Advances in Bioscience and Biotechnology
Vol.5 No.1(2014), Article ID:41659,7 pages DOI:10.4236/abb.2014.51003
Methemoglobinemia—A biomarker and a link to ferric iron accumulation in Alzheimer’s disease
1Department of Environmental Medicine, University of Rijeka School of Medicine, Rijeka, Croatia
2“Lino Rossi” Research Center, Department of Biomedical, Surgical, and Dental Science, University of Milan, Milan, Italy
3Department of Pathology, University of Rijeka School of Medicine, Rijeka, Croatia
4Department of Biology, University of Texas at San Antonio, San Antonio, USA
5Department of Social Medicine and Epidemiology, University of Rijeka School of Medicine, Rijeka, Croatia
6Department of Family Medicine, University of Rijeka School of Medicine, Rijeka, Croatia
7Department of Gynecology and Obstetrics, University of Rijeka School of Medicine, Rijeka, Croatia
Email: *lucijan.mohorovic@pu.t-com.hr
Copyright © 2014 Lucijan Mohorovic et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Lucijan Mohorovic et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 30 September 2013; revised 26 November 2013; accepted 20 December 2013
KEYWORDS
Alzheimer’s Disease (AD); Apoptosis; Blood-Brain Barrier (BBB); Brain Capillary Ferric Iron Deposition; Hemoglobin and Methemoglobin Catabolism; Neurodegenerative Brain Disease; SIDS
ABSTRACT
Understanding the mechanism of oxidative stress is likely to yield new insights regarding the pathogenesis of Alzheimer’s disease (AD). Our earlier work focused on the difference between hemoglobin and methemoglobin degradation, respectively leading to ferrous (Fe2+) iron, or ferric (Fe3+) iron. Methemoglobin has the role of carrier, the donor of cytotoxic and redox-active ferric (Fe3+) iron, which can directly accumulate and increase the rate of capillary endothelial cell apoptosis, and may cross into the brain parenchyma, to the astrocytes, glia, neurons, and other neuronal cells (neurovascular unit). This supposition helps us to understand the transport and neuronal accumulation process of ferric iron, and determine how iron is transported and accumulated intracellularly, identifiable as “Brain rust”. Earlier research found that the incidences of neonatal jaundice (p = 0.034), heart murmur (p = 0.011) and disorders such as dyslalia and learning/memory impairments (p = 0.002) were significantly higher in those children born from mothers with methemoglobinemia. Our hypothesis suggests that prenatal iron abnormalities could lead to greater neuronal death, the disease ageing process, and neurodegenerative disorders such as AD and other neurodegenerative diseases.
1. INTRODUCTION
The mechanisms responsible for redox-active iron accumulation in some regions of the brain in Alzheimer’s disease (AD) are unknown, nor if it is an initial event that causes neuronal death or a consequence of the disease process. However, little is still known about the chemical form of iron associated with neurodegenerative diseases, its role in neurodegeneration (if any) and its origin. Our goal is to understand iron-induced oxidative stress and point out the deleterious effects of redox-active ferric iron as a final product from methemoglobin and heme degradation. Ferric iron has a direct impact on capillary brain endothelial structure and function and in consequence of apoptosis brain parenchyma atrophy finally follows, so pinpointing specific clinical determinants for the onset and progression of AD.
Ferric-Iron Brain Accumulation as a Cause of Neurodegenerative Brain Disease: A New Insight in Understanding the Mechanism of Iron Transport
Continuously inhaling strong oxidants such as NOx (NO and NO2) reversibly oxidizes oxyhemoglobin (Fe2+) to methemoglobin (Fe3+), and irreversible methemoglobinemia results as a disorder from the impacts of the metabolic synergy of nitrogen oxides such as oxidants (RNS) and sulfur dioxide metabolites (RSS) as inhibitors of thiol antioxidants [1].
Methemoglobin by itself, and heme, have prooxidant properties and induce structural and functional changes in the vascular endothelium [2,3]. These changes include cell growth arrest, senescence, morphological alterations and cell apoptosis, and they lead to both vessel thrombosis and endothelial cell denudation under the influence of redox-active ferric iron (Fe3+), as a product of heme-oxygenase, responsible for methemoglobin-heme degradation [4].
There is a striking difference between physiological hemoglobin catabolism, and pathological methemoglobin catabolism, because of the different final products, respectively ferrous and ferric iron, with distinct characteristics (Figures 1A and B). Ferrous iron form has the potential for catalyzing and generating highly cytotoxic hydroxyl radicals shown as Fenton reactions 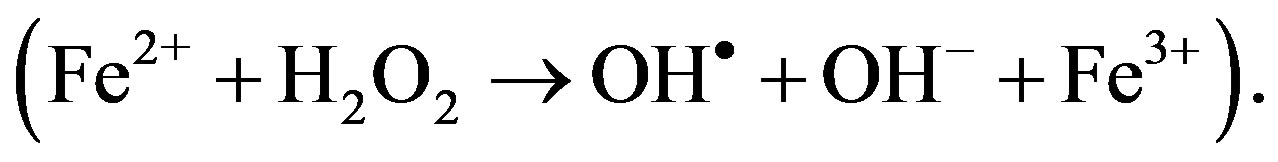 The substantial difference of intracellular ferric iron originates from the physiological Fenton reaction, and ferric iron originates from pathological methemoglobin catabolism occurring as the level of methemoglobin cellular uptake increases, and the resulting methemoglobinemia causes ferric ironinduced oxidative stress injury. We consider that the final product of ferric iron from methemoglobin catabolism is a significant added source to Fenton reaction derived from ferric iron, whose continuous formation impacts the brain and confirms the statement that iron and iron-induced oxidative stress constitutes a common mechanism in the development of neurodegeneration.
