~32% of the GC patients, demonstrating that in some cases of GC RUNX3 expression may actually be induced in the cancer cells [26]. This finding is consistent with other studies showing RUNX3 overexpression in other types of cancers including basal cell carcinoma [28], head and neck cancer [29], ovarian cancer [30] and pancreatic cancer [31], possibly reflecting an oncogenic role of RUNX3. The report that EBNA2-mediated induction of RUNX3 expression in EBV-infected B cells is essential for the proliferation of the transformed cells [32], is also consistent with an oncogenic role of RUNX3 in certain human cancers.
The suggested role of RUNX3 as a TSG was based in part on the reduced frequency of RUNX3 expressing cells in human GC versus normal human gastric epithelium [11] and on the cytoplasmic mislocalization of RUNX3 [25], as detected by IHC using 2 different monoclonal antibodies. Of the RUNX3 IHC studies on human GC versus normal gastric tissue summarized in [11], the frequency of GC patients that were reported as negative for RUNX3 IHC staining in the cancer cells was ~43%. Being a TF that is active in the cell nucleus, the claimed mislocalization of RUNX3 in the cytoplasm of a significant proportion of GC cases [25] was construed as a mechanism for its inactivation. However, IHC staining of RUNX3 in normal human gastric tissue has been reported to be strongest in the region where Chief cells reside and a significant portion of stained cells contained cytoplasmic staining [25]. Furthermore, using another anti-RUNX3 Ab (AS-251) it was shown that while only nuclear RUNX3 was detected in RUNX3 transfected cells, only cytoplasmic RUNX3 staining was detected in the normal human gastric Chief cell region [33]. Therefore, the reported cytoplasmic RUNX3 staining in GC cannot be regarded as a mechanism for its inactivation since it is already “inactivated” in the normal gastric epithelium. Taking into consideration the reservations pointed out above concerning the specificity of RUNX3 IHC staining in gastric tissue and the contradicting results concerning RUNX3 expression in normal gastric epithelium, the inevitable conclusion is that, as in mouse GIT, RUNX3 is not expressed in normal human gastric epithelium and that the reported cytoplasmic retention of RUNX3 in normal gastric epithelium and GC cells reflects an IHC artifact. Thus, both the lack of specific RUNX3 expression in normal human gastric epithelium [26,27] and the reported cytoplasmic staining of RUNX3 in normal gastric epithelium [25,33] indicate that there is no functional RUNX3 in normal gastric epithelium. Therefore, it is highly unlikely that lack of RUNX3 expression in GC is indicative of its function as a TSG, since it is already not expressed in normal GIT epithelium.
5. No Gastric Tumors in Runx3−/− Mice
Loss of TSGs is generally sufficient for induction of tumor development in mice. In some cases loss of a gene function in stromal cells [34,35] or T cells [36] can lead to epithelial tumorigenesis, so a scenario in which RUNX3 activity in GIT leukocytes might protect GIT epithelium against tumorigenesis is a possibility to consider. Indeed, Runx3 is expressed in GIT leukocytes [12, 37] and its absence in Runx3−/− mice is associated with colonic inflammation and epithelial hyperplasia [37], but none of the Runx3−/− mice showed an increased incidence of GIT tumors or any other tumors [24,37]. It is important to note that Runx3−/− mice on a Balb/c background survive to more than a year but do not develop GIT tumors, but peculiarly Runx3+/− mice were reported to develop adenomas in colon and intestine at a very late onset of ~15 months [24]. Such GIT tumors were not detected in Runx3+/− mice on the ICR background [37]. This result is even more puzzling since none of the phenotypes observed in mice deficient for both Runx3 alleles, including ataxia due to loss of TrkC neurons in DRG [18], colitis [37], asthma-like lung inflammation [38] and defective silencing of CD4 expression in CD8+ T cells [39,40], is observed in Runx3+/− mice. This indicates that sufficient amounts of Runx3 are present in Runx3+/− mice to maintain homeostasis. It is thus unlikely that RUNX3 is a bona fide TSG, since loss of only one TSG allele is generally not sufficient to induce tumors and even in cases where tumor development does occur following the loss of one TSG allele (haploinsufficient TSG), the severity of the disease is lesser compared to when both its alleles are lost [41].
6. RUNX3 Mutations and Genomic Alterations Are Not Frequently Found in GC
As indicated earlier, multiple mechanisms were suggested to cause RUNX3 inactivation in GC and other cancers, attempting to justify the claim that RUNX3 is a major TSG. One of these mechanisms was based on the finding of a point mutation in RUNX3 in a single GC patient [10]. To the best of our knowledge, this RUNX3 mutation has not been discovered in other GC cases. Moreover, no other RUNX3 mutations were detected in a recent whole exome sequencing study of 15 GC samples, which detected 718 non-synonymous mutations in 661 genes including frequent mutations in TP53 and several chromatin-remodeling genes [8]. It is a common occurrence in cancer prone pedigrees to detect germline mutations in genes that are also frequently mutated in sporadic cancers. Approximately 10% of gastric cancer cases have a familial predisposition and half of these cases can be attributed to germline mutations in genes including TP53, CHD1, BRCA1, BRCA2, APC, SMAD4 and STK11 [42], but no germline RUNX3 mutations were detected in such pedigrees [43].
