Health
Vol.4 No.10A(2012), Article ID:24241,4 pages DOI:10.4236/health.2012.430143
Antibody administration in experimental influenza increases survival and enhances the effect of oseltamivir
![]()
1Department of Virology, Statens Serum Institute, Copenhagen, Denmark; *Corresponding Author: brit@bsworld.dk
2Research Unite of Clinical Microbiology, Institute of Clinical Research, University of Southern Denmark, Odense, Denmark
Received 16 September 2012; revised 14 October 2012; accepted 26 October 2012
Keywords: Experimental Influenza; Passively Administrated Antibodies; Convalescent Plasma; Passive Immunisation; Oseltamivir; Lethal Infection; Polyclonal IgG; Pandemic Control
ABSTRACT
Anti-viral chemotherapy plays an important part in treating and preventing influenza illness. However, its effectiveness in severe infections can be debated and a reoccurring problem is the emergence of resistant virus. Passive immunisation has for a long time been and is still used for prophylaxis and treatment of a number of infectious diseases. In this experimental study anti-influenza antibodies were passively administrated to mice, subsequently they were infected with influenza virus and treated with oseltamivir. The aim was to investigate, if anti-influenza antibodies influenced the out come of oseltamivir treatment and development of resistance towards oseltamivir. We show, that oseltamivir alone was not able to effectively prevent a fatal outcome, but that oseltamivir administered together with a limited amount of antibodies, resulted in improvement of the clinical condition of the mice. The results also showed that a higher dosage of antibodies alone were able to protect the mice from a lethal dose of virus. These findings suggest that the effectiveness of oseltamivir depends on the host’s immune response to the influenza virus, and that that passive immunization is an option that should be considered in the in control of influenza.
1. INTRODUCTION
Passive immunisation has for a long time been and is still used for prophylaxis and treatment of a number of infectious diseases [1]. Since the 2009 pandemic, the proposal of using convalescent plasma as an early control measurement, in a future influenza pandemic or as treatment for severely ill patients, has been raised by several voices [2-6].
Though there have been recent successful applications of passive immunisation as treatment for human influenza infections, in experimental settings [7-12], further investigations are warranted before it can be implemented in the control of influenza. Support for the passive immunisation strategy also increases, as passive transfer of immune sera followed by lethal challenge in experimental animals, is becoming at standard in confirming new vaccines ability to provide protection [13- 19].
Broadly cross neutralising or even potential universal monoclonal antibody that neutralises several different influenza subtypes has been identified. Passive transfer treatment with such anti-influenza antibodies or convalescent plasma administration, may not only be effective in preventing disease but could also minimize transmission of the virus and therefore have influence on the spread of the virus during a future pandemic [14,20-22]. However, the use of such antibodies is expected to be expensive and escape mutants may develop making such treatment less efficient.
Oseltamivir is used for the treatment of influenza, and the drug remains one of the important strategies in the influenza pandemic preparedness. Thus development of resistance to oseltamivir is an important issue. Initial clinical studies after introduction of the drug revealed that emerging of resistance to oseltamivir during treatment was much more common in children (0% - 27.3%) than in adults (0% - 4%) [23-28]. Immunocompromised patients also showed elevated rates of resistance (13.3% - 33.3%) [29,30].
The increased risk of oseltamivir resistance mutations in children and immunocompromised patients could reflect that they are immunologically naïve. This absence of immunity against influenza A, in the beginning of an infection, leads to a higher level of virus replication, which in turn leads to an augmentation in the number of mutations. This implies an increased risk of oseltamivir resistant mutants, which can be extrapolated to at pandemic situation where the population at large is naive.
Influenza viral infection is not a natural disease in mice, thus these animals have no existing immune response to influenza virus. Influenza infections in these animals are therefore comparable to young children that are infected with influenza for the first time and with adults who get infected with a new pandemic virus, to which they have no prior immunity.
In this work we investigated the dosage dependent effect of administrated antibodies with regard to morbidity and mortality from an otherwise lethal influenza infection. And we investigated the relationship of immune status on the effect of oseltamivir, including development of resistance, in BALB/c mice with a severe influenza infection.
The susceptibility of mice to influenza viruses varies among the different mouse strains, but it also depends on the influenza strain [31,32]. In this model for severe influenza infection we chose BALB/c mice, as they are known to get severe lung infection when infected with mouse pathogenic influenza strains [33]. Furthermore, these mice are also widely used in testing of antiviral drugs against influenza [34-37] and vaccine studies [38- 40]. Ison et al. have previously shown development of oseltamivir resistance in a BALB/c mouse model [41].
2. MATERIALS AND METHODS
All experiments were approved by The National Animal Ethics Committee according to Danish regulations on animal welfare (case nr. 2007/561-1379 and 2009/ 561-1611). They were performed under bio safety level 2 conditions and carried out at the animal facility, Statens Serum Institut with regard to the LD90 experiments, otherwise at the Biomedical laboratory, University of Southern Denmark
2.1. Virus and Antivirals
The influenza A vaccine strain A/Puerto Rico/8/34 (H1N1) (PR8) was propagated in Madin-Darby Canine Kidney (MDCK) cells to a 50% tissue culture infectious dose (TCID50) of 107/ml. Oseltamivir capsules 75 mg (Roche) were used for treatment of the mice. The powder was suspended in sterile water and administered orally, twice daily, to a final dose of 10 mg/kg bodyweight/day. The treatment was commenced 24 h after inoculation and continued for 5 days.