The substantial difference of intracellular ferric iron originates from the physiological Fenton reaction, and ferric iron originates from pathological methemoglobin catabolism occurring as the level of methemoglobin cellular uptake increases, and the resulting methemoglobinemia causes ferric ironinduced oxidative stress injury. We consider that the final product of ferric iron from methemoglobin catabolism is a significant added source to Fenton reaction derived from ferric iron, whose continuous formation impacts the brain and confirms the statement that iron and iron-induced oxidative stress constitutes a common mechanism in the development of neurodegeneration.
Our former research results suggest that methemoglobin plays a particularly important role as a carrier and donor of cytotoxic redox-active ferric (Fe3+) iron, and determines how iron is transported intracellularly. Neuroscience has traditionally focused on the neurons of the central and peripheral nervous systems, and it is now becoming clear that neurons, glia and microvessels are organized into a well-structured neurovascular unit, and recent studies have highlighted the importance of brain endothelial cells in this modular organization [5].
Leung and Moody [6] have demonstrated that ferric heme is significantly more pro-oxidant than ferrous heme. In tandem, this evidence suggests that the MR imagingdetected T1 high signal intensity within the vessel wall is an endogenous biomarker of an intraplaque environment that is highly pro-oxidant and proatherogenic. MR imaging measures showed a T1 relaxivity that was 10 times higher for ferric than for ferrous forms of hemoglobin. Their results support the hypothesis that ferric methemoglobin-generated T1 high signal intensity reflects a pro-oxidant environment that, in the setting of vessel wall disease, might be proatherogenic. This justifies the study of the oxidant effect of methemoglobin catabolic
 A
A 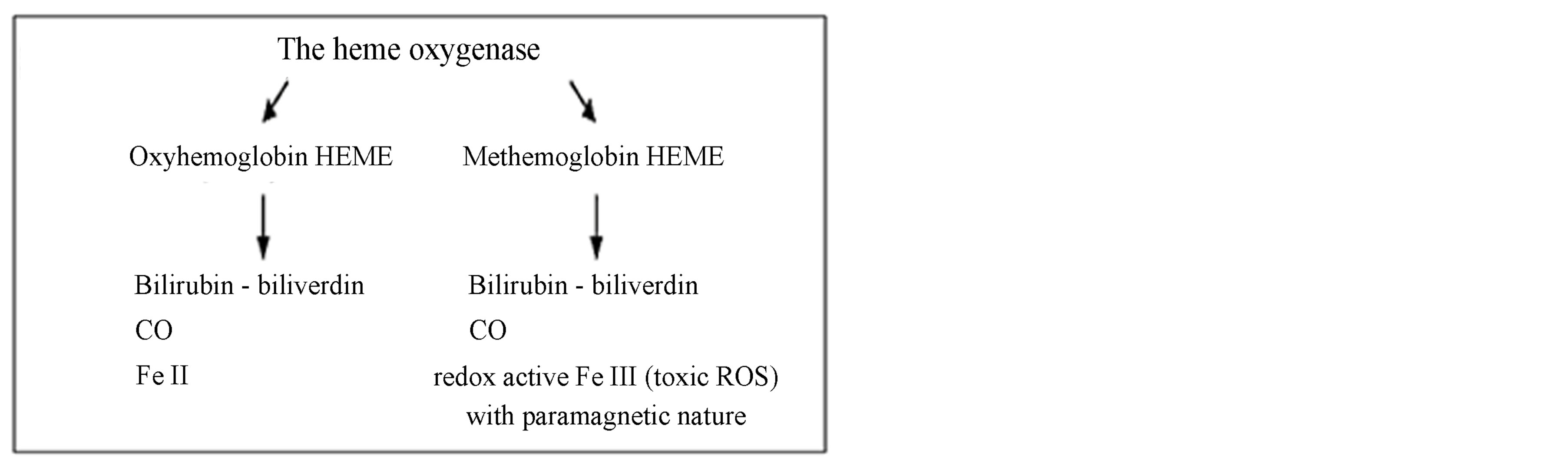 B
B
Figure 1. Difference between physiological hemoglobin catabolism and pathological methemoglobin catabolism.