Association studies of the distribution of polymorphic markers in control and disease populations can identify genomic susceptibility regions to various diseases. One such association study with 10 RUNX3 polymorphic markers on ~300 GC and control Chinese patients reported an association of 3 such markers, located in RUNX3 introns 1, 3 and 4, with an increased risk of GC
[44]. Another larger association study on 583 GC and 1637 control patients with 2 polymorphic markers located in RUNX3 exon 1 and intron 3 revealed that the intron-3 polymorphic marker was associated with H. pylori-induced gastric atrophy but neither of these polymorphic markers was associated with GC [45]. Interestingly, while the same intron-3 polymorphic marker (rs- 760805) was used in both studies [44,45], it was found to be associated with increased risk of GC only in the smaller population size study [44]. Obviously a much larger study is needed to resolve this discrepancy. It is of interest to note that genome-wide association studies (GWAS) conducted on large populations detected that certain RUNX3 polymorphic markers were associated with an increased risk for two GIT diseases, ulcerative colitis [46,47] and celiac disease [48] and also for ankylosing spondylitis [49] and psoriasis [50] (Table 1), all considered to be immune-related diseases.
These findings may suggest that leukocyte-associated RUNX3 functions protect against development of GIT and other inflammatory diseases. As indicated above, Runx3-deficient mice spontaneously develop early onset colitis [37], which can be transferred to immune-suppressed mice by fetal liver hematopoietic precursors (our unpublished results). Although it was initially claimed that the colonic hyperplasia in Runx3−/− mice was unlikely to be due to a leukocyte-specific Runx3 deficiency [24], a later study confirmed our results that transfer of Runx3−/− FL cells into immune-suppressed mice induced colonic hyperplasia [51]. It is thus possible that certain GIT inflammatory diseases may be associated with certain RUNX3 polymorphic markers that might affect RUNX3 functions in GIT leukocytes such as CD8+ T cells, natural killer cells and dendritic cells. Finally, several GWAS studies identified genomic regions located on chromosomes 1q22, 3p13.31, 5p13.1, 8q24 and 20p13 [52-55] that were associated with an increased risk for developing GC in Chinese or Japanese populations, but the RUNX3 harboring region 1p36.11 was not among
Table 1. Association studies implicating RUNX3 in human diseases.
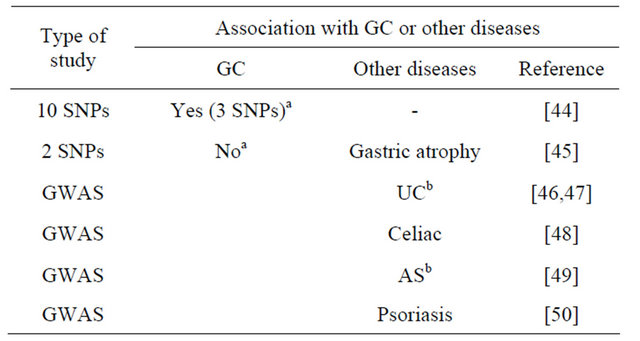
aOne Runx3 SNP (rs760805) was common to both studies [44,45]; bAS: ankylosing spondylitis; UC: ulcerative colitis.
those regions reported to confer susceptibility to GC (Table 2).
GC is associated with many genomic alterations including both amplifications and deletions. Based on fluorescent in-situ hybridization (FISH) analysis it was initially reported that 14 out of 46 GC patients (30%) had a hemizygous deletion of RUNX3, based on a lower than 1:1 ratio of RUNX3 and chromosome 1 centromere- specific signals [10]. However, the finding that in all GC samples the cells had at least 2 RUNX3-specific FISH signals per cell [26] indicated that there was no loss of RUNX3 in GC but rather amplification of regions encompassing the chromosome 1 centromere [26]. Furthermore, two recent high-resolution comparative genomic hybridization (CGH) studies including 193 and 64 GC patients, respectively, revealed that chromosome 1p36, which harbors RUNX3, was not frequently deleted in GC [5,6]. Rather, frequently deleted regions in GC were located on chromosomes 3p, 4pq, 5q, 6q, 8p, 9q, 11q, 14q, 16q, 17p, 18p, 18q, 19p, 21q and 22q, harboring known TSGs such as FHIT, CDKN2A and B, SMAD4, SMAD7 and RB1. Frequently amplified regions were located on chromosomes 1q, 3q, 5p, 6p, 7pq, 8q, 12pq 13q, 18pq, 19p, 20p and 21p, harboring known oncogenes such as KRAS, the receptor tyrosine kinases FGFR2, EGFR, ERBB2 and MET as well as some others including GATA4, GATA6, MYC, CDK6, CCND1 and CCNE1 [5,6] (Table 2). An earlier study on a smaller cohort of 40 GC patients, also described the frequent chromosomal copy number aberrations and although such aberrations were detected across the entire genome, chromosome 1p36 was not among the frequently deleted regions [56]. Thus, although some RUNX3 polymorphic markers may be associated with H. pylori induced gastric changes, possibly due to an altered RUNX3 function in infiltrating inflammatory immune cells, the lack of convincing evidence for frequent 1p36 genomic alterations or association with increased risk for GC development, do not support a major role for RUNX3 as a TSG in GC.
7. RUNX3 Promoter Hypermethylation in GC
Another mechanism initially put forward to support a TSG role for RUNX3 in GC was methylation of the CpG island adjacent to RUNX3 P2 promoter [10]. This methylation was initially detected in 3 samples of GC that did not express RUNX3, as measured by RT-PCR, but no such methylation was detected in 3 samples of normal gastric mucosa, which were reported to express RUNX3 [10]. These results were interpreted as an indication that GC cells do not express RUNX3 due to hypermethylation of its P2 promoter [10].