2.2. Mice
Female BALB/c O1a Hsd mice from Harland; 8 - 9 weeks old at the time of inoculation were used. The mice were challenged by administrating 50 µl of a virus dilution to each nostril. Prior to the intra nasal viral inoculation the mice were anesthetized with isofluran by inhalation for the 90% lethal dose (LD90) experiments. For the rest of the experiments the mice were anesthetized with a mixture of Propofol (0.15 ml per 20 g mice) and Medetomidine (0.02 ml per 20 g mice) given i.p. and their recovery was aided by Antisedan (0.10 ml per 20 g mice) i.p.
There were 8 animals per cage in one of the shorter experiments, otherwise a maximum of 5 animals per cage.
2.3. Surveillance of Clinical Illness and Human Endpoints
After infection the mice were monitored a least every 12th hour. At these time points they were weighed and their temperature was followed using either an electronic rectal probe or a subcutaneous chip and a handheld reader. In addition they were observed for clinical symptoms; ruffled fur, tweaked eyes, hyperventilation and isolation from the group.
A weight loss of 20%, a temperature below 34˚C on two consecutive measurements or a temperature below 32˚C once were used as humane endpoints at which the mouse was terminated.
2.4. LD90
A 10-fold dilution series of A/PR/8/34(H1N1) TCID50 of 107/ml were made with sterile water and 6 groups of 8 - 10 mice were inoculated (group 1: undiluted virus; group 2: 10–1; group 3: 10–2; group 4: 10–3; group 5: 10–4 and group 6: 10–5). A control group of 3 mice received sterile water as inoculum. The mice from group 4 - 6 were bled prior to termination. The LD90 dose was determined by the Reed-Muench method [42].
2.5. Purifications of Antibodies for Passive Immunization
Antibodies from mice infected with PR8 in the LD90 experiment and a subsequent experiment with 30 mice, were purified from serum with the Montage® Antibody Purification Kit and Spin Columns with PROSEP®—A Media from Millipore. IgG concentrations were 1.32 mg/ml and 12.5 mg/ml first and second purification respectively.
2.6. Haemagglutination Inhibition (HAI) Assay
HAI Assay of antibodies was performed according to standard protocols (WHO Manual on Animal Influenza Diagnosis and Surveillance from 2002). Briefly, the antibodies were diluted initially 1:3 in receptor-destroying enzyme from Vibrio cholerae and incubated overnight at 37˚C. The receptor-destroying enzyme was there after inactivated at 56˚C for 30 min. Another six volumes of isotonic saline was added to give a final dilution of antibodies of 1:10. The diluted antibodies were tested for non-specific agglutinins, and were negative.
A two fold serial dilutions of antibodies were pre-incubated with 8 haemagglutination units of virus per well. The content of the plate was mixed by manual agitation and incubated for 15 minutes at room temperature. Guinea pig red blood cells were added to a final concentration of 0.75%, after which the plate was mixed again by manual agitation and incubated at room temperature for 60 min. The HAI titer is the reciprocal of the last dilution of antiserum that completely inhibits haemagglutination.
2.7. Titration of Antibodies in Mice
Three groups of five mice were immunised i.p. 3 days prior to inoculation with PR8. They received an amount of antibodies corresponding to 14 HAI (46.2 μg), 28 HAI (92.4 μg) and 42 HAI (138.6 μg) respectively. The antibodies were diluted in sterile NaCl 0.9%, to a total volume of 100 µl. A fourth group was injected with isotonic saline as a control; this group was not challenged with virus. In a second experiment, a group of 5 mice received antibodies from a second purification corresponding to 21 HAI (82.0 μg) i.p., again 3 days prior to inoculation with A/PR8/34(H1N1). All mice in the challenged groups were inoculated with an LD90 dose of A/PR/ 8/34(H1N1).
2.8. Oseltamivir and Passive Immunisation Experiment
Four groups of mice were all inoculated with an LD90 dose of A/PR/8/34(H1N1). One group of two mice received no further treatment. A second group of two mice were immunised i.p. 3 days prior to inoculation with an amount of antibodies corresponding to 14 HAI. The third and fourth groups contained 13 mice each, which all received oseltamivir treatment, group 4 was in addition immunised i.p. 3 days prior to inoculation like group 2.
Pharyngeal swabs were taken from all mice daily, with Minitip Flocked Swabs (microRheologics) and stored in phosphate buffered saline, in order to quantitate the virus by real-time PCR.
2.9. RNA Extraction
Viral RNA was extracted from 200 μl of mouse pharyngeal swab suspension or from 200 μl of 10 fold dilutions of the cultured A/PR8/34(H1N1) virus, by an automated MagNA Pure LC Instrument applying the MagNA Pure LC Total Nucleic Acid Isolation Kit (Roche Diagnostics), elution volume was 100 μl.
2.10. Real-Time PCR
The real-time RT-PCR was set up using the OneStep RT-PCR Kit (QIAGEN) along with primers and a FAM labelled probe targeting the matrix gene. Five µl purified nucleic acid was added to 20 µl mastermix. The RNA sequence was amplified on an Mx3005p thermocycler (Stratagene) using the following programme: 20 min at 50˚C (RT-step), 15 min at 95˚C (denaturation of the DNA polymerase), 45 cycles of: [15 sec at 95˚C (denaturation), 60 sec at 55˚C (annealing and extension)].
2.11. Sanger Sequencing
A 200 bp sequence around the H274Y mutation site in the NA gene, was amplified using the OneStep RT-PCR Kit (QIAGEN), using the following program: 30 min at 50˚C (RT-step), 15 min at 95˚C (denaturation of the DNA polymerase), 45 cycles of: [30 sec at 94˚C (denaturation), 30 sec at 58˚C (annealing) 2 min at 72˚C (extension)] followed by 10 min at 72˚C.
Five µl purified nucleic acid was added to 45 µl mastermix (F-primer: GGCTGGCCTCGTACAAAAT and R-primer: CCCCACTGCAGATGTATCCT).
