Journal of Modern Physics
Vol. 4 No. 3A (2013) , Article ID: 29334 , 9 pages DOI:10.4236/jmp.2013.43A057
A Density Functional Theory Study of Methoxy and Atomic Hydrogen Chemisorption on Au(100) Surface
1Groupe de Simulations Numériques en Magnétisme et Catalyse, Département de Physique, Faculté des Sciences, Université Marien Ngouabi, Brazzaville, Congo
2Laboratoire des Matériaux, Surfaces et Procédés pour la Catalyse (LMSPC), Université de Strasbourg, Strasbourg, France
Email: *ps_moussounda@yahoo.fr
Received December 26, 2012; revised January 27, 2013; accepted February 7, 2013
Keywords: Chemisorption; Density Functional Calculations; Gold; Methoxy; Hydrogen
ABSTRACT
The adsorption of CH3O and H on the (100) facet of gold was studied using self-consistent periodic density functional theory (DFT-GGA) calculations. The best binding site, energy, and structural parameter, as well as the local density of states, of each species were determined. CH3O is predicted to strongly adsorb on the bridge and hollow sites, with the bridge site as preferred one, with one of the hydrogen atoms pointing toward a fourfold vacancy (bridge-H hollow). The top site was found to be unstable, the CH3O radical moving to the bridge –H top site during geometry optimization. Adsorption of H is unstable on the hollow site, the atom moving to the bridge site during geometry optimization. The 4-layer slab is predicted to be endothermic with respect to gaseous H2 and a clean Au surface.
1. Introduction
The methoxy group CH3O and hydrogen H species have been identified as two intermediates in the decomposition of methanol through an initial O-H bond scission on several transition metal surfaces. For methoxy group CH3O, it appears as an intermediate also in formaldehyde production and in the synthesis of hydrogenated products of CO or its inverse reaction. For catalytic reactions, knowledge of the adsorption geometry of reactants is crucial since it decides on the energy changes during the reaction as well as on the capability of adsorbed species to interact with another one.
Over the last two decades, a number of experimental studies on methoxy over various metal surfaces have been performed using several techniques. Specifically, methoxy on Cu(110) [1-4], Cu(111) [5-7], Cu(100) [8- 13], Ag(111) [14], Ni(111) [15], Ni(110) [16,17], Pt(111) [18] has been studied extensively by using X-ray photoelectron diffraction (XPD), reflection absorption infrared spectroscopy (RAIRS), near-edge X-ray absorption fine structure (NEXAFS), temperature-programmed desorption (TPD), low-energy electron diffraction (LEED), scanning tunneling spectroscopy (STM), high-resolution electron energy-loss spectroscopy (HREELS), energy scanned photoelectron diffraction (PED) and surface extended X-ray absorption fine structure (SEXAFS). We note that the XPD [5], SEXAFS [6] and NEXAFS [6] studies for CH3O/Cu(111) and RAIRS [14] for CH3O/ Ag(111), showed that the CH3O radical resides in threefold hollow site and the C–O bond is normal to the surface. Whereas, in the PED study [7], it was also found that the CH3O radical adopted a geometry in which the C–O bond was close to perpendicular to the surface and the O atom occupied a threefold hollow site, fcc. Despite the existence of a large number of studies, conflicting structural assignments still exist. The NEXAFS study of Outka et al. [9] shows that methoxy C–O axis will be found with an angle of 20˚ - 40˚ relative to the Cu(100) surface normal. Using the infrared spectroscopy [10,11], Ryberg also found that methoxy is tilted when adsorbed on this surface. Camplin et al. [12], using the RAIRS technique, found the C–O methoxy radical bond to be perpendicular to the Cu(100) surface. A later study of Lindner et al. [13] with a combined NEXAFS and photoelectron research concluded that the C–O axis is perpendicular to the surface and that a low symmetry adsorption site between the bridge and the 4-fold hollow site is occupied.
Several theoretical studies have also been done on the methoxy—metal surface interactions [19-28]. A manyelectron embedding theory, at the ab initio configuration interaction level, was used to study the adsorption of methoxy on the Ni(111) surface [20]. That work showed how CH3O is adsorbed at 3-fold hollow sites with the C–O axis tilted 5˚ from the normal to the surface plane. Wang et al. [22] used density functional theory (DFT) to determine CH3O/Ni(111) properties. They found that CH3O interacts with the surface through oxygen and has a binding energy of 2.58 eV for the threefold fcc hollow site. Witko et al. [19,21] have studied the adsorption of CH3O on Cu(111) surface by performing ab initio HFLCAO calculations. Again it was found that CH3O is usually adsorbed at 3-fold hollow sites with a slight preference for fcc sites. On Cu(111), the C–O axis lies perpendicular to the metal surface and the calculated adsorption energy is 2.80 eV. Gomes and coworker [23] have studied the same adsorption of CH3O on Cu(111) surface, using their DFT approach and cluster models. They reported that three-fold hollow sites are the most stable position for methoxy, with fcc and hcp hollows having binding energies of 2.50 eV and 2.18 eV, respectively. Using a DFT approach Greeley and Mavrikakis [24] examined the reaction of CH3O on Pt(111) top site. They evaluated chemisorption energy around 1.54 eV. Recently, Pang et al. [25] have carried out the methoxy adsorption on Ni(111), Ni(110) and Ni(100) surfaces using a DFT method. Very little ab initio and DFT studies have been carried out for methoxy adsorbed on Au. Gomes and Gomes [23] found that CH3O binds at all high symmetric sites of Au (111) with a preference for the hollow fcc site. The corresponding chemisorption energy for this site was found to be 0.89 eV. With a same surface, Chen et al. [26] found that the bridge site is most stable. The corresponding binding energy was calculated to be 0.99 eV. To the best of our knowledge, there is no theoretical study of CH3O adsorption on the Au(100) surface in the literature.
Atomic hydrogen (H) is probably one of the most extensively studied adsorbate in a large number of catalytic processes. Details on the H chemisorption on transition metal surfaces (TMS) can be found in the literature [29, 30]. A series of TPD, LEED, STM, and electron energy loss spectroscopy (EELS) studies of H adsorption have performed by several groups on Ni(111) [31-33]. Schick et al. have applied HREELS to study H on Ir(111) [31]. Techniques such as LEED [34-37], HREELS [38], LERS [39], UPS [40] and calorimetric measurements [41] have been applied to investigate the adsorption of H on the Pt(100), Pt(110), and Pt(111) surfaces. A number of theoretical studies have also performed for H on TMS. Effective medium theory study for H adsorption on Ni(111), Ni(100), W(100) and W(110) has been reported by Nordlander et al. [42]. Jiang and Carter [43] have performed a DFT study of H adsorption on Fe(110). Extensive ab initio calculations and the DFT method have been used to investigate the adsorption of hydrogen on Pt(111) [4447], Pt(110) [47,48], Pt(100) [47,49,50], Ni(100) [49], Ni(111) [51,52] and Cu(001) [53]. To our best knowledge, there are no studies at DFT level of hydrogen adsorption on Au(100) surface.
In this contribution, we present a systematic DFT study of the properties of atomic H and CH3O radical on the Au(100). Our paper is organized as follows. Section 2 gives the details of the computational method. The results and discussions are followed in Section 3. Section 4 concludes with a short summary.
2. Computational Method
We are based our DFT calculations on the DACAPO ab initio package [54]. A (2 × 2) unit cell is used to construct a four or five-layer Au(100) slab. This corresponds to a surface coverage of 1/4 ML when there is only one adsorbate per unit cell. The unit cell is repeated in super cell with successive slabs separated by a vacuum region of 13 Å. Adsorption is allowed on only one of the two exposed surfaces. The top layers of the slab and the adsorbate were allowed to relax. The maximum force criterion of 0.05 eV/Å was considered for convergence. The surface irreducible Brillouin zone was sampled by 18 special k-points using the 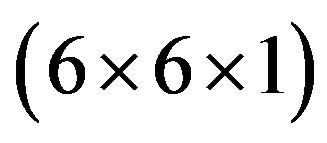 Monkhorst-Pack grids.
Monkhorst-Pack grids.
The Kohn-Sham one-electron valence states are expanded in a basis of plane waves with kinetic energies up to 400 eV, and ionic cores were described by ultra soft pseudo potentials [55]. All calculations were performed non-spin polarized. The exchange-correlation potential and energy are described self-consistently using GGAPW91 functional [56]. The electron density is determined by iterative diagonalization of the Kohn-Sham Hamiltonian, Fermi-population of the Kohn-Sham states  of the resulting electron density. Total energies are extrapolated to
of the resulting electron density. Total energies are extrapolated to .