Source: Balla J. et al. Proc Natl Acad Sci USA 1993, 90:9285-9; Denicola A. et al. Proc Natl Acad Sci USA 1998, 95:3566-71; Jeney V. et al. Blood 2002; Umbreit J. Am J Hematol 2007, 100:879-87.
products on vital organs and the CNS, resulting in their dysfunction. The research MRI data showed that extracellular methemoglobin generates significantly more lipid oxidation than intracellular products. However methemoglobin in both these environments has similar measures of r1. Therefore, the T1 high signal intensity due to methemoglobin is not solely restricted to an environment that causes lipid oxidation. Thus, the ability of this high signal intensity to reflect at-risk plaque may be diminished. However, it is known that in the absence of any chemical modifications, ferric heme substantially degrades the integrity of red blood cells (RBC) and the membrane, and the eventual fate of static RBC is lysis. Thus, intracellular methemoglobin is destined to rapidly become extracellular, thereby adding to oxidative drive [7]. The cellular and intercellular iron transport mechanisms in the CNS are still poorly understood, while accumulating evidence suggests that impaired iron metabolism is an initial cause of neurodegeneration [8]. Brar et al. [9] concluded that the development of parkinsonism during the course of AD appears to be associated with the accumulation of iron, which in turn may contribute to the pathogenesis of neurologic decline.
2. METHODS AND RESULTS
The present standpoint derived from literature and proper research results showed that continuous exposure to inhaled toxic substances in addition to oxidants by food, water, drugs and their associated cummulative effects, causes increased oxidative stress. It postulates the role of environmental toxic factors on the endothelial small vessels of the brain, increasing the rate of endothelial cell apoptosis and making possible the transport of methemoglobin and heme derived ferric iron across the bloodbrain-barrier (BBB), and its accumulation in the brain parenchyma.
Our results point out the consequence of toxic environmental oxidants, caused by brain damage with a view to the role of methemoglobin catabolism in pregnancy as the source of ferric (Fe3+) iron concentrated in various brain regions.
Methemoglobin and hemolysis both occur as a result of oxidative stress, but the prevalent difference between them is that methemoglobin is a reversible phenomenon (oxidant-antioxidant balance) whereas hemolysis, which occurs as a result of oxidative stress on the erythrocyte membrane, is an irreversible event. Methemoglobinemia can additionally exacerbate an existing anemia, stimulating hypoxia that may be additionally dangerous. Our prospective study of methemoglobin in pregnancy, revealed a significant rise in the level of methemoglobin >1.5 g/L (r = 0.72, p < 0.01) in the air-polluted exposure period, which can be explained on the basis of an oxidant-antioxidant imbalance, resulting in methemoglobinemia [10]. Methemoglobinemia and stillbirth recorded throughout the exposure period are significantly higher than those recorded in the control period (p = 0.0205) and the frequencies of reproductive loss were significantly lower in the control than in the exposure period (p < 0.05) [11].
As we have found no evidence of the consequences of maternal methemoglobinemia on the fetus, the second objective was to direct attention to methemoglobin. As oxidants possess the capacity to cross the damaged fetomaternal placental barrier, for instance from the methemoglobin catabolism ferric iron placental chorionic deposition appearance (Figure 2), “fetal preeclampsia” is an expected manifestation. Under these adverse conditions, ferric iron positivity in capillary endothelial cells of the BBB in the fetus rises, resulting in preterm birth, stillbirth or early neonatal death (Figure 3). As an early
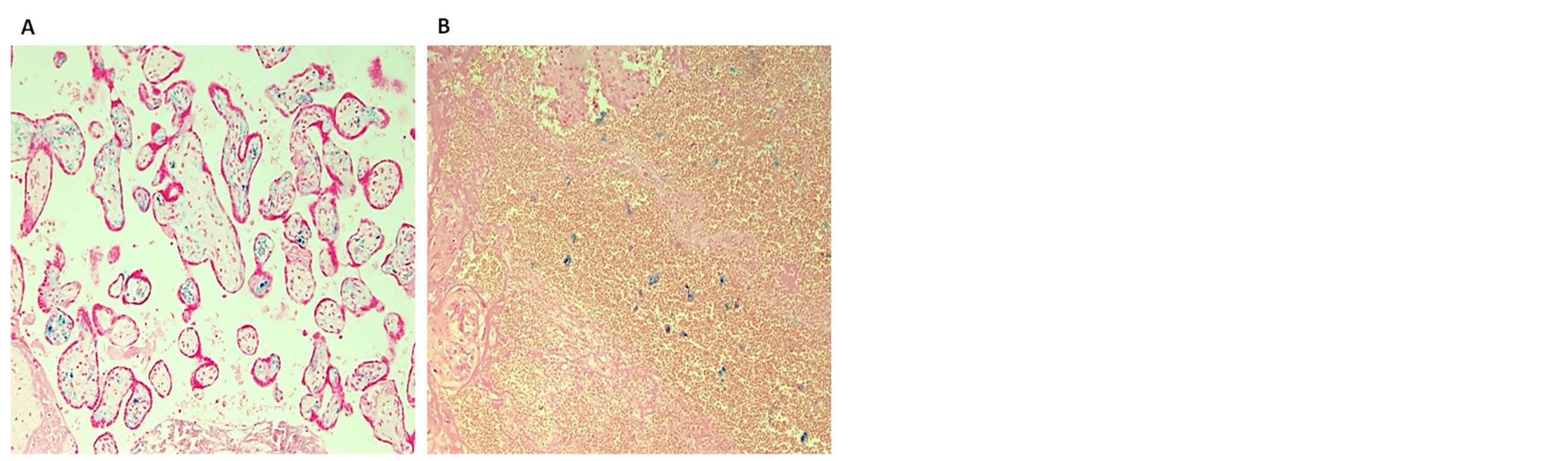
Figure 2. Positive ferric iron deposits in an intervillous hemorrhage (A) and in placental chorionic villi stroma (B). Placenta from a stillbirth after 38 weeks of gestation. Mother MetHb > 1%. Staining: Perl’s Prussian Blue reaction for ferric iron—Magnification: (A) 4×; (B) 20×.