PCR products were purified using the GFX™ PCR DNA or Gel Band Purification Kit (GE Healthcare) prior to sequencing. For some of the samples it was necessary to perform a nested PCR, to get sufficient product.
The first PCR was performed as above with the following primers;
Forward: TATTACGCGTCAGGAGCAAAAGCAGGAGT
Reverse: ATATGGCGCCGTATTAGTAGAAACAAGGAGTTTTTT.
The second PCR was performed on 5 µl of the first PCR reactions as template and was run like above.
The sequencing reaction was performed by ABI PRISM® BigDye™ Terminators v1.1 Cycle Sequencing Kit (Applied Biosystems) using 25 ng template, according to the following program: 2 min at 96˚C (denaturation), 25 cycles of: [10 sec at 96˚C (denaturation), 5 sec at 50˚C (annealing) 4 min at 60˚C (extension)]. They were developed on an automatic ABI PRISM® 3130 genetic analyzer (Applied Biosystems) with 80 cm capillaries.
The basecalling in the electropherograms was verified in Sequence Scanner software (Applied Biosystems). Sequence assembly, multiple alignment and alignment trimming were performed with the BioEdit software v.7.0.0.
2.12. Statistics
The p-values for the survival data, was calculated using Fisher’s exact one tailed probability test. α < 0.05 was considered significant.
3. RESULTS
3.1. Lethal Infection
A lethal infection model with influenza A virus A/PR/ 8/34(H1N1) was established in BALB/c mice, without prior adaption. The LD90 titer was established using the humane endpoints; 20% weight loss, temperature below 34˚C on two consecutive measurements or temperature below 32˚C, as surrogates for death. Survival curves from titration of influenza virus in the mice can be seen in Figure 1, along with the changes in body weight for three of the groups. Infection with undiluted virus (TCID50: 107/ml) gave a very rapid onset of clinical ill-
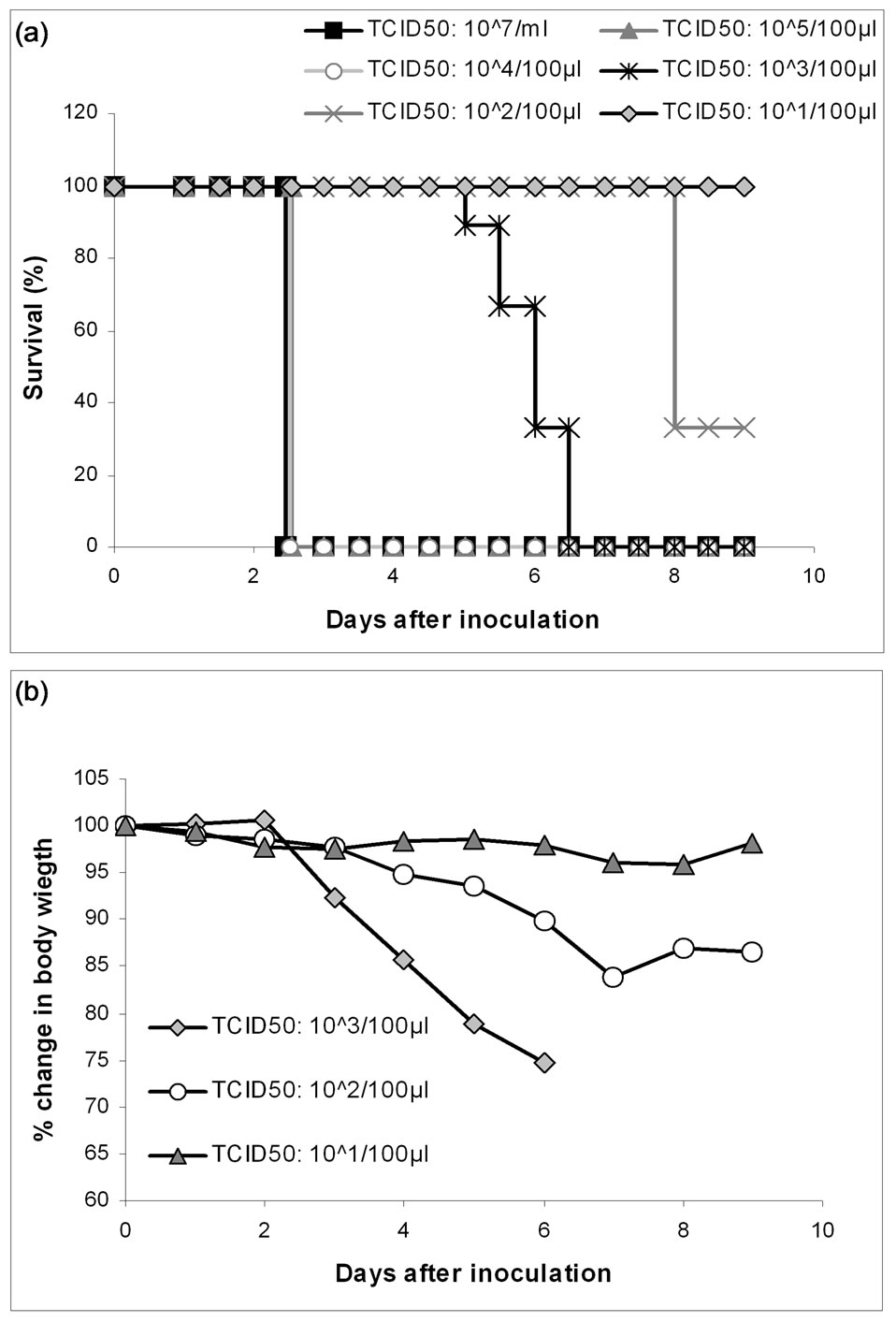
Figure 1. LD90 experiment. 6 groups of mice were inoculated intranasally with a 10 fold titration from the undiluted TCID50: 107/ml A/PR/8/34(H1N1) to TCID50: 101/100 µl respectively, to determine the LD90 dosage. Panel A pictures the survival of the mice from the 6 groups and panel B presents the average weight loss in percent for the three groups receiving the lowest dosages.