.
Using DFT as described above yields a bulk lattice constant of Au of 4.18 Å, to be compared with the experimental value of 4.08 Å [56]. Our rather high result is in perfect agreement with other theoretical evaluation using similar methods (4.17 Å [58] or 4.19 Å [59]).
3. Results and Discussion
In this section we describe the properties of the adsorbates studied, including the binding energies, site preferences, geometries, and local density of states (LDOS), and we compare these results with theoretical or experimental data available on transition and noble metal surfaces.
3.1. CH3O Chemisorption on Au(100)
3.1.1. Chemisorption Energies
As usual, the adsorption energy (Eads) is evaluated as:
 (1)
(1)
where 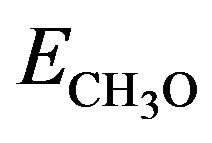 is the total energy of the free
is the total energy of the free  radical in the gas phase, Eslab is the total energy of the clean Au slab and
radical in the gas phase, Eslab is the total energy of the clean Au slab and 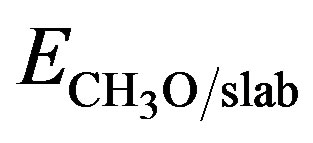 is the total energy of the
is the total energy of the 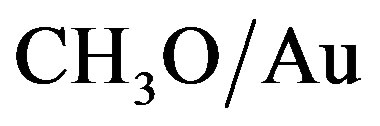 system. With this definition, a positive Eads corresponds to a stable adsorption on the slab. The energy of the isolated
system. With this definition, a positive Eads corresponds to a stable adsorption on the slab. The energy of the isolated radical was determined by performing calculations on a single molecule in a cubic cell with 20 Å parameter.
radical was determined by performing calculations on a single molecule in a cubic cell with 20 Å parameter.
The adsorption of  on Au(100) at high-symmetry top, bridge and hollow sites, as shown in Figure 1, were investigated. For the bridge site, two different hydrogen orientations were considered: one of the H atoms in the CH3 group points either toward the nearest-neighbor Au atom (bridge-H top) or toward a fourfold vacancy (bridge-H hollow).
on Au(100) at high-symmetry top, bridge and hollow sites, as shown in Figure 1, were investigated. For the bridge site, two different hydrogen orientations were considered: one of the H atoms in the CH3 group points either toward the nearest-neighbor Au atom (bridge-H top) or toward a fourfold vacancy (bridge-H hollow).
Adsorption energies for CH3O radical at the hollow, bridge-H top and bridge-H hollow, calculated using different numbers of layers in the slab for methoxy coverage of 0.25 ML are listed in Table 1. The top site was found to be unstable, the CH3O radical moving to the hollow site during geometry optimization. From Table 1, it can be found that the adsorption energy of CH3O increases by 0.586 eV from 4 to 5-layer slab, for all adsorption sites. In addition, one can see from this table that the most stable site for CH3O adsorption on Au(100)
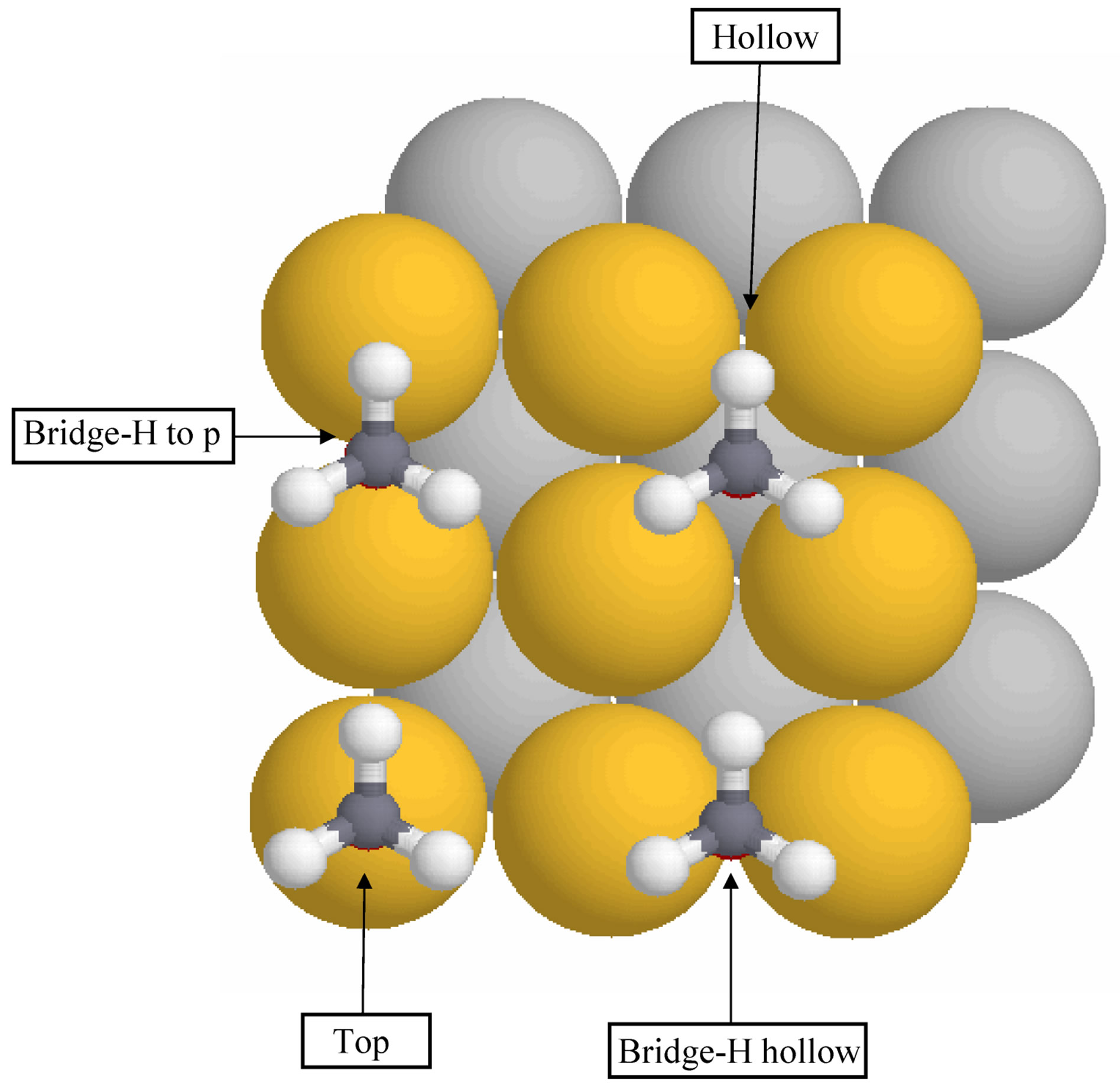
Figure 1. Top view of three high-symmetry adsorption geometries of CH3O on the Au(100) surface. The indicated adsorption sites are the top, bridge and hollow sites. For the bridge site, two different hydrogen orientations were considered: one of the H atoms in the CH3 group points either toward the nearest-neighbor Au atom (bridge-H top) or toward a fourfold vacancy (bridge-H hollow) (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article).
is the bridge site with one of the hydrogen atoms pointing toward a fourfold vacancy (bridge-H hollow), at four or five layers of Au. The corresponding adsorption energy for this site was found 1.727 eV (4-layer slab) and 2.314 eV (5-layer slab). The bridge-H top is significantly less stable by 0.153 eV. The adsorption energy difference between bridge-H hollow and hollow sites is only 30 meV. No calculations of CH3O radical adsorption on Au(100) have been published. There are theoretical results for adsorption in the bridge site on the Au(111), Ni(111), Ni(110), Ni(100), Cu(111), Cu(110) and Cu(100) surfaces. Gomes and Gomes [23] used a cluster model to study the adsorption of CH3O on Au(111) surface and predicted a chemisorption energy of 0.471 eV. For the same surface, Chen et al. [26], using ab initio DFT-GGA calculations with three-layer slab, reported adsorption energy of 0.999 eV for coverage of 1/6 ML. The discrepancy in binding energies may be due to the model effect and the computational methodology (slab vs. cluster). Pang et al. [25] have described the methoxy adsorption on Ni(111), Ni(100) and Ni(110) with non-spin-polarized calculations elaborately, and found the adsorption energy of 2.292 eV, 2.478 eV, 2.978 eV (in the short-bridge) and 2.281 eV (in the long-bridge) at 1/6 ML, respectively. In the case of methoxy adsorption on Cu(111) surface, Gomes et al. [23] and Chen et al. [27] found the adsorption energies of 2.036 eV and 2.363 eV, respectively. For the Cu(100) and Cu(110) surfaces, Pick [28], using ab initio DFT-GGA calculations with fivelayer Cu slabs, obtained the binding energies of 2.540 eV, 2.690 eV (in the short-bridge) and 2.350 eV (in the longbridge), respectively. These calculated chemisorption energies are much higher than our results.