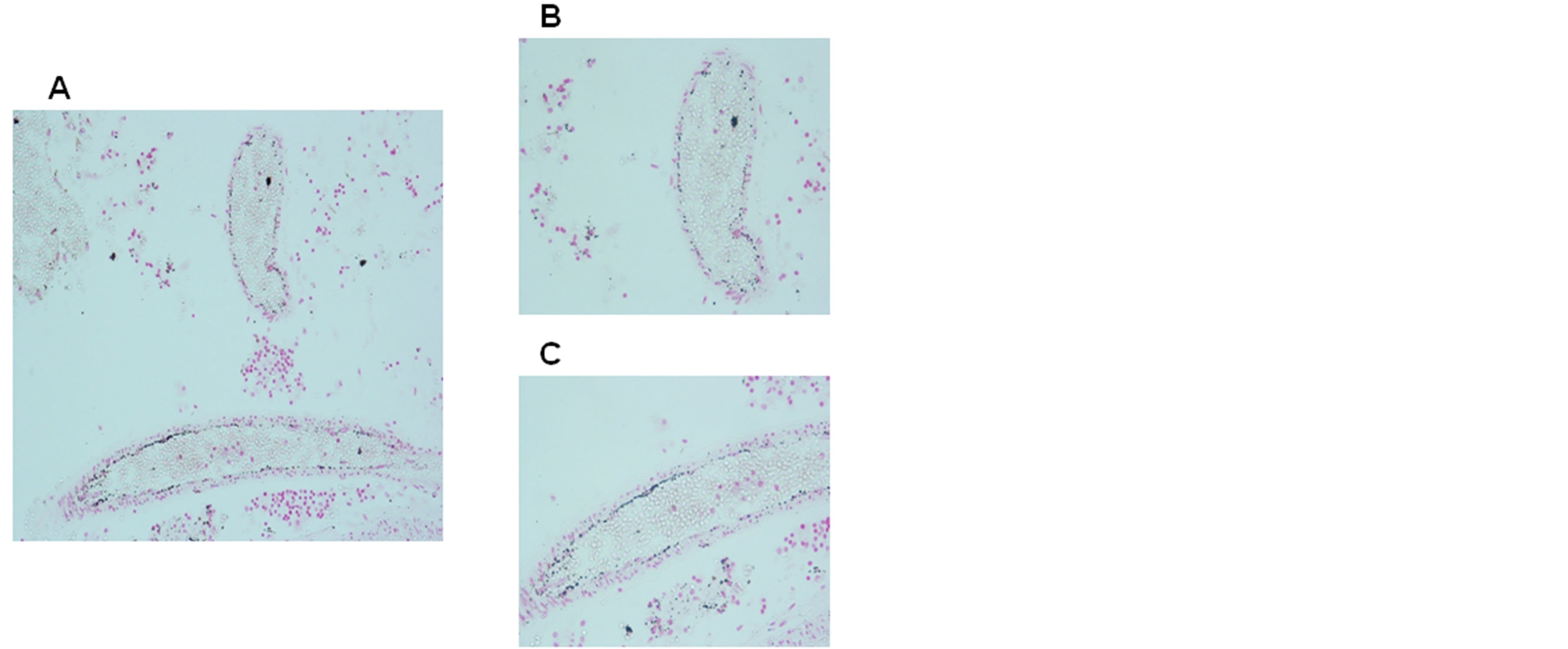
Figure 3. Iron positivity in capillary endothelial cells of the blood-brain barrier in a victim of sudden intrauterine unexplained death (SIUD) aged 36 gestational weeks. Staining: Perl’s Prussian Blue reaction for ferriciron—magnification: A) 10×; B) and C) 20×)
biomarker of environmental toxic and oxidative stress effects, this puts pregnancy at risk and may later impair the health of newborns, children and adolescents. Obtained proper research found an incidence of neonatal jaundice (p = 0.034), heart murmur at a later age (p = 0.011), as well as child and adult mild disorders such as dyslalia and learning/memory impairments (p = 0.002) significantly higher than in the children and adults of control mothers without pregnancy methemoglobinemia [12]. Lavezzi, Mohorovic et al. recently presented findings, confirmed by pathohistological techniques, that under adverse conditions, ferric iron positivity in capillary endothelial cells of the BBB in the fetus rises, also resulting in preterm birth, stillbirth or early neonatal death [13]. The application of the Blue Prussian method highlighted accumulations of blue granulations, indicative of nonheme Fe3+-positive reactions in the brainstem and cerebellum of 12 (33%) of the 36 sudden intrauterine unexplained death (SIUD) and sudden infant death syndrome (SIDS) victims and in none of the control group. In positive cases, iron deposits were widespread in the brain parenchyma or localized in specific areas showing variable extent and intensity (Figures 4 and 5). The positivity of free iron in the cerebral parenchyma of a patient with Alzheimer’s disease is represented in Figure 6. In the same cases of their previous study [13], Lavezzi, Mohorovic et al., by applying the TUNEL method, highlighted in the same cases showing iron deposition in the capillary endothelial cells of the BBB and in the brain parenchima, high percentages of apoptotic cells in the ventricular barriers, and precisely in the ependyma and in choroid plexus epithelial cells (Figures 7 and 8).
