Advances in Bioscience and Biotechnology
Vol.4 No.7A1(2013), Article ID:34628,6 pages DOI:10.4236/abb.2013.47A1003
Rethinking cytokine function during hepatitis A and hepatitis C infections
![]()
1Unidad de Inmunovirologia, Hospital Civil de Guadalajara Fray Antonio Alcalde, Guadalajara, Mexico
2Universidad de Guadalajara, Guadalajara, Mexico
3Servicio de Biologia Molecular en Medicina, Hospital Civil de Guadalajara Fray Antonio Alcalde, Guadalajara, Mexico
Email: *apanduro@prodigy.net.mx
Copyright © 2013 Nora A. Fierro et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 15 May 2013; revised 15 June 2013; accepted 26 June 2013
Keywords: hepatitis A virus; hepatitis C virus; cytokines
ABSTRACT
Hepatitis A virus (HAV) and hepatitis C virus (HCV) are both viruses with hepatotropic lifestyles. HAV induces an acute infection that results in the elimination of the virus by the host whereas HCV is typically able to establish a persistent infection that may result in cirrhosis and hepatocellular carcinoma. The mechanisms responsible for this difference are unknown. However, given HAV and HCV are both non-cytophatic viruses, the observed symptoms and liver injury during the infections are the result of specific immune responses under the control of cytokines. Thus, the production of cytokines during hepatotropic viral infections may constitute a mechanism leading to different outcomes. Therefore, understanding the differences in the cytokine patterns induced in response to HAV and HCV is likely to provide important insights into the cytokine-mediated mechanisms underlying the long-term persistence of HCV, the broad spectrum of clinical manifestations induced by HAV and the resolution of HAV infection during the acute phase. Herein, we focus on discoveries that hold promise in identifying cytokines as therapeutic targets for the treatment of viral hepatitis.
1. INTRODUCTION
Hepatitis A virus (HAV) and hepatitis C virus (HCV) are hepatotropic, non-cytopathic viruses that induce inflammatory liver disease. Over the last several years, research efforts have focused on HCV because it causes frequent chronic infections that may result in cirrhosis and hepatocellular carcinoma development. HAV causes only acute hepatitis and is not associated with chronic liver disease. The incidence of hepatitis A has fallen since the introduction of vaccines. However, HAV remains the primary cause of viral hepatitis in pediatric patients through the world [1] and may cause dramatic disease outbreaks. In addition, HAV isolated from adult patients with severe cases of fulminant hepatitis with fatal outcomes has been reported [2-6].
An infection with a hepatotropic virus activates the immune system so that it can defend the host. The immune response involves a broad range of cells and molecular components, which include the specific production of cytokines by immune and liver cells. HCV successfully evades the immune response in 60% - 80% of cases, resulting in chronic liver disease. Conversely, the immune response to HAV is robust and extremely effective in eliminating the virus and results in a naturally acute disease that in most cases resolves spontaneously [6,7].
Currently, it is accepted that cytokines are key components of efficient immune responses and play a significant role during viral infections [8,9]. Given both, HAV and HCV are non-cytophatic viruses, meaning that the viruses do not by themselves cause hepatocyte damage; instead, it is the immune response that is responsible for the observed symptoms and liver injury. It may be plausible that differences in cytokine profiles during HAV and HCV infections dictate the distinct outcomes observed during the infections.
2. IMMUNE RESPONSE DURING HEPATIC VIRAL INFECTION
Multiple mechanisms to control innate and adaptive immune responses are activated as a result of hepatitis A or hepatitis C viral infections to defend the host. Virusspecific cytotoxic T lymphocytes and helper CD4 T cells play key effector and regulatory roles in immune responses against hepatotropic viruses. Specific CD4 T cells are key components in the adaptive immune response because they help activate cytotoxic and humoral mechanisms. Additionally, CD4 T cells can secrete Type 1 (Th1) cytokines including interferon-gamma (IFN-γ). This favors neutrophil and macrophage recruitment and leads to an inflammatory response. Specific CD4 T cells may also release Type 1 (Th2) cytokines such as IL-10, which favor the development of humoral response and limit Th1 cytokine-mediated responses [8,9].