3.1.2. Geometric Parameters
Now turn our attention to the geometric parameters. First of all, we checked that the properties of the isolated CH3O were accurately reproduced. Table 2 compares our calculated and previous theoretical [25] bond lengths and bond angles of CH3O. Our results are in good agreement with previous DFT results [26]. Second, we examined the structural parameters, upon adsorption. It can be seen from Table 1 that the geometric parameters are independent of number of layers in the slab. Similar structural parameters were calculated for the hollow and bridge-H hollow sites. The H–C–H angle is increased from 106.4˚ (shortest H–C–H angle) in isolated methoxy to 108.5˚ (longest H–C–H angle) corresponding to methoxy adsorbed at a bridge-H hollow site. On the bridge-H top site the value of this longest H–C–H angle is 108.7˚. On the bridge-H hollow and bridge-H top sites the longest values of C–O–Au angle are about 120.7˚ and 134.3˚, respectively. Our values are significantly smaller than that the value (179.2˚) obtained par Chen et al. [26] for
Table 1. Adsorption energies (Eads) and geometries for CH3O radical at the hollow, bridge-H top and bridge-H hollow, calculated using different numbers of layers in the slab for methoxy coverage of 0.25 ML. Numbers in parentheses represent the number of bonds with that length or the number of equal angles.
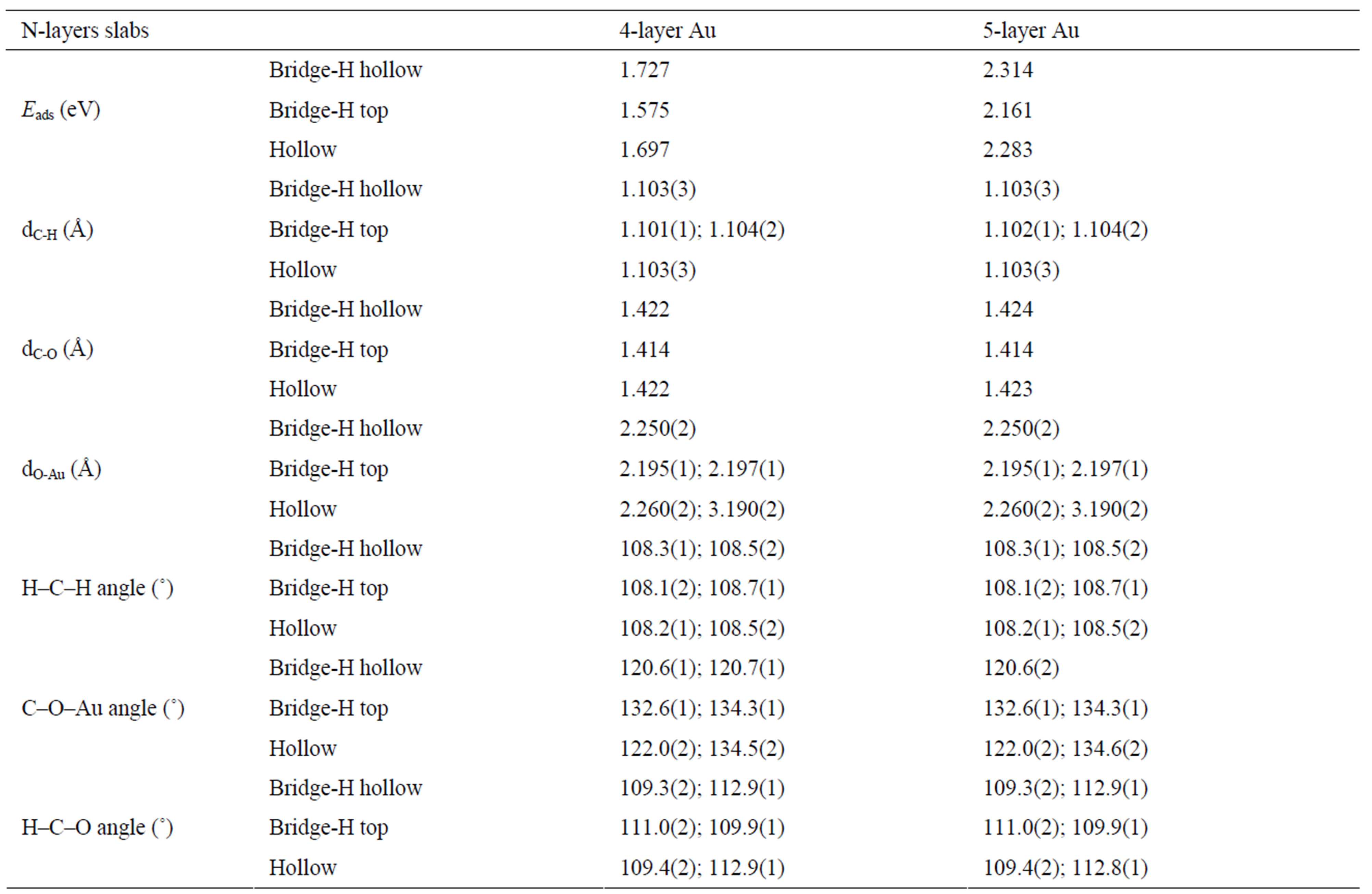
Table 2. Comparison between our calculations for geometric parameters for free CH3O and previous calculated results. Numbers in parentheses represent the number of bonds with that length or the number of equal angles.
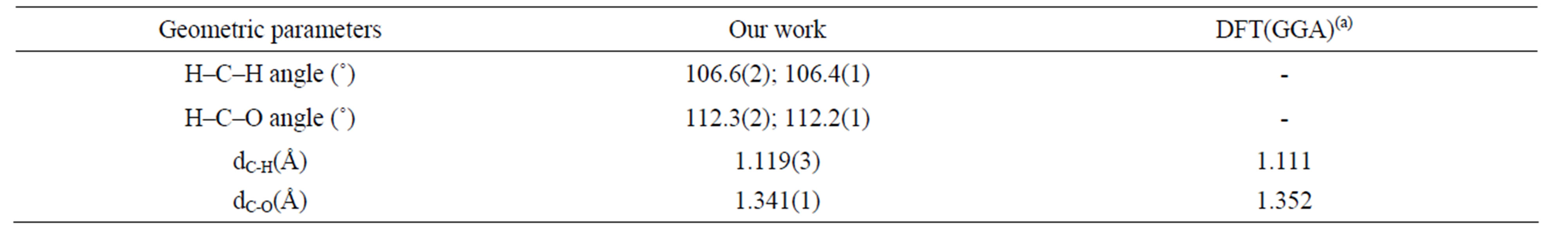
(a)Reference [26].
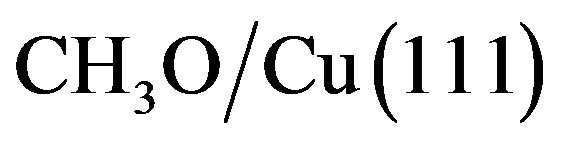 on the bridge site. For CH3O on the bridge H-top site, the H-C-O angle, in the range of 109.9˚ - 111.0˚, is smaller than that the free methoxy (112.3˚), and in good agreement with the theoretical results by Pick (109.9˚ - 110.0˚) for the adsorbed CH3O on Cu(100) on the bridge site [28].
on the bridge site. For CH3O on the bridge H-top site, the H-C-O angle, in the range of 109.9˚ - 111.0˚, is smaller than that the free methoxy (112.3˚), and in good agreement with the theoretical results by Pick (109.9˚ - 110.0˚) for the adsorbed CH3O on Cu(100) on the bridge site [28].