3. DISCUSSION
Proper research results suggest that methemoglobin plays an important role as a biomarker, a carrier and a donor of cytotoxic and redox-active ferric iron, and determines how iron is transported and accumulated intracellularly, identifiable as “Brain rust”.
We highlight the role of methemoglobin and heme catabolism to produce ferric iron with cytotoxic and paramagnetic properties. We propose that ferric iron and ferric iron-induced oxidative stress constitutes a common mechanism in the development of neurodegeneration, and also suggests an initial cause of neuronal death. The experiments showed that ferric and ferrous iron can enter cells via different pathways; they do not indicate which pathway is dominant in humans [14].
Smith et al. suggest that iron is able to participate in “in situ” oxidation and readily catalyzes an H2O2-dependent oxidation, and indicate that iron accumulation could be an important contributor to the oxidative damage of Alzheimer’s disease [15]. Our work, from our standpoint, supports the above statement about the importance of disturbed heme metabolism. Heme oxygenase-1, an en-
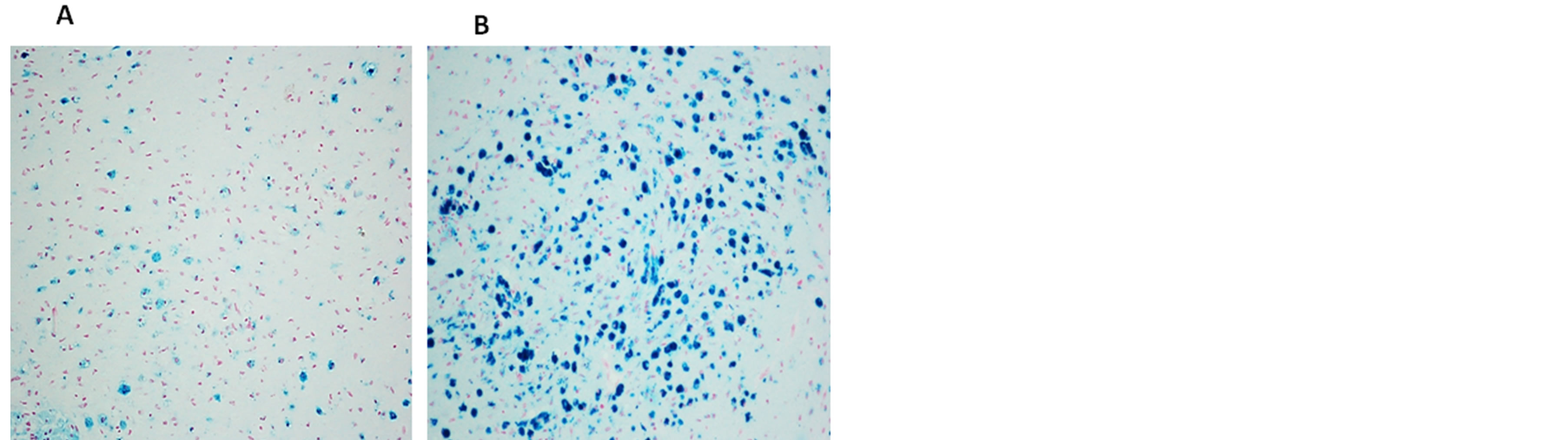
Figure 4. Free iron positivity in the medullary parenchyma of a 7 month-old victim of SIDS. (A) widespread distribution of positive neurons. (B) accumulation of strong iron-positive neurons. Staining: Perl’s Prussian Blue reaction for ferric ironMagnification: (A) and (B) 20×.
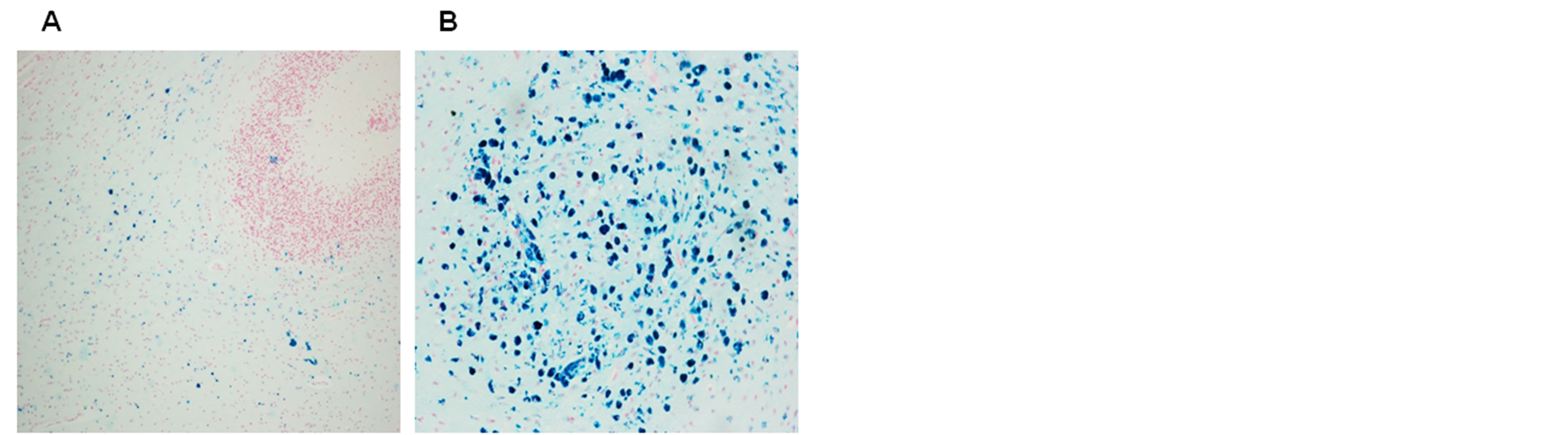
Figure 5. Free iron positivity in the subcortical region (A) and parenchyma (B) of the cerebellum of a 6 month-old victim of SIDS (case no.9). Staining: Perl’s Prussian Blue reaction for ferric iron—Magnification 20×.