In many of the following studies on RUNX3 hypermethylation in GC (summarized in [11]) the average frequency of GC samples with RUNX3 promoter methyllation was ~58% and the average frequency of GC samples with reduced RUNX3 IHC staining was ~54%. However, most of these studies measured either only RUNX3 promoter methylation or only RUNX3 by IHC staining, so it cannot be determined to what extent were RUNX3 methylation and its reduced expression related in each of the GC samples. More recently, another study on 123 GC and 111 healthy patients that determined both RUNX3 methylation and RUNX3 expression by IHC or RT-PCR reported that 55% of the GC cases showed RUNX3 methylation [57]. However, while 68% of the GC patients that did not have RUNX3 methylation did express RUNX3, 41% of patients that did have methylation also expressed RUNX3 [57]. These results suggest that there is no good correlation between RUNX3 methylation and RUNX3 expression.
Since both IHC and the bisulfite-modified methylation-specific PCR (MSP) are not quantitative techniques
Table 2. Association studies implicating other genes and/or genomic regions in GC.
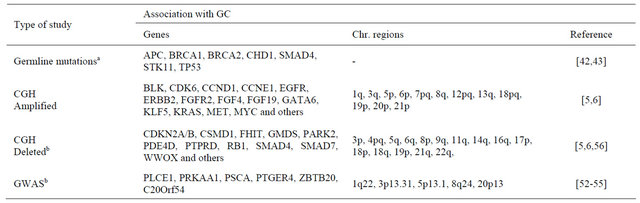
aNo RUNX3 germ-line mutations were detected in familial GC [43]. No RUNX3 mutations were detected in sporadic GC [8]. A single RUNX3 point mutation in one GC patient was described [10]; bChromosome region 1p36.1, in which RUNX3 resides, is not frequently deleted in GC [5,6,56]. This region was also not detected as a GC-associated region in GWAS studies [52-55].
they cannot accurately measure the degree of RUNX3 methylation or its level of expression. RUNX3 methylation was recently measured using a quantitative technique involving methyl-specific DNA microarray and found that only 23% of GC patients showed RUNX3 methylation as compared to 46% with the conventional MSP technique [58]. It is thus reasonable to assume that the actual degree of RUNX3 methylation in GC is lower than originally proposed. Furthermore, it was reported that RUNX3 methylation in GC is mostly monoallelic [59], so even if this could theoretically completely silence expression of just one allele, the remaining allele would still be expressed and functional. In addition, even when RUNX3-P2 promoter is methylated, the remaining unmethyltated P1 promoter could still allow RUNX3 expression to occur.
Finally, as indicated earlier, GC is associated with epigenetic alterations in many genes [9] and previous studies have noted [60-62] that several hundred genes undergo methylation in tumor cell genomes, most of which are not expressed in the normal tissue of origin of these cancers. Therefore, a demonstration of promoter methylation, on its own, does not and cannot represent a proof or even a credible indication/suggestion that methylation is synonymous with silencing and that a methylated gene is a TSG.
8. Does the 1p36 Region Harbor Any TSG?
Human chromosome 1p36 large deletions occur frequently in a variety of cancers including various neural, epithelial and hematopoietic malignancies, suggesting the possible location of one or more TSG in this region [63]. The putative TSG harboring region deleted in many of these cancers was narrowed down to a 4.3 Mb of DNA in the syntenic mouse chromosome 4 region that corresponds to human chromosome 1p36. Using chromosome engineering it was found that an extra copy of this 4.3 Mb region suppressed proliferation and enhanced apoptosis and senescence of cultured cells, whereas a heterozygous deficiency of the region enhanced proliferation and suppressed senescence [64]. This finding is consistent with the possibility that it harbored a TSG [64]. To determine which of the genes that reside in the 4.3 Mb region could be responsible for suppressing proliferation when overexpressed, 11 candidate genes were selected according to their Gene Ontology terms. Knocking down of 10 of these genes did not rescue the suppressed proliferation but knockdown of CHD5, phenocopied the effect of deletion one copy of the 4.3 Mb region. Accordingly, mice heterozygous for the loss of Chd5 spontaneously developed tumors including squamous cell carcinoma, hybernoma (mesenchymal neoplasm) and lymphoma [64] but no GC or other GIT tumors were observed. Thus it appears that while several TSG candidates are located on human chromosome 1p36, including TP73, DNAJC1, and CAMTA1, the locus encoding CHD5 is the major TSG in this region.
9. The Role of RUNX Family Genes in Cancer
Although here we have focused specifically on the issue of RUNX3 in GC, it is worth mentioning that all 3 RUNX genes were found to have cancer connections. The association of RUNX1 to cancer development in humans is multifaceted, including predisposition to development of AML in families with germline RUNX1 point mutations or deletions [65,66], frequent point mutations in AML [67-69], chromosomal rearrangements that create fusion proteins with a variety of partners in various leukemias [70-72] and deletions in breast cancer [73]. Recently it was also reported that epithelium-specific conditional deletion of RUNX1 in mice induces the development of adenomas in the duodenum and significantly enhances development of GIT tumors in the colon, cecum and intestine in APCmin mice [74]. These results are consistent with the finding that Runx1 is expressed in GIT epithelium [12,17] and with the notion that RUNX1 can function as a TSG. However, other studies have shown that RUNX1 acts as an oncogene and when overexpressed plays an important role in leukemia [75] and tumor initiation in various epithelial tumors [76,77].