As Tables 1 and 2 show, the C–H bond is reduced from 1.119 Å of the isolated CH3O radical to 1.101 Å. The optimized C–H bond is very similar to the C–H bond reported by Pick [28]. The calculated C–O bond length for the adsorbed CH3O, in the range of 1.414 - 1.424 Å, is longer than that of the free species (1.341 Å), and in excellent agreement with the theoretical results by Pick (1.420 Å) [28] and Chen et al. (1.408 Å) [26], and with the experimental results by Hoffmann [7],
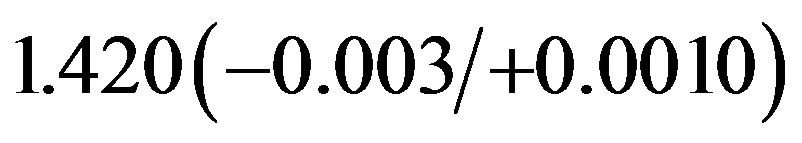 Å and Amemiya et al. [6], 1.460 ± 0.005 Å. Compared with the bridge-H top site, the bridge-H hollow geometry has longer O-Au bonds. In addition, the calculated dO-Au values for the both bridge sites are smaller than the corresponding ones obtained by using DFT/B3LYP for Au7 cluster [23]. Comparing with the ionic radius of Au+ and O2–, which are 1.37 and 1.32 Å, respectively, [60]; the values of dO-Au are smaller than the ionic radius sum, indicating the strong interaction between O of CH3O and the surface Au atom.
Å and Amemiya et al. [6], 1.460 ± 0.005 Å. Compared with the bridge-H top site, the bridge-H hollow geometry has longer O-Au bonds. In addition, the calculated dO-Au values for the both bridge sites are smaller than the corresponding ones obtained by using DFT/B3LYP for Au7 cluster [23]. Comparing with the ionic radius of Au+ and O2–, which are 1.37 and 1.32 Å, respectively, [60]; the values of dO-Au are smaller than the ionic radius sum, indicating the strong interaction between O of CH3O and the surface Au atom.
3.1.3. Analysis of Local Density of States (LDOS)
We now comment on the evolution of the local electronic structure of the molecule and the surface upon adsorption for the most stable adsorption site. Figure 2(a) compares the LDOS of O in CH3O, adsorbed on a preferred bridge-H hollow site, (continuous curve) to the LDOS of O in CH3O, when it is in a gas phase (dashed curve), i.e. in the case of a free CH3O radical. The graph obviously falls into two main regions. The first region, from −13 eV to −9 eV, has essentially a mixed  character for O atom. Here, the O density of states can be clearly recognized as they are essentially shifted to lower binding energies by ca. 1.42 eV with respect to the free CH3O radical. The second region of the graph is the energy range from −9 eV to + 2 eV. The states represented in this region all contain O 2s and O 2p characters. One can see from Figure 2(a) that a dramatic change in these orbitals occurs upon adsorption. There is a strong mixing between these orbitals and the 5d states of Au, particularly the dxz-band of the Au surface.
character for O atom. Here, the O density of states can be clearly recognized as they are essentially shifted to lower binding energies by ca. 1.42 eV with respect to the free CH3O radical. The second region of the graph is the energy range from −9 eV to + 2 eV. The states represented in this region all contain O 2s and O 2p characters. One can see from Figure 2(a) that a dramatic change in these orbitals occurs upon adsorption. There is a strong mixing between these orbitals and the 5d states of Au, particularly the dxz-band of the Au surface.
The Au surface is affected by the CH3O adsorption. By examining the components of the Au-d orbitals, we found that dxz is the most affected. Figure 2(b) shows the LDOS at a surface gold atom of the bridge-H hollow site (solid line) with the LDOS for a Au atom in the clean surface (dotted line) superimposed. The characters of the two curves are not similar. The LDOS curve for the chemisorption system has a small peak of resonance at ~ −11.36 eV which is due to density in oxygen 2s and 2p-derived orbitals lying within the region of the cut. The other notable differences are the presence of two peaks of
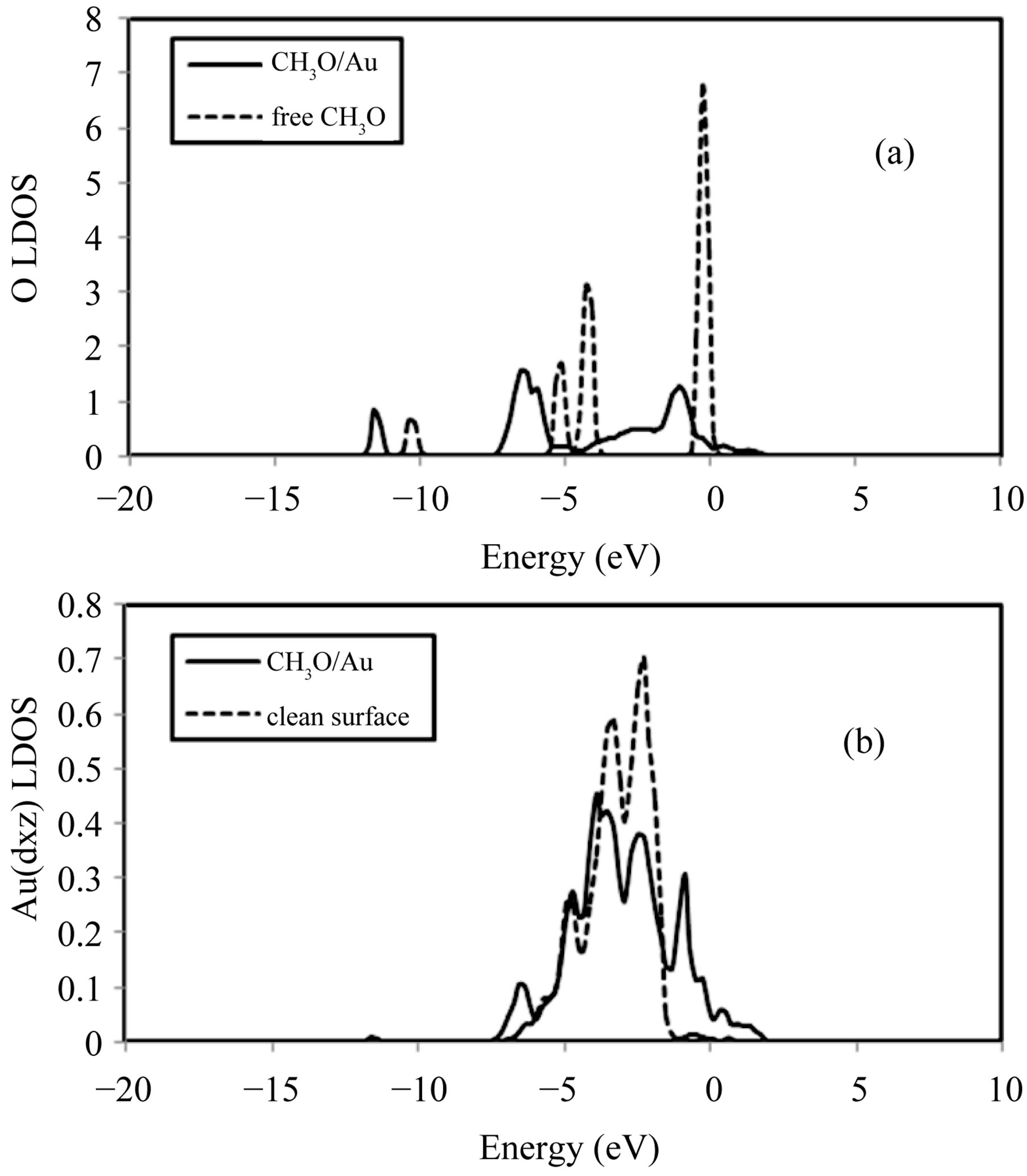
Figure 2. LDOS curves for: (a) O in the free CH3O and ; (b) dxz orbital of Au. The zero energy corresponds to the Fermi level.
; (b) dxz orbital of Au. The zero energy corresponds to the Fermi level.
resonance at about −6.31 eV and −0.86 eV in the chemisorption system curve. These peaks of resonance correspond to states with O 2s and O 2p characters as described previously. In addition, on can notice there is a clear intensity decrease in the −3.68 eV to −1.46 eV region, and the appearance of continuously distributed states from −0.25 eV up to the Fermi level.