Figure 6. Free iron positivity in the cerebral parenchyma of a 77 year old patient with Alzheimer’s disease A) Focal distribution of positive glia and neurons; B) accumulation of strong ironpositive perivascular regions. Staining: Perl’s Prussian Blue reaction for ferric iron—Magnification: 40×.
zyme that catalyzes the conversion of methemoglobin and heme to ferric iron, is increased in Alzheimer’s disease, suggesting heme turnover as a source of redoxactive iron. Perry et al. have found that while mitochondrial DNA and cytochrome C oxidase protein are increased in Alzheimer’s disease, the number of mitochon-

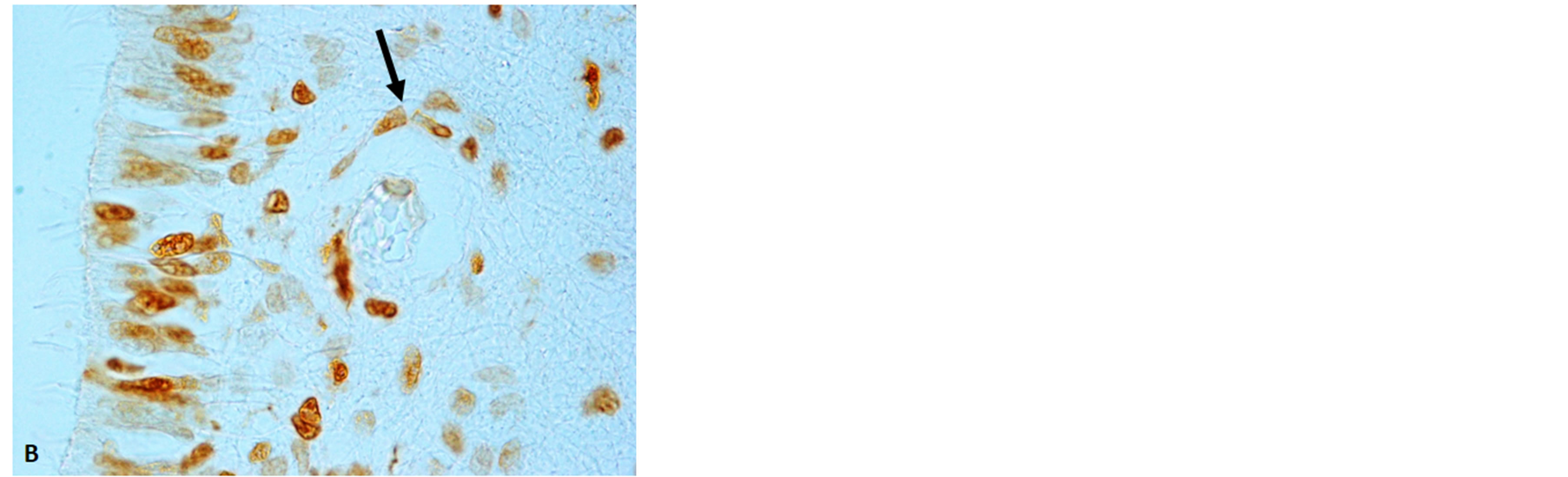
Figure 7. Floor of the fourth ventricle showing high expression of apoptosis in the ependyma and subependimal parenchima. The framed area in (A) is represented at higher magnification in (B). The arrow indicates apoptotic endothelial cells of a blood vessel. Immunostaining: TUNEL method. Magnification: (A) 10×; (B) 100×.