ness, and after only 30 h most of the mice were terminally ill. In the mice inoculated with virus diluted to TCID50: 105/100 µl or 104/100 µl the symptoms were delayed with approximately 18 h, but all together these infections resulted in LD100 after only 55 h. Group 4 received virus diluted to TCID50: 103/100 µl. The mice in this group did not begin to display signs of clinical illness until after 56 h and LD100 was reached on day 6. The onset of illness was delayed even further for group 5, where mice were challenged with virus diluted to TCID50: 102/100 µl. In this group 3 mice recovered and lived at day 9, when the experiment ended. The last group received virus diluted to TCID50: 101/100 µl. These mice did, with the exception of 2, not show any signs of clinical illness, and all survived to day 9. LD90 titer of the virus was determined to be 103.33/ml.
3.2. Passive Immunisation
In an initial immunisation experiment antibodies were titrated in mice, 15 control mice were challenged without prior immunisation. Of these 15, one survived to day 11 (7%). In the immunisation groups of 5 mice, 14, 28 and 42 HAI units of antibodies against the virus were given respectively, three days before viral inoculation. From the first group none (0%) of the mice survived past day 9 and the severity of the illness was similar to the group without any prior immunisation. In the groups receiving double and triple dose 4 and 5 mice, respectively, survived to day 11 (80% and 100%). Survival curves and the changes in body weight, from the antibody titration is illustrated in Figure 2. These differences are statistically significant with a P-value of 0.0049 for the double dose group and 0.0003 for the triple dose group, compared to the control group respectively. Some of these mice did show symptoms of influenza illness, but apart from the one that was terminated all recovered.
The mice in an immunisation control group showed no adverse effects with regard to weight or temperature nor with regard to the other clinical symptoms described above.
In the subsequent experiment all (100%) of the mice immunised with 21 HAI units of antibodies, from the second purification, survived to day 13 (see Figure 2) and they showed no signs of illness, whereas none of the mice that, in this second experiment, were challenged without prior immunisation survived past day 5 (0%) (data not shown). This is a statistically significant difference with a P-value of 0.0003, compared to the control group.
3.3. Oseltamivir and Passive Immunisation Experiment
In the “virus only” control group two mice were infected with A/PR/8/34(H1N1). Both of these were ter-
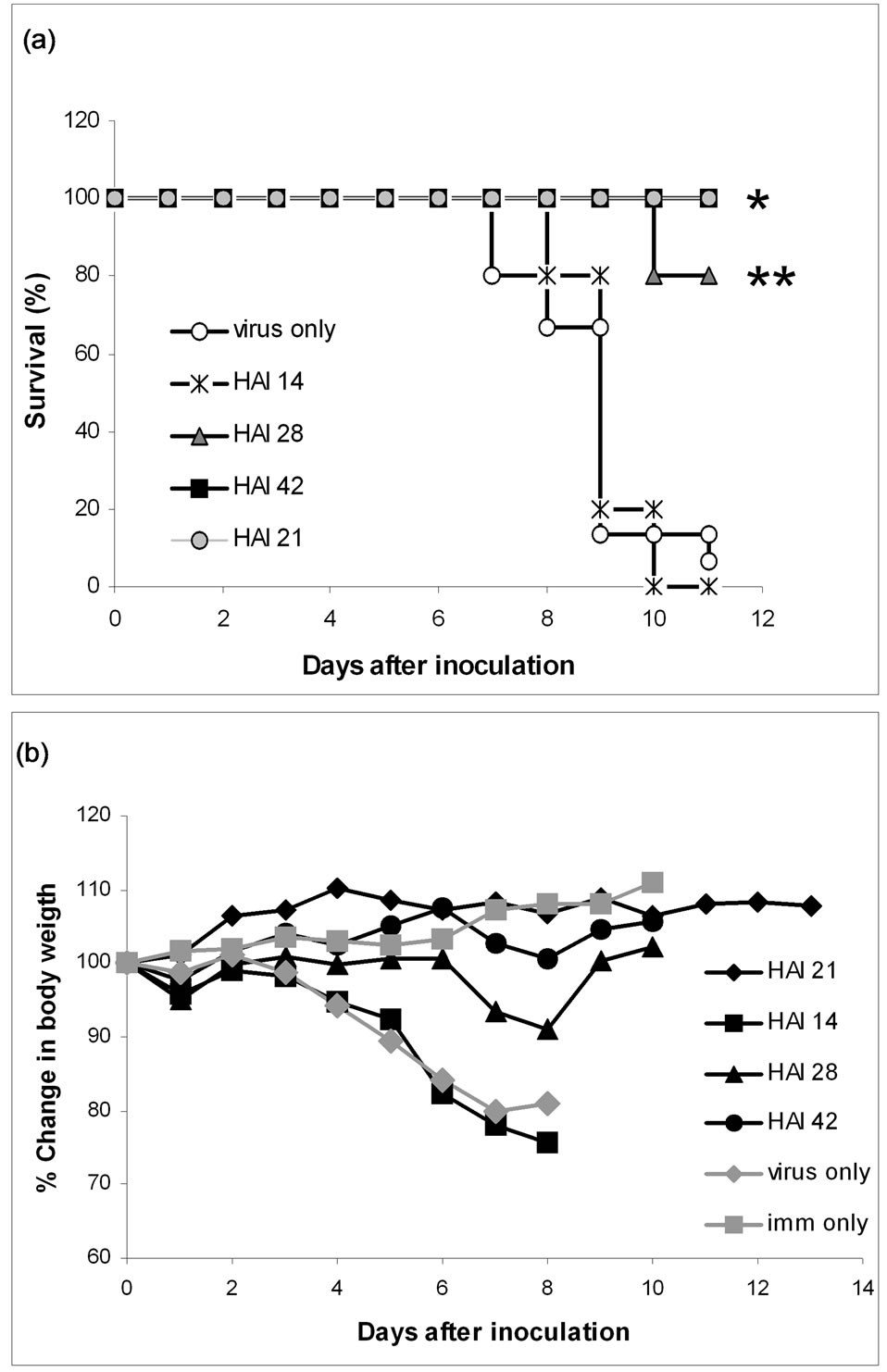
Figure 2. Passive immunisation experiments. Four groups of mice were respectively immunised with 14, 28 and 42 HAI units of antibodies against A/PR/8/34(H1N1), three days prior to inoculation with the same virus. Two control groups were included; one group (imm only) was injected with isotonic saline at the time of immunisation and was not challenged with virus. The other control group (virus only) was challenged with virus but not immunised. The data summarises two experiments and the antibodies used to immunise the 21 HAI units group was from a different purification and the immunisation was performed in the 2 experiments. Panel A pictures the survival of the mice from the 6 groups and panel B presents the average weight loss in percent. *P = 0.0003, **P = 0.0049.