3.2. H Adsorption on Au(100) Surface
3.2.1. Chemisorption Energies and Geometrical Parameters
In this subsection, we investigate the adsorption of H on Au(100) surface. The adsorption energy for atomic hydrogen is calculated by the equation:
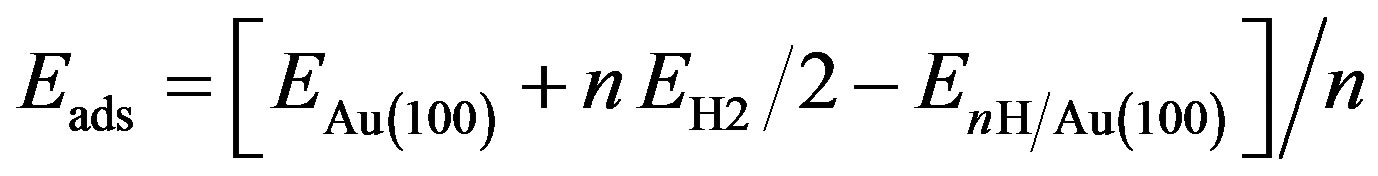 (2)
(2)
where n is the number of H atoms in the surface unit, 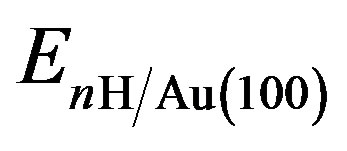 and EAu(100) are the energies of the Au(100) system with and without nH absorbates, while EH2 is the ground state energy of a free H2 molecule. The first and last terms are calculated with the same parameters (ksampling, energy cutoff, etc.). The second term is calculated with only the Γ-point for the Brillouin zone sampling. With this definition, a positive Eads corresponds to a stable adsorption on the slab.
and EAu(100) are the energies of the Au(100) system with and without nH absorbates, while EH2 is the ground state energy of a free H2 molecule. The first and last terms are calculated with the same parameters (ksampling, energy cutoff, etc.). The second term is calculated with only the Γ-point for the Brillouin zone sampling. With this definition, a positive Eads corresponds to a stable adsorption on the slab.
The energy of the free hydrogen molecule was determined from calculations performed on a single hydrogen molecule in a cubic cell with an edge of 15 Å. We made sure that the properties of the free H2 molecule were accurately reproduced. Table 3 compares the calculated and experimental bond lengths dH-H for gas-phase hydrogen. Our calculated result (0.7600 Å) can be compared with the experimental value of 0.7414 Å [57]. Our bigger result is however in excellent agreement with other theoretical evaluation using similar methods [43,61,62].
We start by considering the on-top and bridge adsorption of H on Au(100) for coverages of 0.25 ML and 0.5 ML. The hollow site was found to be unstable, the atom moving to the bridge site during geometry optimization. This phenomenon was also observed by Moussounda et al. [49] and Saad et al. [47] on the adsorption of H on Pt(100). The adsorption energies for different number of layers in the slab and different coverages are reported in Table 4. It might be surprising to find a desorption energy for H in the case of the 4-layer slab for the two coverages, in other terms, the 4-layer slab is predicted to be endothermic with respect to gaseous H2 and a clean Au surface. These observations are consistent with the theoretical results reported by Sundell et al. [53], and Jiang et al. [63] and Fabiani et al. [64] for 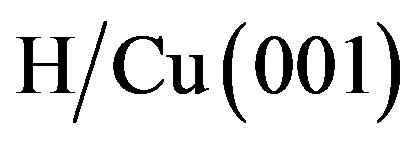 and
and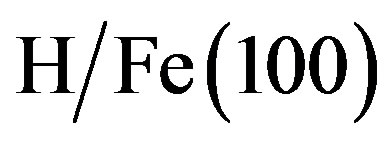 , respectively. Our results reveal that hydrogen preferred adsorbs at the bridge site on Au(100) for the 5-layer slab. The adsorption energy of bridged bonded
, respectively. Our results reveal that hydrogen preferred adsorbs at the bridge site on Au(100) for the 5-layer slab. The adsorption energy of bridged bonded
Table 3. Comparison between the calculated and experimental bond lengths dH-H of free hydrogen molecule.

(a)Reference [61], (b)Reference [50], (c)Reference [43], (d)Reference [62], (e) Reference [57].
Table 4. Adsorption energies (Eads) and bond lengths (dH-Au) obtained for hydrogen adsorbed at the top and bridge sites of Au(100).

H decreases significantly with the increasing H coverage. At the coverage of 0.25 ML, we found an adsorption energy of 0.568 eV. No calculations of hydrogen adsorption on Au(100) have been published. However, comparative values calculated at the same H coverage of 0.25 ML for adsorption in bridge site on Pt(100) and Pt(110) are 0.610 eV [49] and 0.640 eV [64], respectively. Lai et al. [50] obtained an adsorption energy of 0.542 eV for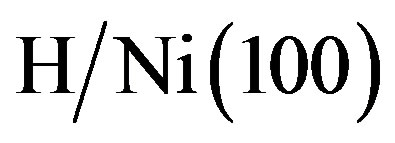 . Kresse et al. [62] reported an adsorption energy of 0.567 eV for
. Kresse et al. [62] reported an adsorption energy of 0.567 eV for , which is in good agreement with our value.
, which is in good agreement with our value.
From Table 4, we note the dH-Au bond lengths for both sites. The H–Au distance does not depend on the coverage and increases as usual with the adsorption surface coordination of the adsorbate (1.610 Å and 1.790 Å for top and bridge sites, respectively). Theoretical calculations using similar method for H/Pt(100) [49] found the same result for top and bridge sites (1.572 Å and 1.763 Å, respectively). DFT calculations, using DACAPO package [46], obtained a similar H-Pt bond length for adsorption of H on-top of Pt(111) (1.570 Å). With hydrogen in the unstable top site, our H-Au bond length (1.61 Å) is somewhat higher than the theoretical result published by Haroun et al. [52] who have reported 1.46 Å for  .
.
3.2.2. LDOS Calculations
In order to obtain a deeper understanding of the properties of , we have also analyzed their local density of states (LDOS), in the most stable energetically site for 0.25 ML. The H LDOS for an adsorbed H on Au(100) for the bridge site is shown in Figure 3 as well as the corresponding LDOS of a free H atom. We can see that the state situated at ~ –5.30 eV initially is found dispersed on a largest domain of energy (~10 eV). The H 1s band has a strong interaction with Au bands, as seen by
, we have also analyzed their local density of states (LDOS), in the most stable energetically site for 0.25 ML. The H LDOS for an adsorbed H on Au(100) for the bridge site is shown in Figure 3 as well as the corresponding LDOS of a free H atom. We can see that the state situated at ~ –5.30 eV initially is found dispersed on a largest domain of energy (~10 eV). The H 1s band has a strong interaction with Au bands, as seen by
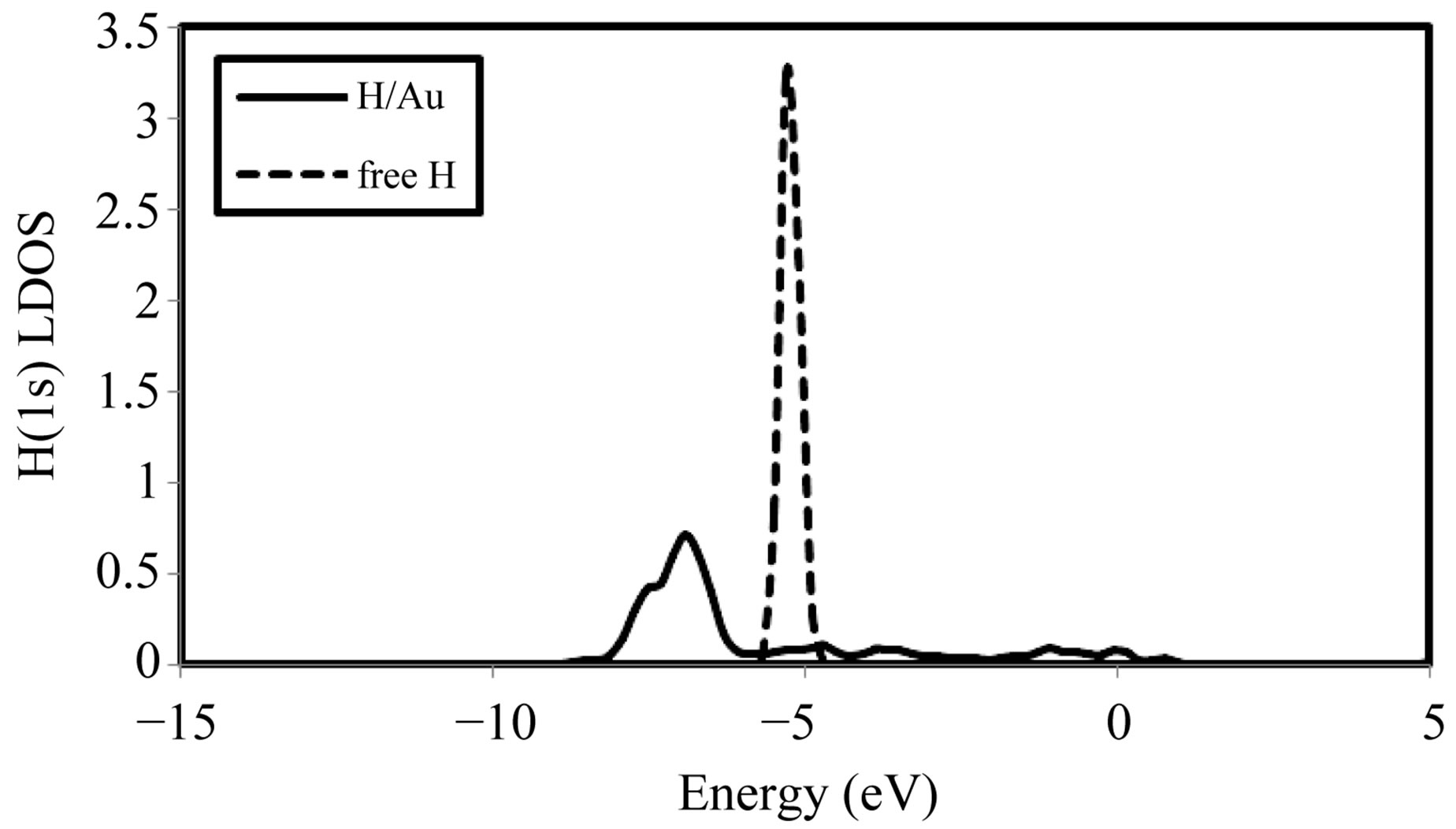
Figure 3. LDOS of H-1s orbital. The zero energy corresponds to the Fermi level.