RUNX2 appears to have an oncogenic role in human breast and prostate cancers as well in mouse leukemia and lymphoma models [78]. RUNX3 has been suggested to act as a TSG in gastrointestinal cancer [10], by regulating the anti-oncogenic TGFb/BMP pathway and attenuating the Wnt/b-catenin pathway [24,79]. However, the finding of RUNX3 overexpression in different types of cancers including GC [26,28-31] may actually reflect an oncogenic role of RUNX3. This possibility is supported by several findings including amplification of the 1p36.11 region in B-cell lymphoma [80], the essential role of EBNA2-mediated induction of RUNX3 expression in EBV-infected B cells for proliferation of the transformed cells [32] and the ability of RUNX3 to enhance leukemia and lymphoma development [81,82]. These findings have recently led to the postulated suggestion that RUNX3 might switch from TSG to oncogenic activity [83]. In sharp contrast to the findings that are consistent with an oncogenic role of RUNX3 in certain human cancers, its role as TSG remains highly dubious. Accordingly RUNX3 is not mentioned in “Cancer Gene Census” http://www.sanger.ac.uk/genetics/CGP/Census/, which lists all the cancer genes.
10. Conclusions
GC is a major cause of worldwide cancer mortality. Thus, studies aimed at identification of genes that contribute to GC development and progression are of utmost importance. Such studies could identify promising gene-targets for potential new therapies. These targets can be either TSG if they can be reactivated or oncogenes if they can be inactivated. RUNX3 has been suggested a decade ago to be a major TSG in GC. Hundreds of following studies involving thousands of GC patients have invested much effort attempting to verify and extend this RUNX3 TSG paradigm. The first and foremost premise of a TSG is that it should be expressed in the normal tissue from which the cancer has arisen. As outlined above, this basic requirement concerning RUNX3 expression in normal GIT has turned out to be false and has been refuted. In addition, other premises inferring RUNX3 TSG function in GC including RUNX3 inactivation by point mutations or LOH are not supported by many genome wide analyses. GWAS have failed to reveal an association of GC with RUNX3 and loss of the 1p36 chromosome 1 region that harbors RUNX3 is not a common event in GC.
The frequent TP53 mutations and the deletions of regions harboring many other well-known TSGs, make them and not RUNX3 the more relevant TSGs in GC biology. In contrast, RUNX3 appears to be overexpressed in various cancers, it can promote malignancy in insertional mutagenesis studies and its activation is essential for the proliferation of EBV-transformed B cells. The frequent amplification of chromosomal regions harboring known oncogenes, especially the RAS/RTK family, suggests that these genes could be promising targets for GC treatment.
11. Acknowledgements
The work was supported by grants from the Israel Science Foundation (ISF) individual grants and the ISF Bio-Med program and by the Commission of the EU (AnEUploidy grant).
REFERENCES
- H. Brenner, D. Rothenbacher and V. Arndt, “Epidemiology of Stomach Cancer,” Methods in Molecular Biology, Vol. 472, 2009, pp. 467-477.
- H. H. Hartgrink, E. P. Jansen, N. C. van Grieken and C. J. van de Velde, “Gastric Cancer,” Lancet, Vol. 374, No. 9688, 2009, pp. 477-490. doi:10.1016/S0140-6736(09)60617-6
- S. Nagini, “Carcinoma of the Stomach: A Review of Epidemiology, Pathogenesis, Molecular Genetics and Chemoprevention,” World Journal of Gastrointestinal Oncology, Vol. 4, No. 7, 2012, pp. 156-169. doi:10.4251/wjgo.v4.i7.156
- M. Fukayama, “Epstein-Barr Virus and Gastric Carcinoma,” Pathology International, Vol. 60, No. 5, 2010, pp. 337-350. doi:10.1111/j.1440-1827.2010.02533.x
- N. Deng, L. K. Goh, H. Wang, et al., “A Comprehensive Survey of Genomic Alterations in Gastric Cancer Reveals Systematic Patterns of Molecular Exclusivity and CoOccurrence among Distinct Therapeutic Targets,” Gut, Vol. 61, No. 5, 2012, pp. 673-684. doi:10.1136/gutjnl-2011-301839
- B. Fan, S. Dachrut, H. Coral, et al., “Integration of DNA Copy Number Alterations and Transcriptional Expression Analysis in Human Gastric Cancer,” PLoS One, Vol. 7, No. 4, 2012, Article ID: e29824. doi:10.1371/journal.pone.0029824
- C. Greenman, P. Stephens, R. Smith, et al., “Patterns of Somatic Mutation in Human Cancer Genomes,” Nature, Vol. 446, No. 7132, 2007, pp. 153-158. doi:10.1038/nature05610
- Z. J. Zang, I. Cutcutache, S. L. Poon, et al., “Exome Sequencing of Gastric Adenocarcinoma Identifies Recurrent Somatic Mutations in Cell Adhesion and Chromatin Remodeling Genes,” Nature Genetics, Vol. 44, No. 5, 2012, pp. 570-574. doi:10.1038/ng.2246
- C. Zhao and X. Bu, “Promoter Methylation of Tumor-ReLated Genes in Gastric Carcinogenesis,” Histology and Histopathology, Vol. 27, No. 10, 2012, pp. 1271-1282.