dria is decreased, indicating accelerated mitochondria turnover, and suggests mitochondrial dysfunction as a potentially inseparable component of the initiation and progression of Alzheimer’s disease [16]. It was also found that oxidative damage occurs primarily within the cytoplasm rather than in mitochondria. From this originates the hypothesis that mitochondria, as a source of hydrogen peroxide, an intermediate, once in the cytoplasm, will be converted into highly reactive hydroxyl radicals through the Fenton reaction in the presence of metal ion, causing damage to the cytoplasm [17,18]. Various pathologies can result from oxidative stress-induced apoptotic signaling, consequently leading to ROS increases and/or antioxidant decreases, the disruption of intracellular redox homeostasis, and an irreversible impact on oxidative modifications of lipid, protein, or DNA [19]. Furthermore, iron participates in different pathological processes through the Fenton reaction. To test the hypothesis that this reaction accelerates apoptosis, Jacob et al. used human umbilical vein endothelial cells (HUVECs) as surrogates for the microvasculature in vivo. HUVECs were loaded with Fe [III] (ferric chloride and ferric ammonium citrate), causing apoptosis after heat shock stimulus [20]. Accordingly, in our studies on vic-


Figure 8. Choroid plexus of the fourth ventricle showing high expression of apoptosis in the epithelial cells. The framed area in (A) is represented at higher magnification in (B). Immunostaining: TUNEL method. Magnification: (A) 10×; (B) 100×.
tims of sudden perinatal death (SIUD and SIDS), we observed that, under adverse hypoxic conditions, iron deposition was associated to apoptotic degeneration of the brainstem lining. Brain iron is a major contributor to the MRI contrast in normal gray matter. Non-heme brain iron is present mainly in the form of ferritin. The quantitation of non-heme brain ferric iron indicated by MRI helps in the diagnosis and monitoring of different neurological diseases [21]. Most brain non-heme iron is believed to be present as a storage pool, consisting of ferritin or hemosiderin, and also as a product of methemoglobin catabolism [22]. This fact suggests considering the role of methemoglobin catabolism as the source of ferric iron (FeIII) form concentrated in various brain regions. Nowdays, non-heme-bound Fe3+ is quantified using MRI, thanks to its paramagnetic properties. It is believed that most non-heme-bound iron is deposited in the form of ferritin, haemosiderin, or methemoglobin catabolic products, whereas transferrin-bound iron concentration is always low and can not be detected by MRI [23]. Recent research results indicate a ferrihydrite-magnetite coreshell ferritin structure. It was also found that magnetite in the ferritin iron core is not a source of free toxic ferrous iron, as previously believed. Therefore, the presence of magnetite in the ferritin cores of patients with Alzheimer’s disease is not a cause of their increased brain ferrous iron(II) concentration [24].
4. CONCLUSION
A proper statement should emphasize the role of methemoglobin and heme which by themselves have prooxidant properties. An abundant source of cytotoxic and redox-active ferric (Fe3+) iron, without ferrous-ferric inversions “in situ”, as a cause of iron-induced oxidative stress has a direct and specific impact on the endothelial small vessels of the brain, and increases the rate of endothelial cell apoptosis, making it possible to cross over from methemoglobin and heme derived ferric iron and accumulate in the brain parenchyma. In conclusion we point out the importance of methemoglobinemia not only as a biomarker and precursor of environmental oxidants’ noticeable effects, but also as a carrier and donor of redox-active ferric iron form. We point out ferric iron as an originator of brain parenhyma accumulation, having an important role in crossing the brain microvessels to the neurons (neurovascular unit), causing neuronal death, continuous ageing process, and leading finally to hard neurodegenerative disorders such as AD, parenchyma and other diseases. Nevertheless our findings in relation to environmental oxidants and the pathogenesis of neurodegenerative diseases need further research confirmation.
ACKNOWLEDGEMENTS
The author “declares a competing financial interest because the Croatia Electricity Company, PP Plomin Ltd, the management company for Plomin 2, provided funding for the study.
This study was also supported by the Convention with the Provincia autonoma di Trento, Italy (Conv 7436/555-12).
REFERENCES
- Mohorovic, L., Materljan, E., Micovic, V., Malatestinic, D., Stifter, S. and Lavezzi, A.M. (2012) Impacts of exogenously derived nitrogen oxide and sulfur compounds on the structure and function of the vascular endothelium—The link between pregnancy hypertension and later life hypertension. Journal of Hypertension Open Access, 1, 103.
- Balla, J., Vercellotti, G.M., Jeney, V., Yachie, A., Varga, Z., Jacob, H.S., Eaton, J.W. and Balla, G. (2007) Heme, heme oxygenase, and ferritin: How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal, 9, 2119-2137. http://dx.doi.org/10.1089/ars.2007.1787
- Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G.M., Eaton, J.W. and Balla, G. (2002) Pro-oxidant and cytotoxic effects of circulating heme. Blood, 100, 879- 887. http://dx.doi.org/10.1182/blood.V100.3.879
- Maisels, M.J. and Kring, E. (2006) The contribution of hemolysis to early jaundice in normal newborns. Pediatrics, 118, 276-279. http://dx.doi.org/10.1542/peds.2005-3042
- Abbott, N.J., Rönnbäck, L. and Hansson, E. (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nature Reviews Neuroscience, 7, 41-53. http://dx.doi.org/10.1038/nrn1824
- Leung, G. and Moody, A.R. (2010) MR imaging depicts oxidative stress induced by methemoglobin. Radiology, 257, 470-476. http://dx.doi.org/10.1148/radiol.10100416
- Baysal, E., Monteiro, H.P., Sullivan, S.G. and Stern, A. (1990) Desferrioxamine protects human red blood cells from hemin-induced hemolysis, Free Radic Biol Med, 9, 5-10. http://dx.doi.org/10.1016/0891-5849(90)90043-I
- Mills, E., Dong, X.P., Wang, F. and Xu, H. (2010) Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Medicinal Chemistry, 2, 51. http://dx.doi.org/10.4155/fmc.09.140
- Brar, S., Henderson, D., Schenck, J. and Zimmerman, E.A. (2009) Iron accumulation in the substantia nigra of patients with Alzheimer’s disease and parkinsonism. JAMA Neurology, 66, 371-374. http://dx.doi.org/10.1001/archneurol.2008.586
- Mohorović, L. (2003) The level of maternal methemoglobin during pregnancy in an air-polluted environment, Environmental Health Perspectives, 111, 1902-1905. http://dx.doi.org/10.1001/archneurol.2008.586
- Mohorović, L. and Micovic, V. (2012) The importance of first blood circulation stage, a new insight into the pathogenesis of clinical manifestations of preeclampsia, Advances in Bioscience and Biotechnology , 3, 945-950. http://dx.doi.org/10.4236/abb.2012.327116
- Mohorovic, L., Materljan, E. and Brumini, G. (2008) Are neonatal jaundice, heart murmur, dyslalia and learning/ memory impairments the consequences of maternal exposure to environmental oxidants? XXIV International Con- gress Fetus as a Patients, Frankfurt.