the significant change of the Au s, py, dxx-yy and dyz LDOS for the bridge site (Figures 4 and 5). Several peaks of resonance appear on the Au py LDOS (Figure 4(b)), the peaks increase in intensity and we can see in Figures 4(a) and 5(b) the strong shift of s and dyz centre of gravity, respectively. This is clearly due to the hybridization between 1s state of H and s-p-d Au states after H adsorption.
4. Conclusion
We performed all-electron periodic DFT-GGA calculations of the adsorption of CH3O and H on Au(100). For methoxy radical, we found that the CH3O adsorption energy depends strongly on number of layers in the slab and increases with increases in number of layers in the slab. The electronic structure analysis of the adsorbed CH3O shows that there is a pronounced hybridization between the O 2s, 2p orbitals and dxz-band of the Au surface. For atomic hydrogen, the desorption is found to be favorable for the 4-layer slab. The local density of states curves around H of the adsorbed hydrogen show dispersed states below the metal Fermi level indicating an
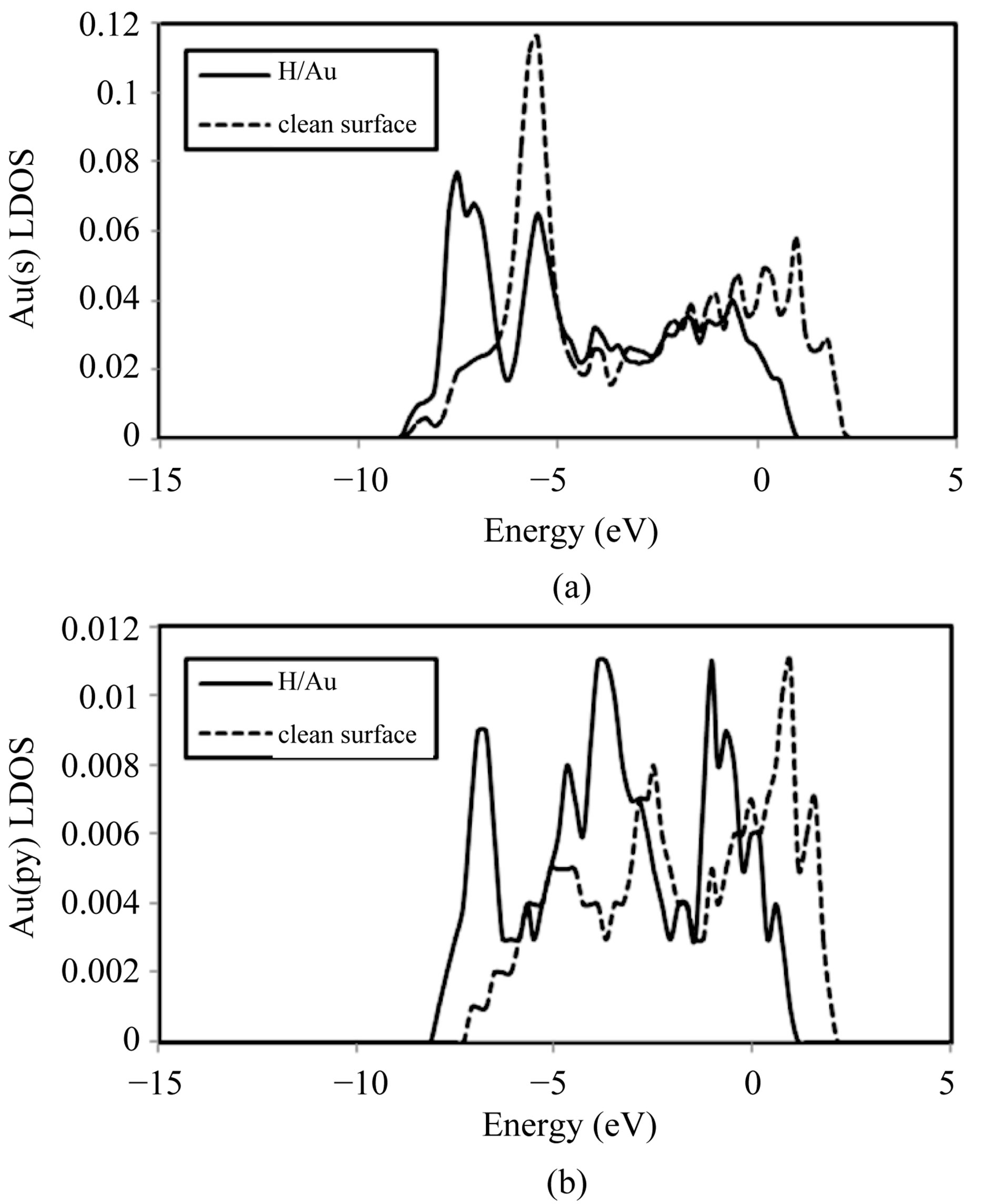
Figure 4. LDOS of Au band after H adsorption on Au(100): (a) s; (b) py [solid curves]. The dashed curves indicate the Au orbitals before adsorption of H. The Fermi level lies at 0 eV.

Figure 5. LDOS of Au band after H adsorption on Au(100): (a) dxx-yy, (b) dyz [solid curves]. The dashed curves indicate the Au orbitals before adsorption of H. The Fermi level lies at 0 eV.
H–Au mixing demonstrating a chemical interaction.
5. Acknowledgements
PS Moussounda acknowledge the “Ecole de Chimie, Polymères et Matériaux de Strasbourg (ECPM) and the “Université de Strasbourg (UDS)” for their financial support and the “Laboratoire des Matériaux, Surfaces et Procédés pour la Catalyse” of the “Université de Strasbourg (UDS)” for access to computational resources.