- Q. L. Li, K. Ito, C. Sakakura, et al., “Causal Relationship between the Loss of RUNX3 Expression and Gastric Cancer,” Cell, Vol. 109, No. 1, 2002, pp. 113-124. doi:10.1016/S0092-8674(02)00690-6
- M. M. Subramaniam, J. Y. Chan, K. G. Yeoh, et al., “Molecular Pathology of RUNX3 in Human Carcinogenesis,” Biochimica et Biophysica Acta, Vol. 1796, No. 2, 2009, pp. 315-331.
- D. Levanon, Y. Bernstein, V. Negreanu, et al., “Absence of Runx3 Expression in Normal Gastrointestinal Epithelium Calls into Question Its Tumour Suppressor Function,” EMBO Molecular Medicine, Vol. 3, No. 10, 2011, pp. 593-604. doi:10.1002/emmm.201100168
- D. Levanon and Y. Groner, “Structure and Regulated Expression of Mammalian RUNX Genes,” Oncogene, Vol. 23, No. 24, 2004, pp. 4211-4219. doi:10.1038/sj.onc.1207670
- D. Levanon, V. Negreanu, Y. Bernstein, et al., “AML1, AML2, and AML3, the Human Members of the Runt Domain Gene-Family: cDNA Structure, Expression, and Chromosomal Localization,” Genomics, Vol. 23, No. 2, 1994, pp. 425-432. doi:10.1006/geno.1994.1519
- K. B. Avraham, D. Levanon, V. Negreanu, et al., “Mapping of the Mouse Homolog of the Human Runt Domain Gene, AML2, to the Distal Region of Mouse Chromosome 4,” Genomics, Vol. 25, No. 2, 1995, pp. 603-605. doi:10.1016/0888-7543(95)80073-U
- C. Bangsow, N. Rubins, G. Glusman, Y. Bernstein, et al., “The RUNX3 Gene-Sequence, Structure and Regulated Expression,” Gene, Vol. 279, No. 2, 2001, pp. 221-232. doi:10.1016/S0378-1119(01)00760-0
- D. Levanon, O. Brenner, V. Negreanu, et al., “Spatial and Temporal Expression Pattern of Runx3 (Aml2) and Runx1 (Aml1) Indicates Non-Redundant Functions during Mouse Embryogenesis,” Mechanisms of Development, Vol. 109, No. 2, 2001, pp. 413-417. doi:10.1016/S0925-4773(01)00537-8
- D. Levanon, D. Bettoun, C. Harris-Cerruti, et al., “The Runx3 Transcription Factor Regulates Development and Survival of TrkC Dorsal Root Ganglia Neurons,” The EMBO Journal, Vol. 21, No. 13, 2002, pp. 3454-3463. doi:10.1093/emboj/cdf370
- K. Ito, K. I. Inoue, S. C. Bae and Y. Ito, “Runx3 Expression in Gastrointestinal Tract Epithelium: Resolving the Controversy,” Oncogene, Vol. 28, No. 10, 2009, pp. 1379- 1384. doi:10.1038/onc.2008.496
- P. Soriano, “Generalized lacZ Expression with the ROSA26 Cre Reporter Strain,” Nature Genetics, Vol. 21, No. 1, 1999, pp. 70-71. doi:10.1038/5007
- S. Srinivas, T. Watanabe, C. S. Lin, et al., “Cre Reporter Strains Produced by Targeted Insertion of EYFP and ECFP into the ROSA26 Locus,” BMC Developmental Biology, Vol. 1, 2001, p. 4. doi:10.1186/1471-213X-1-4
- C. A. Yoshida, H. Yamamoto, T. Fujita, et al., “Runx2 and Runx3 Are Essential for Chondrocyte Maturation, and Runx2 Regulates Limb Growth through Induction of Indian Hedgehog,” Genes & Development, Vol. 18, No. 8, 2004, pp. 952-963. doi:10.1101/gad.1174704
- D. Normile, “Cancer Research. Dispute over Tumor Suppressor Gene Runx3 Boils over,” Science, Vol. 334, No. 6055, 2011, pp. 442-443. doi:10.1126/science.334.6055.442
- K. Ito, A. C. Lim, M. Salto-Tellez, et al., “RUNX3 Attenuates Beta-Catenin/T Cell Factors in Intestinal Tumorigenesis,” Cancer Cell, Vol. 14, No. 3, 2008, pp. 226- 237. doi:10.1016/j.ccr.2008.08.004
- K. Ito, Q. Liu, M. Salto-Tellez, et al., “RUNX3, a Novel Tumor Suppressor, Is Frequently Inactivated in Gastric Cancer by Protein Mislocalization,” Cancer Research, Vol. 65, No. 17, 2005, pp. 7743-7750.