minated after 4 days. In the other control group “immunisation” the mice had received antibodies prior to infection; these two mice were terminated after 5.5 days, so the immunisation dosage had the desired effect of conveying some protection without hindering the course of infection. Comparing the two oseltamivir treated groups “oseltamivir” and “immunisation + oseltamivir” we observed a clear effect of the antibodies, as 10 out of 13 mice (76.9%) in the immunised the group survived to day 9, opposed to 3 out of 13 (23%) in the group which only received oseltamivir (see survival curves in Figure 3). The difference between the two groups is statistically
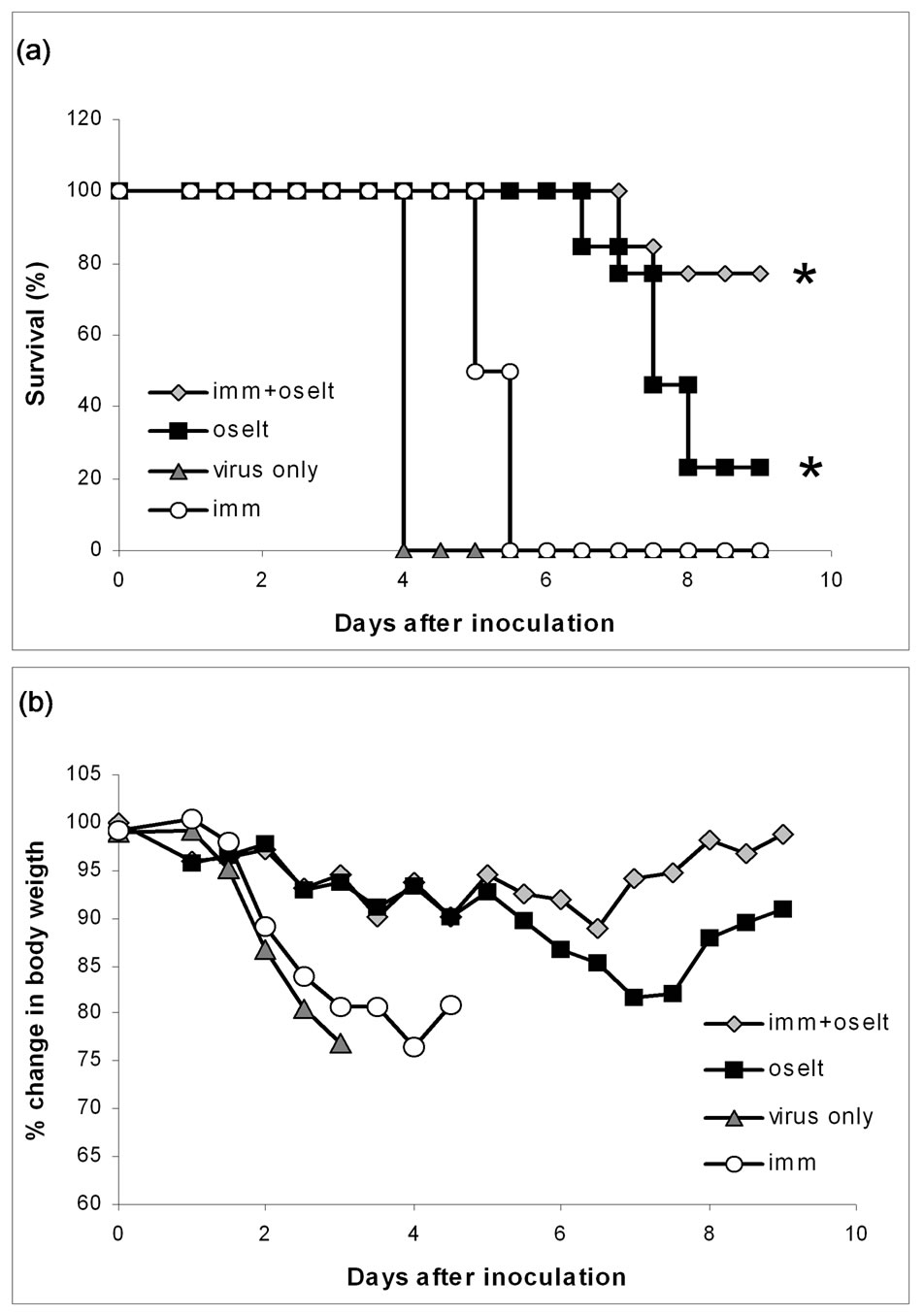
Figure 3. Oseltamivir/passive immunisation experiment. Two investigate whether circulating anti A/PR/8/34(H1N1) antibodies prior to challenge with A/PR/8/34(H1N1) virus could enhance the effect of oseltamivir; two groups of mice were inoculated with A/PR/8/34(H1N1). Tree days earlier, one of the groups had been immunised passively with 14 HAI units of antibodies against the virus (imm + oselt) the other group (oselt) was not immunised. Both groups received oseltamivir treatment for 5 days, starting at day one. Two control groups were included; one group (imm) was also immunised passively with 14 HAI units of antibodies against the virus tree day before challenge. The other control group (virus only) was challenged with virus, but not immunised. The control groups were not treated with oseltamivir. Panel A pictures the survival of the mice from the 4 groups and panel B presents the average weight loss in percent. *Statistical significant difference, between the two groups (P = 0.0085).