REFERENCES
- C. Barnes, P. Pudney, Q. Guo and M. Bowker, “Molecular-Beam Studies of Methanol Partial Oxidation on Cu(110),” Journal of the Chemical Society, Faraday Transactions, Vol. 86, No. 15, 1990, pp. 2693-2699. doi:10.1039/ft9908602693
- S. M. Francis, F. M. Leibsle, S. Haq, N. Xiang and M. Bowker, “Methanol Oxidation on Cu(110),” Surface Science, Vol. 315, No. 3, 1994, pp. 284-292. doi:10.1016/0039-6028(94)90132-5
- M. Bader, A. Puschmann and J. Haase, “Orientation of CH3O on Cu(110) as Examined by Near-Edge X-Ray Absorption Fine Structure Spectroscopy,” Physical Review B, Vol. 33, No. 10, 1986, pp. 7336-7338. doi:10.1103/PhysRevB.33.7336
- E. Holub-Krappe, K. C. Prince, K. Horn and D. P. Woodruff, “X-Ray Photoelectron Diffraction Determination of the Molecular Orientation of CO and Methoxy Adsorbed on Cu(110),” Surface Science, Vol. 173, No. 1, 1986, pp. 176-193. doi:10.1016/0039-6028(86)90115-9
- A. V. de Carvalho, M. C. Asensio and D. P. Woodruff, “Determination of the Orientation of Methoxy on Cu(111) Using X-Ray Photoelectron Diffraction,” Surface Science, Vol. 273, No. 3, 1992, pp. 381-384. doi:10.1016/0039-6028(92)90075-H
- K. Amemiya, Y. Kitajima, Y. Yonamoto, S. Terada, H. Tsukabayashi, T. Yokoyama and T. Ohta, “Oxygen K-Edge X-Ray-Absorption Fine-Structure Study of Surface Methoxy Species on Cu(111) and Ni(111),” Physical Review B, Vol. 59, No. 3, 1999, pp. 2307-2312. doi:10.1103/PhysRevB.59.2307
- P. Hofmann, K. M. Schindler, S. Bao, V. Fritzsche, D. E. Ricken, A. M. Bradshaw and D. P. Woodruff, “The Geometric Structure of the Surface Methoxy Species on Cu(111),” Surface Science, Vol. 304, No. 1-2, 1994, pp. 74-84. doi:10.1016/0039-6028(94)90754-4
- R. Ryberg, “The Oxidation of Methanol on Cu(100),” Journal of Chemical Physics, Vol. 82, No. 1, 1985, pp. 567- 573. doi:10.1063/1.448729
- D. A. Outka, R. J. Madix and J. Stoehr, “Structural Studies of Formate and Methoxy Groups on the Cu(100) Surface NEXAFS and SEXAFS,” Surface Science, Vol. 164, No. 1, 1985, pp. 235-259. doi:10.1016/0039-6028(85)90710-1
- R. Ryberg, “Symetry and Orientation of CH3O on Cu(100),” Physical Review B, Vol. 31, No. 4, 1985, pp. 2545-2547. doi:10.1103/PhysRevB.31.2545
- R. Ryberg, “CH Stretch Vibrations of Adsorbed Molecules Studied by Infrared Spectroscopy: CH3O on Cu(100),” Chemical and Physics Letters, Vol. 83, No. 3, 1981, pp. 423-426. doi:10.1016/0009-2614(81)85493-0
- J. P. Camplin and E. M. McCash, “ARAIRS Study of Methoxy and Ethoxy on Oxidized Cu(100),” Surface Science, Vol. 360, No. 1-3, 1996, pp. 229-241. doi:10.1016/0039-6028(96)00641-3
- T. Lindner, J. Somers, A. M. Bradshaw, A. L. D. Kilcoyne and D. P. Woodruff, “A Photoelectron Diffraction and NEXAFS Study of the Structure of the Methoxy Species (CH3O-) on Cu(100),” Surface Science, Vol. 203, No. 3, 1988, pp. 333-352. doi:10.1016/0039-6028(88)90087-8
- W. S. Sim, P. Gardner and D. A. King, “Structure and Reactivity of the Surface Methoxy Species on Ag(111),” Journal of Physical Chemistry, Vol. 99, No. 43, 1995, pp. 16002-16010. doi:10.1021/j100043a046
- L. J. Richer and W. Ho, “Reactive Adsorption of H2CO on Ni(110) at 95K,” Journal of Chemical Physics, Vol. 83, No. 5, 1985, pp. 2165-2169. doi:10.1063/1.449308
- H. E. Dastoor, P. Gardner and D. A. King, “Identification of Two Telted Adsorbed μ2-Methoxy Species on Ni(110 Using RAIRS,” Chemical Physics Letters, Vol. 209, No. 5-6, 1993, pp. 493-498.
- A. Emundts, G. Pirug, J. Werner and H. P. Bonzel, “Large Solid Angle X-Ray Photoelectron Intensity Distributions from CO, Methoxy and Formate Adsorbed on Ni(110),” Surface Science, Vol. 410, No. 2-3, 1998, pp. L727-L735. doi:10.1016/S0039-6028(98)00233-7
- B. Sexton, “Methanol Decomposition on Platinum(111),” Surface Science, Vol. 102, No. 1, 1981, pp. 271-281. doi:10.1016/0039-6028(81)90321-6
- M. Witko, K. Hermann, D. Ricken, W. Stenzel, H. Conrad and A. M. Bradshaw, “The Electron Structure of the Surface Methoxy Species on Cu(111),” Chemical Physics, Vol. 177, No. 2, 1993, pp. 363-371. doi:10.1016/0301-0104(93)80018-5
- H. Yang, J. L. Whitten and C. M. Friend, “Adsorption of CH3O on Ni(111),” Surface Science, Vol. 313, No. 3, 1994, pp. 295-307. doi:10.1016/0039-6028(94)90050-7
- M. Witko and K. Hermann, “Site-Dependent Binding of Methoxy on Cu(111): Cluster Model Studies,” Journal of Chemical Physics, Vol. 101, No. 11, 1994, pp. 10173- 10180. doi:10.1063/1.468006
- G. C. Wang, Y. H. Zhoua, Y. Morikawa, J. Nakamura, Z. S. Cai and X. Z. Zhao, “Kinetic Mechanism oh Methanol Decomposition on Ni(111) Surface: A Theoretical Study,” Journal of Physical Chemistry B, Vol. 109, No. 25, 2005, pp. 12431-12442. doi:10.1021/jp0463969
- J. R. B. Gomes and J. A. N. F. Gomes, “Comparative Study of Geometry and Bonding Character for Methoxy Radical Adsorption on Noble Metals,” Journal of Molecular Structure (Theochem), Vol. 503, No. 3, 2000, pp. 189-200. doi:10.1016/S0166-1280(99)00286-9
- J. Greeley and M. Mavrikakis, “Competitive Paths for Methanol Decomposition on Pt(111),” Journal of American Chemical Society, Vol. 126, No. 10, 2004, pp. 3910- 3919. doi:10.1021/ja037700z
- X. Y. Pang, C. Wang, Y. H. Zhou, J. M. Zhao and G. C. Wang, “DFT Study of the Structure Sensitivity for the Adsorption of Methyl, Methoxy, and Formate on Ni(111), Ni(100), and Ni(110) Surfaces,” Journal of Molecular Structure (Theochem), Vol. 948, No. 1-3, 2010, pp. 1-10. doi:10.1016/j.theochem.2010.01.034
- W. K. Chen, S. H. Liu, M. J. Cao, Q. G. Yan and C. H. Lu, “Adsorption and Dissociation of Methanol on Au(111) Surface: A First-Principles Periodic Density Functional Study,” Journal of Molecular Structure (Theochem), Vol. 770, No. 1-3, 2006, pp. 87-91. doi:10.1016/j.theochem.2006.05.040
- W. K. Chen, S. H. Liu, M. J. Cao, C. H. Lu, Y. Xu and J. Q. Li, “Adsorption of Methanol and Methoxy on Cu(111) Surface: A First-Principles Periodic Density Functional Theory Study,” Chinical Journal of Chemistry, Vol. 24, No. 7, 2006, pp. 872-876. doi:10.1002/cjoc.200690166
- S. Pick, “Density-Functional Study of the Methoxy Intermediates at Cu(111), Cu(110) and Cu(001) Surfaces,” Journal of Physics: Condensed Matterials, Vol. 22, No. 39, 2010, Article ID: 395002.
- K. Christmann, “Interaction of Hydrogen with Solid Surfaces,” Surface Science, Vol. 9, No. 1-3, 1988, pp. 1-163. doi:10.1016/0167-5729(88)90009-X
- G. A. Somorjai, “Introduction to Surface Chemistry and Catalysis,” Wiley-Interscience, New York, 1994.