- R. Carvalho, A. N. Milne, M. Polak, et al., “Exclusion of RUNX3 as a Tumour-Suppressor Gene in Early-Onset Gastric Carcinomas,” Oncogene, Vol. 24, No. 56, 2005, pp. 8252-8258. doi:10.1038/sj.onc.1208963
- M. J. Friedrich, R. Rad, R. Langer, et al., “Lack of RUNX3 Regulation in Human Gastric Cancer,” The Journal of Pathology, Vol. 210, No. 2, 2006, pp. 141-146. doi:10.1002/path.2042
- M. Salto-Tellez, B. K. Peh, K. Ito, et al., “RUNX3 Protein Is Overexpressed in Human Basal Cell Carcinomas,” Oncogene, Vol. 25, No. 58, 2006, pp. 7646-7649. doi:10.1038/sj.onc.1209739
- Y. Kudo, T. Tsunematsu and T. Takata, “Oncogenic Role of RUNX3 in Head and Neck Cancer,” Journal of Cellular Biochemistry, Vol. 112, No. 2, 2011, pp. 387-393. doi:10.1002/jcb.22967
- C. W. Lee, L. S. Chuang, S. Kimura, et al., “RUNX3 Func- Tions as an Oncogene in Ovarian Cancer,” Gynecologic Oncology, Vol. 122, No. 2, 2011, pp. 410-417. doi:10.1016/j.ygyno.2011.04.044
- J. Li, J. Kleeff, A. Guweidhi, et al., “RUNX3 Expression in Primary and Metastatic Pancreatic Cancer,” Journal of Clinical Pathology, Vol. 57, No. 3, 2004, pp. 294-299. doi:10.1136/jcp.2003.013011
- G. Brady, H. J. Whiteman, L. C. Spender and P. J. Farrell, “Downregulation of RUNX1 by RUNX3 Requires the RUNX3 VWRPY Sequence and Is Essential for Epstein-Barr Virus-Driven B-Cell Proliferation,” Journal of Virology, Vol. 83, No. 13, 2009, pp. 6909-6916. doi:10.1128/JVI.00216-09
- M. Osaki, M. Moriyama, K. Adachi, et al., “Expression of RUNX3 Protein in Human Gastric Mucosa, Intestinal Metaplasia and Carcinoma,” European Journal of Clinical Investigation, Vol. 34, No. 9, 2004, pp. 605-612. doi:10.1111/j.1365-2362.2004.01401.x
- N. A. Bhowmick, E. G. Neilson and H. L. Moses, “Stromal Fibroblasts in Cancer Initiation and Progression,” Nature, Vol. 432, No. 7015, 2004, pp. 332-337. doi:10.1038/nature03096
- P. Katajisto, K. Vaahtomeri, N. Ekman, et al., “LKB1 Signaling in Mesenchymal Cells Required for Suppression of Gastrointestinal Polyposis,” Nature Genetics, Vol. 40, No. 4, 2008, pp. 455-459. doi:10.1038/ng.98
- B. G. Kim, C. Li, W. Qiao, et al., “Smad4 Signalling in T Cells Is Required for Suppression of Gastrointestinal Cancer,” Nature, Vol. 441, No. 7096, 2006, pp. 1015-1019. doi:10.1038/nature04846
- O. Brenner, D. Levanon, V. Negreanu, et al., “Loss of Runx3 Function in Leukocytes Is Associated with Spontaneously Developed Colitis and Gastric Mucosal Hyperplasia,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 101, No. 45, 2004, pp. 16016-16021. doi:10.1073/pnas.0407180101
- O. Fainaru, E. Woolf, J. Lotem, et al., “Runx3 Regulates Mouse TGF-Beta-Mediated Dendritic Cell Function and Its Absence Results in Airway Inflammation,” The EMBO Journal, Vol. 23, No. 4, 2004, pp. 969-979. doi:10.1038/sj.emboj.7600085
- I. Taniuchi, M. Osato, T. Egawa, et al., “Differential Requirements for Runx Proteins in CD4 Repression and Epigenetic Silencing during T Lymphocyte Development,” Cell, Vol. 111, No. 5, 2002, pp. 621-633. doi:10.1016/S0092-8674(02)01111-X
- E. Woolf, C. Xiao, O. Fainaru, et al., “Runx3 and Runx1 Are Required for CD8 T Cell Development during Thymopoiesis,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 13, 2003, pp. 7731-7736. doi:10.1073/pnas.1232420100
- A. H. Berger, A. G. Knudson and P. P. Pandolfi, “A Continuum Model for Tumour Suppression,” Nature, Vol. 476, No. 7359, 2011, pp. 163-169. doi:10.1038/nature10275
- N. Chun and J. M. Ford, “Genetic Testing by Cancer Site: Stomach,” The Cancer Journal, Vol. 18, No. 4, 2012, pp. 355-363. doi:10.1097/PPO.0b013e31826246dc
- G. Keller, H. Vogelsang, I. Becker, et al., “Germline Mutations of the E-Cadherin(CDH1) and TP53 Genes, rather than of RUNX3 and HPP1, Contribute to genetic Predisposition in German Gastric Cancer Patients,” Journal of Medical Genetics, Vol. 41, No. 6, 2004, p. e89. doi:10.1136/jmg.2003.015594
- D. Wu, Y. Tian, W. Gong, et al., “Genetic Variants in the Runt-Related Transcription Factor 3 Gene Contribute to Gastric Cancer Risk in a Chinese Population,” Cancer Science, Vol. 100, No. 9, 2009, pp. 1688-1694. doi:10.1111/j.1349-7006.2009.01229.x
- A. Hishida, K. Matsuo, Y. Goto, et al., “Significant Association of RUNX3 T/A Polymorphism at Intron 3 (rs760805) with the Risk of Gastric Atrophy in Helicobacter Pylori Seropositive Japanese,” Journal of Gastroenterology, Vol. 44, No. 12, 2009, pp. 1165-1171. doi:10.1007/s00535-009-0118-7