- Lavezzi, A.M., Mohorovic, L., Alfonsi, G., Corna, M.F. and Matturri, L.L. (2011) Brain iron accumulation in unexplained fetal and infant death victims with smoker mothers—The possible involvement of maternal methemoglobinemia, BMC Pediatrics, 11, 62-67. http://dx.doi.org/10.1186/1471-2431-11-62
- Conrad, M.E., Umbreit, J.N., Moore, E.G., Hainsworth, L.N., Porubcin, M., Simovich, M.J., Nakada, M.T., Dolan, K. and Garrick, M.D. (2000) Separate pathways for the cellular uptake of ferric and ferrous iron. American Journal of Physiology: Gastrointestinal and Liver Physiology, 279, 767-774.
- Smith, M.A., Harris, P.L.R., Sayre, L.M. and Perry, G. (1997) Iron accumulation in Alzheimer’s disease is a source of redox-generated free radicals, Proceedings of the National Academy of Sciences of the United State of America, 94, 9866-9868. http://dx.doi.org/10.1073/pnas.94.18.9866
- Perry, G., Taddeo, M.A., Petersen, R.B., Castellani, R.J., Harris, P.L., Siedlak, S.L., Cash, A.D., Liu, Q., Nunomura, A., Atwood, C.S. and Smith, M.A. (2003) Iron and copper are at the center of oxidative damage in Alzheimer’s disease. Biometals, 16, 77-81. http://dx.doi.org/10.1023/A:1020731021276
- Marlatt, M., Lee, H.G., Perry, G., Smith, M.A. and Zhu, X. (2004) Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer’s disease. Acta Neurobiologiae Experimentalis (Warsaw), 64, 81-87.
- Honda, K., Smith, M.A.,, Zhu, X., Baus, D., Merrick, W.C., Tartakoff, A.M., Hattier, T., Harris, O.L. Siedlak, S-L., Fujioka, H., Liu, K., Moreira, P.I., Miller, F.P., Nunomura, A., Shimohama, S. and Perry, G., (2005) Ribosomal RNA in Alzheimer’s disease is oxidized by bound redox-active iron. The Journal of Biological Chemistry, 280, 20978-20986. http://dx.doi.org/10.1074/jbc.M500526200
- Circu, M.L. and Aw, T.Y. (2010) Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology & Medicine, 48, 749-762. http://dx.doi.org/10.1016/j.freeradbiomed.2009.12.022
- Jacob, A.K., Hotchkis, R.S., DeMeester, S.L., Hiramatsu, M., Karl, I.E., Swanson, P.E., Cobb, J.P. and Buchman, T.G. (1997) Endothelial cell apoptosis is accelerated by inorganic iron and heat via an oxygen radical dependent mechanism, Surgery, 122, 243-253. http://dx.doi.org/10.1016/S0039-6060(97)90015-5
- Vymazal, J., Brooks, R.A., Patronas, N., Hajek, M., Bulte, J.W. and Di Chiro, G. (1995) Magnetic resonance imaging of brain iron in health and disease. Neurological Sciences, 134, 1926.
- Schenk, J.F. and Zimmerman, E.A. (2004) High-field magnetic resonance imaging of brain iron: Birth of a biomarker? NMR in Biomedicine, 17, 433-445. http://dx.doi.org/10.1002/nbm.922
- Rouault, T.A. and Cooperman, S.S, (2006) Brain iron metabolism, Seminars in Pediatric Neurology, 13, 142- 148. http://dx.doi.org/10.1016/j.spen.2006.08.002
- Gálvez, N., Fernández, B., Sánchez, P., Cuesta, R., Ceolín, M., Clemente-León, M., Trasobares, S., López-Haro, M., Calvino, J.J., Stéphan, O. and Domínguez-Vera J.M. (2008) Comparative structural and chemical studies of ferritin cores with gradual removal of their iron contents. Journal of the American Chemical Society, 130, 8062- 8068. http://dx.doi.org/10.1021/ja800492z
NOTES
*Corresponding author.