- M. Schick, J. Lauterbach and W. Weinberg, “The CO Adsorption of Hydrogen and Potassium on Ir(111),” Surface Science, Vol. 360, No. 1-3, 1996, pp 255-260. doi:10.1016/0039-6028(96)00666-8
- H. Okuyama, T. Ueda, T. Aruga and M. Nishijima, “Overtones of H Vibrations at Ni(111): Formation of Delocalized States,” Physical Review B, Vol. 63, No. 23, 2001, pp. 233403-233406. doi:10.1103/PhysRevB.63.233403
- G. X. Cao, E. Nabighian and X. D. Zhu, “Diffusion of Hydrogen on Ni(111) over Wide Range of Temperature: Exploring Quantum Diffusion on Metals,” Physial Review Letters, Vol. 79, No. 19, 1997, pp. 3696-3699. doi:10.1103/PhysRevLett.79.3696
- A. E. Morgan and G. A. Somorjai, “LEED Studies of Gas Adsorption on the Platinum (100) Single Crystal Surface,” Surface Science, Vol. 12, No. 3, 1968, pp. 405-425. doi:10.1016/0039-6028(68)90089-7
- R. W. McCabe and L. D. Schmidt, “Adsorption on H2 and CO on Clean and Oxidized (110) Pt,” Surface Science, Vol. 60, No. 1, 1976, pp. 85-98. doi:10.1016/0039-6028(76)90008-X
- K. Christmann, G. Ertl and T. Pignet, “Adsorption of Hydrogen on a Pt(111) Surface,” Surface Science, Vol. 54, No. 2, 1976, pp. 365-392. doi:10.1016/0039-6028(76)90232-6
- J. R. Engstrom, W. Tsai and W. H. Weinberg, “The Chemisorption of Hydrogen on the Pt(111) and (110)-(1x2) Surfaces of Iridium and Platinum,” Journal of Chemical Physics, Vol. 87, No. 5, 1987, pp. 3104-3119. doi:10.1063/1.453048
- L. J. Richter, “Vibrational Spectroscopy of H on Pt(111): Evidence for Universally Soft Parallel Modes,” Physical Review B, Vol. 36, No. 18, 1987, pp. 9797-9800. doi:10.1103/PhysRevB.36.9797
- B. J. J. Koelman, S. T. de Zwart, A. L. Boers, B. Poelsema and L. K. Verheij, “Information on Adsorbate Positions from Low-Energy Recoil Scattering: Adsorption of Hydrogen on Pt,” Physical Review Letters, Vol. 56, No. 11, 1986, pp. 1152-1155. doi:10.1103/PhysRevLett.56.1152
- W. Di, K. E. Smith and S. D. Kevan, “Angle-Resolved Photoemission Study of the Clean and Hydrogen-Covered Pt(111) Surface,” Physical Review B, Vol. 45, No. 7, 1992, pp. 3652-3658. doi:10.1103/PhysRevB.45.3652
- B. E. Spiewak, R. D. Cotright and J. A. Dumesic, “Microcalorimetric Studies of H2, C2H4 and C2H2 Adsorption on Pt Powder,” Journal of Catalysis, Vol. 176, No. 2, 1998, pp. 405-414. doi:10.1006/jcat.1998.2047
- P. Nordlander, S. Holloway and J. K. Nørskov, “Hydrogen Adsorption on Metal Surfaces,” Surface Science, Vol. 136, No. 1, 1984, pp. 59-81. doi:10.1016/0039-6028(84)90655-1
- D. E. Jiang and E. A. Carter, “Adsorption and Diffusion Energetics of Hydrogen Atoms on Fe(110) from First Principles,” Surface Science, Vol. 547, No. 1-2, 2003, pp. 85-98. doi:10.1016/j.susc.2003.10.007
- P. Légaré, “A Theoretical Study of H Surface and Subsurface Species on Pt(111),” Surface Science, Vol. 559, No. 2-3, 2004, pp. 169-178. doi:10.1016/j.susc.2004.04.013
- F. Faglioni and W. A. Goddard III, “Energetics of Hydrogen Coverage on Group VIII Transition Metal Surfaces and Kinetic Model for Adsorption/Desorption,” Journal of Chemical Physics, Vol. 122, No. 1, 2005, Article ID: 014704. doi:10.1063/1.1814938.
- D. C. Ford, Y. Xu and M. Mavrikakis, “Atomic and Molecular Adsorption on Pt(111),” Surface Science, Vol. 587, No. 3, 2005, pp. 159-174. doi:10.1016/j.susc.2005.04.028
- F. Saad, M. Zemerli, M. Bennaki and S. Bouarab, “AbInitio Study of Coadsorption of Li and H on Pt(001), Pt(110) and Pt(111) Surfaces,” Physica B, Vol. 407, No. 4, 2012, pp. 698-704. doi:10.1016/j.physb.2011.12.005
- M. A. Petersen, S. J. Jenkins and D. A. King, “Theory of Methane Dehydrogenation on Pt{110} (1x2). Part I: Chemisorption of CHx (x = 0 - 3),” Journal of Physical Chemistry B, Vol. 108, No. 19, 2004, pp. 5909-5919. doi:10.1021/jp037880z
- P. S. Moussounda, M. F. Haroun, G. Rakotovelo and P. Légaré, “A Theoretical Study of CH4 Dissociation on Pt(100),” Surface Science, Vol. 601, No. 18, 2007, pp. 3697-3701. doi:10.1016/j.susc.2007.04.014
- W. Lai, D. Xie and D. H. Zhang, “First-Principles Study of Adsorption of Methyl, Coadsorption of Methyl and Hydrogen, and Methane Dissociation on Ni(100),” Surface Science, Vol. 594, No. 1-3, 2005, pp. 83-92. doi:10.1016/j.susc.2005.07.012
- J. Greeley and M. Mavrikakis, “A First-Principles Study of Surface and Subsurface H on and in Ni(111): Diffusional Properties and Coverage—Dependent Behavior,” Surface Science, Vol. 540, No. 2-3, 2003, pp. 215-229. doi:10.1016/S0039-6028(03)00790-8
- M. F. Haroun, P. S. Moussounda, P. Légaré and J. C. Parlebas, “Adsorption and Co-Adsorption of CH3 and H on Flat and Defective Nickel (111) Surfaces,” European Physical Journal B, Vol. 7, No. 3, 2010, pp. 353-358. doi:10.1140/epjb/e2010-10680-0
- P. G. Sundell and G. Wahnström, “Hydrogen Tunneling on a Metal Surface: A Density-Functional Study of H and D Atoms on Cu(001),” Surface Science, Vol. 593, No. 1-3, 2005, pp. 102-109. doi:10.1016/j.susc.2005.06.051
- B. Hammer, L. B. Hansen and J. K. Nørkov, “Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals,” Physical Review B, Vol. 59, No. 11, 1999, pp. 7413-7421. doi:10.1103/PhysRevB.59.7413
- D. Vanderbilt, “Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism,” Physical Review B, Vol. 41, No. 11, 1990, pp. 7892-7895. doi:10.1103/PhysRevB.41.7892
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, “PW91: Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation,” Physical Review B, Vol. 46, No. 11, 1992, pp. 6671-6687. doi:10.1103/PhysRevB.46.6671
- D. R. Lide, “CRC Handbook of Chemistry and Physics,” CRC Press, Boca Raton, 1996.
- B. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, “Theoretical Study of O-Assisted Selective Coupling of Methanol on Au(111),” Journal of Physical Chemistry, Vol. 115, No. 5, 2011, pp. 3703-3708. doi:10.1021/jp110835w
- F. Mehmood, A. Kara, T. S. Rahman and C. R. Henry, “Comparative Study of CO Adsorption on Flat, Stepped and Kinked Au Surfaces Using Density Functional Theory,” Physical Review B, Vol. 79, No. 7, 2009, Article ID: 075422.
- F. Herzberg, “Molecular Spectra and Molecular Structure. II. Infrared and Raman Spectra of Polyatomic Molecules,” D. Van Nostrand Company, New York, 1945.
- P. S. Moussounda, “Adsorption et Activation du Méthane et du Méthanol sur la Surface (100) du Platine: Une Etude par la Fonctionnelle de la Densité,” Ph.D. Thesis, University of Louis Pasteur Strasbourg 1, Strasbourg, 2006.
- G. Kresse and J. Hafner, “First Principles Study of the Adsorption of Atomic H on the Ni(111), (100), (110) Surfaces,” Surface Science, Vol. 459, No. 3, 2000, pp. 287- 302. doi:10.1016/S0039-6028(00)00457-X
- D. E. Jiang and E. A. Carter, “Diffusion of Interstitial Hydrogen into and through bcc Fe from First Principles,” Physical Review B, Vol. 70, No. 6, 2004, Article ID: 064102.
- F. C. Fabiani, G. Fratesi and G. P. Brivio, “Adsorption of H2S, HS, S and H on a Stepped Fe(310) Surface,” European Physical Journal B, Vol. 78, No. 4, 2010, pp. 455- 460.
NOTES
*Corresponding author.