- C. Guo, F. Yao, K. Wu, et al., “Chromatin Immunoprecipitation and Association Study Revealed a Possible Role of Runt-Related Transcription Factor 3 in the Ulcerative Colitis of Chinese Population,” Clinical Immunology, Vol. 135, No. 3, 2010, pp. 483-489. doi:10.1016/j.clim.2010.01.004
- R. K. Weersma, L. Zhou, I. M. Nolte, et al., “Runt-Related Transcription Factor 3 Is Associated with Ulcerative Colitis and Shows Epistasis with Solute Carrier Family 22, Members 4 and 5,” Inflammatory Bowel Diseases, Vol. 14, No. 12, 2008, pp. 1615-1622. doi:10.1002/ibd.20610
- P. C. Dubois, G. Trynka, L. Franke, et al., “Multiple Com- mon Variants for Celiac Disease Influencing Immune Gene Expression,” Nature Genetics, Vol. 42, No. 4, 2010, pp. 295-302. doi:10.1038/ng.543
- D. M. Evans, C. C. Spencer, J. J. Pointon, et al., “Interaction between ERAP1 and HLA-B27 in Ankylosing Spondylitis Implicates Peptide Handling in the Mechanism for HLA-B27 in Disease Susceptibility,” Nature Genetics, Vol. 43, No. 8, 2011, pp. 761-767. doi:10.1038/ng.873
- L. C. Tsoi, S. L. Spain, J. Knight, et al., “Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity,” Nature Genetics, Vol. 44, No. 12, 2012, pp. 1341-1348. doi:10.1038/ng.2467
- M. Sugai, K. Aoki, M. Osato, et al., “Runx3 Is Required for Full Activation of Regulatory T Cells to Prevent Colitis-Associated Tumor Formation,” The Journal of Immunology, Vol. 186, No. 11, 2011, pp. 6515-6520. doi:10.4049/jimmunol.1001671
- Y. Shi, Z. Hu, C. Wu, et al., “A Genome-Wide Association Study Identifies New Susceptibility Loci for NonCardia Gastric Cancer at 3q13.31 and 5p13.1,” Nature Genetics, Vol. 43, No. 12, 2011, pp. 1215-1218. doi:10.1038/ng.978
- C. C. Abnet, N. D. Freedman, N. Hu, et al., “A Shared Susceptibility Locus in PLCE1 at 10q23 for Gastric Adenocarcinoma and Esophageal Squamous Cell Carcinoma,” Nature Genetics, Vol. 42, No. 9, 2010, pp. 764- 767. doi:10.1038/ng.649
- H. Sakamoto, K. Yoshimura, N. Saeki, et al., “Genetic Variation in PSCA Is Associated with Susceptibility to Diffuse-Type Gastric Cancer,” Nature Genetics, Vol. 40, No. 6, 2008, pp. 730-740. doi:10.1038/ng.152
- L. D. Wang, F. Y. Zhou, X. M. Li, et al., “Genome-Wide Association Study of Esophageal Squamous Cell Carcinoma in Chinese Subjects Identifies Susceptibility Loci at PLCE1 and C20orf54,” Nature Genetics, Vol. 42, No. 9, 2010, pp. 759-763. doi:10.1038/ng.648
- Y. Tsukamoto, T. Uchida, S. Karnan, et al., “GenomeWide Analysis of DNA Copy Number Alterations and Gene Expression in Gastric Cancer,” The Journal of Pathology, Vol. 216, No. 4, 2008, pp. 471-482. doi:10.1002/path.2424
- S. L. Hu, D. B. Huang, Y. B. Sun, et al., “Pathobiologic Implications of Methylation and Expression Status of Runx3 and CHFR Genes in Gastric Cancer,” Medical Oncology, Vol. 28, No. 2, 2011, pp. 447-454. doi:10.1007/s12032-010-9467-6
- G. Tamura, K. So, H. Miyoshi, et al., “Quantitative Assessment of Gene Methylation in Neoplastic and NonNeoplastic Gastric Epithelia Using Methylation-Specific DNA Microarray,” Pathology International, Vol. 59, No. 12, 2009, pp. 895-899. doi:10.1111/j.1440-1827.2009.02458.x
- H. J. Song, K. N. Shim, Y. H. Joo, et al., “Methylation of the Tumor Suppressor Gene RUNX3 in Human Gastric Carcinoma,” Gut and Liver, Vol. 2, No. 2, 2008, pp. 119- 125. doi:10.5009/gnl.2008.2.2.119
- D. Sproul, C. Nestor, J. Culley, et al., “Transcriptionally Repressed Genes Become Aberrantly Methylated and Distinguish Tumors of Different Lineages in Breast Cancer,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 108, No. 11, 2011, pp. 4364-4369. doi:10.1073/pnas.1013224108
- E. N. Gal-Yam, G. Egger, L. Iniguez, et al., “Frequent Switching of Polycomb Repressive Marks and DNA Hypermethylation in the PC3 Prostate Cancer Cell Line,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 105, No. 35, 2008, pp. 12979-12984. doi:10.1073/pnas.0806437105
- I. Keshet, Y. Schlesinger, S. Farkash, et al., “Evidence for an Instructive Mechanism of De Novo Methylation in Cancer Cells,” Nature Genetics, Vol. 38, No. 2, 2006, pp. 149-153. doi:10.1038/ng1719
- A. Bagchi and A. A. Mills, “The Quest for the 1p36 Tumor Suppressor,” Cancer Research, Vol. 68, No. 8, 2008, 2551-2556. doi:10.1158/0008-5472.CAN-07-2095
- A. Bagchi, C. Papazoglu, Y. Wu, et al., “CHD5 Is a Tumor Suppressor at Human 1p36,” Cell, Vol. 128, No. 3, 2007, pp. 459-475. doi:10.1016/j.cell.2006.11.052
- C. Preudhomme, D. Warot-Loze, C. Roumier, et al., “High Incidence of Biallelic Point Mutations in the Runt Domain of the AML1/PEBP2 Alpha B Gene in Mo Acute Myeloid Leukemia and in Myeloid Malignancies with Acquired Trisomy 21,” Blood, Vol. 96, No. 8, 2000, pp. 2862-2869.