significant with a P-value of 0.0085. All the mice in the “oseltamivir” and “immunisation + oseltamivir” groups tested positive for influenza A virus by real-time PCR analyses on one or several days. We tried to monitor the viral load in the mice over the 9 days the experiment lasted, but probably due to technical difficulties in obtaining comparable material from the pharyngeal swabs, this was not possibly in a reliably way (Data not show).
The sequence analysis on the virus specimens from the pharyngeal swabs showed no evidence of the oseltamivir resistance mutation H274Y in either group.
3.4. Humane Endpoints
A review of the criteria for termination shows that in the two passive immunisation experiments and in the oseltamivir/passive immunisation experiment only three mice were terminated on the basis of a temperature below 34˚C on two consecutive measurements and none on the basis of a temperature below 32˚C once. All three were at the point of termination very close to the weight loss limit.
In the LD90 experiments with the three highest dosages the temperature was more often the basis for the decision to terminate.
4. DISCUSSION
In the 1918 pandemic plasma from convalescent patients was in some cases used to treat other patients with severe illness. In a meta-analysis of these studies Luke et al. [43] concludes that the patients “may have experienced a clinical important reduction in the risk of death”. Since the idea of passive immunisation as treatment for severe influenza has continued to re-emerge from time to time without really being put to the test or offered as a general treatment. Antiviral treatment of the avian influenza H5N1 with oseltamivir was reported to decrease the fatalities from 88% (49/56) in patients not receiving antiviral treatment to 53% (100/188) [44]. However, no randomized controlled study exists and these groups of patients are hardly comparable.
The issue of emergence of oseltamivir resistance during treatment of H5N1 is not addressed in any large study. de Jong et al. describes that despite treatment with oseltamivir at least two of eight developed the H274Y resistance mutation in a Vietnamese study [45]. Giving us an indication that development of oseltamivir resistance might occur frequently in H5N1 infections in man. Despite the fact that oseltamivir may not be very effective in severe infections, it is still of great concern that resistance towards it seem to develop rather rapidly. An alternate treatment option lies in passive immunisation or a combination of passive immunotherapy and antiviral treatments. Wang et al. points out the need for more research into such combination strategies after one off their patients recovered after receiving oseltamivir and postvaccination plasma [11]. Other recent settings where experimental treatment with IgG or convalescent plasma were given to humans, also found it to be protective [7, 8,10-12].
Several monoclonal antibodies against the stem loop of the haemagglutinin have lately been produced. These antibodies react against conserved epitopes of the influenza virus surface protein and have shown to be effective in animal challenge models [46]. However, if such antibodies are to be used in humans they will probably needed to be humanized and as we have seen with other therapeutic antibodies they will be very expensive. For therapeutics the monoclonal antibodies may have to be combined in order to reduce the likelihood of antigenic drift and thereby resistance towards the effect of such antibodies. Polyclonal antibodies are easy to obtain and produce from reconvalesent persons, are relative inexpensive, easy to administrate and are not likely to cause significant resistance towards its effect.
With this study we aimed to examine the influence of immune status on the effect of oseltamivir and the development of resistant mutants, by administering polyclonal reconvalesent anti-influenza antibodies, in an experimental model of severe influenza infection.
Our results show that we were able to protect mice from death and even clinical illness, by passive immunisation, with IgG from mice previously infected with the same strain, in a dosage dependant manner. No adverse effects of the immunisation were detected in the control group. This finding is in agreement with several studies that have been conducted in animals, investigating the prophylactic and therapeutic effects of passive immunisation in cases of severe influenza infection [47-49]. The amount of antibodies needed to convey 80% and 100% protection in our study were considerably lower, than the ones described by Király et al. [50].
An immunisation dosage (14HAI) that had the desired effect of conveying some protection, without hindering the course of infection and in this way simulating a limited immunity was determined. We showed that even this very limited antibody protection, which on its own was not sufficient to rescue the mice from a fatal outcome, enhanced the clinical effect of oseltamivir treatment and increased the survival of mice. These findings suggest that the effectiveness of oseltamivir depends on the host’s immunological memory.
A combination therapy of convalescent polyclonal anti-influenza IgG and oseltamivir also reduced mortality in patients with severe influenza A infection, in a prospective cohort study by Hung et al. [9], during the 2009 pandemic.
In our experiments we did not detect oseltamivir resistant viruses in our animals. However, from studies in humans we know that immunocompromised patients and immunological naïve patients are at risk of developing resistant viruses. We think that a timely administration of polyclonal antibodies may help to reduce this risk.
5. CONCLUSIONS
In our study we show that passive administration of antibodies can protect mice from a lethal infection with influenza A viruses. Furthermore, we were able to show that sub-protective dosages of antibody in combination with oseltamivir were more effective in saving the animals than one of these treatment options alone.
Passive immunization with polyclonal anti-influenza IgG is an option that should be considered in a pandemic situation for severe cases of influenza infection, especially for high risk patients in combination with antiviral treatment.
In a future experiment we suggest to evaluate the protection level in a study were IgG are administered therapeutically for example one or two days post infection.
Also the prophylactic use of polyclonal anti-influenza antibodies to patients that due to immunosuppression don’t respond normal influenza vaccines should be explored.
REFERENCES
- Luke, T., et al. (2010) Hark back: Passive immunotherapy for influenza and other serious infections. Critical Care Medicine, 38, e66-e73. doi:10.1097/CCM.0b013e3181d44c1e
- Hoi, K.W., et al. (2010) Practical limitations of convalescent plasma collection: a case scenario in pandemic preparation for influenza A (H1N1) infection. Transfusion, 50, 1967-1971. doi:10.1111/j.1537-2995.2010.02651.x
- Kreil, T.R., et al. (2012) Preparation of commercial quantities of a hyperimmune human intravenous immunoglobulin preparation against an emerging infectious disease: The example of pandemic H1N1 influenza. Transfusion, 52, 803-809. doi:10.1111/j.1537-2995.2011.03347.x
- Parry, R.P., et al. (2012) Strategies for screening blood donors to source convalescent H1N1v plasma for intervention therapy. Vox Sanguinis, 103, 107-112. doi:10.1111/j.1423-0410.2012.01599.x
- Wu, J.T., et al. (2010) Logistical feasibility and potential benefits of a population-wide passive-immunotherapy program during an influenza pandemic. Proceedings of the National Academy of Sciences, 107, 3269-3274. doi:10.1073/pnas.0911596107
- Leider, J.P., et al. (2010) Convalescent transfusion for pandemic influenza: Preparing blood banks for a new plasma product? Transfusion, 50, 1384-1398. doi:10.1111/j.1537-2995.2010.02590.x
- Kong, L. and Zhou, B. (2006) Successful treatment of avian influenza with convalescent plasma. Hong Kong Medical Journal, 12, 489.
- Zhou, B., et al. (2007) Treatment with convalescent plasma for influenza A (H5N1) infection. New England Journal of Medicine, 357, 1450-1451. doi:10.1056/NEJMc070359
- Hung, I.F., et al. (2011) Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clinical Infectious Diseases, 52, 447-456. doi:10.1093/cid/ciq106
- Gordon, C.L., et al. (2010) Association between severe pandemic 2009 influenza A (H1N1) virus infection and immunoglobulin G2 subclass deficiency. Clinical Infectious Diseases, 50, 672-678. doi:10.1086/650462
- Wang, H., et al. (2008) Probable limited person-to-person transmission of highly pathogenic avian influenza A (H5N1) virus in China. The Lancet, 371, 1427-1434. doi:10.1016/S0140-6736(08)60493-6
- Yu, H., et al. (2008) Clinical characteristics of 26 human cases of highly pathogenic avian influenza A (H5N1) virus infection in china. PLoS ONE, 3, e2985. doi:10.1371/journal.pone.0002985
- Boon, A.C.M., et al. (2010) Cross-reactive neutralizing antibodies directed against pandemic H1N1 2009 virus are protective in a highly sensitive DBA/2 mouse influenza model. Journal of Virology, 84, 7662-7667. doi:10.1128/JVI.02444-09
- Corti, D., et al. (2011) A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science, 333, 850-856. doi:10.1126/science.1205669
- Hessel, A., et al. (2010) A pandemic influenza H1N1 live vaccine based on modified vaccinia ankara is highly immunogenic and protects mice in active and passive immunizations. PLoS ONE, 5, e12217. doi:10.1371/journal.pone.0012217
- Howard, M.K., et al. (2011) H5N1 whole-virus vaccine induces neutralizing antibodies in humans which are protective in a mouse passive transfer model. PLoS ONE, 6, e23791. doi:10.1371/journal.pone.0012217
- Kistner, O., et al. (2010) A whole virus pandemic influenza H1N1 vaccine is highly immunogenic and protective in active immunization and passive protection mouse models. PLoS ONE, 5, e9349. doi:10.1371/journal.pone.0009349
- Nguyen, H.H., et al. (2010) Prophylactic and therapeutic efficacy of avian antibodies against influenza virus H5N1 and H1N1 in mice. PLoS ONE, 5, e10152. doi:10.1371/journal.pone.0010152
- Shahzad, M., et al. (2008) Passive immunization against highly patho-genic Avian Influenza Virus (AIV) strain H7N3 with antiserum generated from viral polypeptides protect poultry birds from lethal viral infection. Virology Journal, 5, 144. doi:10.1186/1743-422X-5-144
- Ekiert, D.C., et al. (2009) Antibody recognition of a highly conserved influenza virus epitope. Science, 324, 246-251. doi:10.1126/science.1171491
- Friesen, R.H.E., et al. (2010) New class of monoclonal antibodies against severe influenza: Prophylactic and therapeutic efficacy in ferrets. PLoS ONE, 5, e9106. doi:10.1371/journal.pone.0009106
- Oh, H.L.J., et al. (2010) An antibody against a novel and conserved epitope in the hemagglutinin 1 subunit neutralizes numerous H5N1 influenza viruses. Journal of Virology, 84, 8275-8286. doi:10.1128/JVI.02593-09