- W. J. Song, M. G. Sullivan, R. D. Legare, et al., “Haploin- sufficiency of CBFA2 Causes Familial Thrombocytopenia with Propensity to Develop Acute Myelogenous Leukaemia,” Nature Genetics, Vol. 23, No. 2, 1999, pp. 66-75. doi:10.1038/13793
- M. Osato, “Point Mutations in the RUNX1/AML1 Gene: Another Actor in RUNX Leukemia,” Oncogene, Vol. 23, No. 24, 2004, pp. 4284-4296. doi:10.1038/sj.onc.1207779
- C. Roumier, V. Eclache, M. Imbert, et al., “M0 AML, Clinical and Biologic Features of the Disease, Including AML1 Gene Mutations: A Report of 59 Cases by the Groupe Francais d’Hematologie Cellulaire (GFHC) and the Groupe Francais de Cytogenetique Hematologique (GFCH),” Blood, Vol. 101, No. 4, 2003, pp. 1277-1283. doi:10.1182/blood-2002-05-1474
- S. Schnittger, F. Dicker, W. Kern, et al., “RUNX1 Mutations Are Frequent in De Novo AML with Noncomplex Karyotype and Confer an Unfavorable Prognosis,” Blood, Vol. 117, No. 8, 2011, pp. 2348-2357. doi:10.1182/blood-2009-11-255976
- J. R. Downing, “The Core-Binding Factor Leukemias: Lessons Learned from Murine Models,” Current Opinion in Genetics & Development, Vol. 13, No. 1, 2003, pp. 48-54. doi:10.1016/S0959-437X(02)00018-7
- S. M. Hart and L. Foroni, “Core Binding Factor Genes and Human Leukemia,” Haematologica, Vol. 87, No. 12, 2002, pp. 1307-1323.
- J. D. Rowley, “The Role of Chromosome Translocations in Leukemogenesis,” Seminars in Hematology, Vol. 36, No. 4, 1999, pp. 59-72.
- S. Banerji, K. Cibulskis, C. Rangel-Escareno, et al., “Sequence Analysis of Mutations and Translocations across Breast Cancer Subtypes,” Nature, Vol. 486, No. 7403, 2012, pp. 405-409. doi:10.1038/nature11154
- R. J. Fijneman, R. A. Anderson, E. Richards, et al., “Runx1 Is a Tumor Suppressor Gene in the Mouse Gastrointestinal tract,” Cancer Science, Vol. 103, No. 3, 2012, pp. 593- 599. doi:10.1111/j.1349-7006.2011.02189.x
- E. R. Cameron and J. C. Neil, “The Runx Genes: Lineage-Specific Oncogenes and Tumor Suppressors,” Oncogene, Vol. 23, No. 24, 2004, pp. 4308-4314. doi:10.1038/sj.onc.1207130
- C. S. Hoi, S. E. Lee, S. Y. Lu, et al., “Runx1 Directly Pro-Motes Proliferation of Hair Follicle Stem Cells and Epithelial Tumor Formation in Mouse Skin,” Molecular and Cellular Biology, Vol. 30, No. 10, 2010, pp. 2518- 2536. doi:10.1128/MCB.01308-09
- C. J. Scheitz, T. S. Lee, D. J. McDermitt and T. Tumbar, “Defining a Tissue Stem Cell-Driven Runx1/Stat3 Signalling Axis in Epithelial Cancer,” The EMBO Journal, Vol. 31, No. 21, 2012, pp. 4124-4139 doi:10.1038/emboj.2012.270
- K. Blyth, F. Vaillant, A. Jenkins, et al., “Runx2 in Normal Tissues and Cancer Cells: A Developing Story,” Blood Cells, Molecules and Diseases, Vol. 45, No. 2, 2010, pp. 117-123. doi:10.1016/j.bcmd.2010.05.007
- K. Ito, “RUNX3 in Oncogenic and Anti-Oncogenic Signaling in Gastrointestinal Cancers,” Journal of Cellular Biochemistry, Vol. 112, No. 5, 2011, pp. 1243-1249. doi:10.1002/jcb.23047
- F. Kreisel, S. Kulkarni, R. T. Kerns, et al., “High Resolution Array Comparative Genomic Hybridization Identifies Copy Number Alterations in Diffuse Large B-Cell Lymphoma That Predict Response to Immuno-Chemotherapy,” Cancer Genetics, Vol. 204, No. 3, 2011, pp. 129-137. doi:10.1016/j.cancergen.2010.12.010
- K. Blyth, E. R. Cameron and J. C. Neil, “The RUNX Genes: Gain or Loss of Function in Cancer,” Nature Reviews Cancer, Vol. 5, No. 5, 2005, pp. 376-387. doi:10.1038/nrc1607
- E. R. Cameron, K. Blyth, L. Hanlon, et al., “The Runx Genes as Dominant Oncogenes,” Blood Cells, Molecules and Diseases, Vol. 30, No. 2, 2003, pp. 194-200. doi:10.1016/S1079-9796(03)00031-7
- L. S. Chuang and Y. Ito, “RUNX3 Is Multifunctional in Carcinogenesis of Multiple Solid Tumors,” Oncogene, Vol. 29, No. 18, 2010, pp. 2605-2615. doi:10.1038/onc.2010.88
NOTES
*Corresponding author.