- Ward, P., et al. (2005) Oseltamivir (Tamiflu®) and its potential for use in the event of an influenza pandemic. Journal of Antimicrobial Chemotherapy, 55, 5-21. doi:10.1093/jac/dki018
- Gubareva, L.V., et al. (2001) Selection of influenza virus mutants in experimentally infected volunteers treated with oseltamivir. Journal of Infectious Diseases, 183, 523-531. doi:10.1086/318537
- Kiso, M., et al. (2004) Resistant influenza A viruses in children treated with oseltamivir: Descriptive study. The Lancet, 364, 759-765. doi:10.1016/S0140-6736(04)16934-1
- Stephenson, I., et al. (2009) Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clinical Infectious Diseases, 48, 389- 396. doi:10.1086/596311
- Whitley, R., et al. (2001) Oral oseltamivir treatment of influenza in children. The Pediatric Infectious Disease Journal, 20, 127-133. doi:10.1097/00006454-200102000-00002
- Roche (2001) Clinical trial result information (JV16284). http://www.rochetrials.com/studyResultGet.action?studyResultNumber=JV16284&productName=Tamiflu& gener ic Name=Oseltamivir
- Tramontana, A.R., et al. (2010) Oseltamivir resistance in adult oncology and hematology patients infected with pandemic (H1N1) 2009 virus, Australia. Emerging Infectious Diseases, 16, 1068-1075. doi:10.3201/eid1607.091691
- Carr, S., et al. (2011) Oseltamivir-resistant influenza A and B viruses preand postantiviral therapy in children and young adults with cancer. The Pediatric Infectious Disease Journal, 30, 284-288. doi:10.1097/INF.0b013e3181ff863b
- Srivastava, B., et al. (2009) Host genetic background strongly influences the response to influenza A virus infections. PLoS ONE, 4, e4857. doi:10.1371/journal.pone.0004857
- van der Laan, J.W., et al. (2008) Animal models in influenza vaccine testing. Expert Review of Vaccines, 7, 783- 793. doi:10.1586/14760584.7.6.783
- Barnard, D.L. (2009) Animal models for the study of influenza pathogenesis and therapy. Antiviral Research, 82, A110-A122. doi:10.1016/j.antiviral.2008.12.014
- Sidwell, R.W., et al. (1998) Inhibition of influenza virus infections in mice by GS4104, an orally effective influenza virus neuraminidase inhibitor. Antiviral Research, 37, 107-120. doi:10.1016/S0166-3542(97)00065-X
- Mendel, D.B., et al. (1998) Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob Agents Chemother, 42, 640-646. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC105511/pdf/ac000640.pdf
- Yen, H.L., et al. (2005) Virulence may determine the necessary duration and dosage of oseltamivir treatment for highly pathogenic A/Vietnam/1203/04 influenza virus in mice. Journal of Infectious Diseases, 192, 665-672. doi:10.1086/432008
- Sidwell, R.W., et al. (2007) Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob Agents Chemother, 51, 845- 851. doi:10.1128/AAC.01051-06
- Suguitan, A.L., et al. (2006) Live, attenuated influenza A H5N1 candidate vaccines provide broad cross-protection in mice and ferrets. PLOS Medicine, 3, e360. doi:10.1371/journal.pmed.0030360
- Tamura, S.I. (2010) Studies on the usefulness of intranasal inactivated influenza vaccines. Vaccine, 28, 6393- 6397. doi:10.1016/j.vaccine.2010.05.019
- Min, J.Y., et al. (2010) Classical swine H1N1 influenza viruses confer cross protection from swine-origin 2009 pandemic H1N1 influenza virus infection in mice and ferrets. Virology, 408, 128-133. doi:10.1016/j.virol.2010.09.009
- Ison, M.G., et al. (2006) Comparative activities of oseltamivir and A-322278 in immunocompetent and immunocompromised murine models of influenza virus infection. Journal of Infectious Diseases, 193, 765-772. doi:10.1086/500464
- Reed, L. and Muench, H. (1938) A simple method of estimating fifty percent endpoints. The American Journal of Hygiene, 27, 493-497. http://aje.oxfordjournals.org/content/27/3/493.short
- Luke, T.C., et al. (2006) Meta-analysis: Convalescent blood products for spanish influenza pneumonia: A future H5N1 treatment? Annals of Internal Medicine, 145, 599. http://annals.org/article.aspx?articleid=729754
- WHO (2008) Update on avian influenza A (H5N1) virus infection in humans. New England Journal of Medicine, 358, 261-273. doi.10.1056/NEJMra0707279
- de Jong, M.D., et al. (2005) Oseltamivir resistance during treatment of influenza A (H5N1) infection. New England Journal of Medicine, 353, 2667-2672. doi.10.1056/NEJMoa054512
- De Marco, D., et al. (2012) A non-VH1-69 heterosubtypic neutralizing human monoclonal antibody protects mice against H1N1 and H5N1 viruses. PLoS ONE, 7, e34415. doi:10.1371/journal.pone.0034415
- Mozdzanowska, K., et al. (1997) A pulmonary influenza virus infection in SCID mice can be cured by treatment with hemagglutinin-specific antibodies that display very low virus-neutralizing activity in vitro. Journal of Virology, 71, 4347-4355. http://jvi.asm.org/content/71/6/4347.abstract
- Prabakaran, M., et al. (2009) Combination therapy using chimeric monoclonal antibodies protects mice from lethal H5N1 infection and prevents formation of escape mutants. PLoS ONE, 4, e5672. doi:10.1371/journal.pone.0005672
- Simmons, C.P., et al. (2007) Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLOS Medicine, 4, e178. doi:10.1371/journal.pmed.0040178
- Király, J., et al. (2011) Evaluation of anti-influenza efficiency of polyclonal IgG antibodies specific to the ectodomain of M2 protein of influenza A virus by passive immunization of mice. Acta Virologica, 55, 261-265. doi:10.4149/av_2011_03_261