Currently, it is accepted that liver damage and viral replication are mainly caused by a fail in the specific immune response, which results in the recruitment of nonspecific inflammatory infiltrates to the liver [6,10]. The infected liver secretes chemokines such as CXCL9 and CXCL10 to promote the migration of nonspecific mononuclear cells, which result in sustained low-grade inflammation and liver injury. A positive correlation has been reported between the expression levels of CXCL9, CXCL10, their receptors and histological damage [11- 13], supporting the influence of chemokines in liver damage. In addition, cytokines such as IL-6, TNF-alpha (TNF-α), TGF-beta (TGF-β) and growth factors play a significant role in the regeneration process conducted during chronic inflammation. Moreover, hepatotropic viruses are able to block the expression of chemokine receptors associated with cellular responses to avoid the migration of cells toward the liver. Thus, inhibiting chemokines and chemokines-receptors interactions may limit nonspecific cell migration and reduce inflammation during hepatotropic viral infections [14,15].
3. HEPATITIS C VIRUS AND CYTOKINE PRODUCTION
Infection with HCV is the major cause of the chronic liver injury worldwide. Liver fibrosis is primarily due to a disproportion between the production and degradation of the extracellular matrix as a result of hepatocyte necrosis, mainly mediated by inflammation. Liver fibrosis is characterized by the release of cytokines, such as IL-6, IFN-γ, TGF-β1, platelet-derived growth factor (PDGF), TNF-α, insulin-like growth factor (IGF), hepatocyte growth factor (HGF), and IL-10 [16-18]. The relationship among specific immune response, viral clearance and inflammatory processes, is very complicated. Thus, the role of cell-mediated immune responses in hepatic fibrosis is not clear yet. However, it is known that changes in various cytokine and chemokine activities might favor viral persistence. By binding CD81, the HCVe2 protein has been shown to induce RANTESCCL5 expression by T cells, whereas overexpressed RANTES-CCL5 binds the CCR5 receptor, which results in receptor internalization. This reduction in the levels of chemokine receptors associated with cytotoxic responses in T cells may impair the chemotaxis of these cells to the liver and allow for viral persistence [19,20].
The primary cellular defense mechanism active during HCV infection is the synthesis of antiviral cytokines such as type I interferon (IFN α/β). Upon binding its receptor, this cytokine activates a number of intracellular mechanisms that can prevent viral replication and the spread of the virus to other liver cells. In vitro, HCV can block type I IFN induction, but this effect is not observed in vivo. Although HCV is a good inducer of IFN α/β expression, HCV seems to be unresponsive to the effects of IFN α/β The HCV NS5A protein has been shown to induce the expression of the pro-inflammatory chemokine IL-8, which is associated with IFN-α inhibition both in vitro and in vivo [21,22]. Thus, high IL-8 levels in the presence of IFN during HCV infection may explain the effective replication of HCV in the liver despite IFN α/β induction. In addition, in vitro studies have shown that HCV structural proteins can induce IL-10 production, which eventually inhibits IL-12 production by myeloid cells. This correlates with a reduction in IL-12 reported in HCV patients and a Th1-to-Th2 shift in the immune response observed in chronic hepatitis C patients [23]. Once the virus infects the liver, the innate immune response is mounted. Immune response is mediated by the specific production of inflammatory and antiviral cytokines by macrophages, natural killer cells (NK cells) and neutrophils which release perforin, granzyme B, IFN-α and TNF-α. Simultaneously, immune cells express FasL, inducing death of infected hepatocytes [10]. Adaptive immune response against HCV is mediated by the induction of naïve T cell differentiation into virus-specific CD4 and CD8 T cells. Dendritic cells (DCs) present antigenic viral components to CD8 T and CD4 T cells. Th2 CD4 T cells induce B cells to produce antiviral antibodies, Th1 CD4 T cells secrete IFN-γ and induce the proliferation of cytotoxic CD8 cells, which represent a crucial element during viral infections through the destruction of infected cells and specific secretion of proinflammatory cytokines [16-18] (Figure 1).
A multi-specific, strong, sustained CD4-T-cell-Th1 response is often observed during infections with hepatotropic viruses that are progressing toward resolution [18,24]. However, when an infection becomes chronic, a weak CD4-specific response with limited type I cytokine production is observed. Inadequate activation mediated by antigen-presenting cells in the setting of Th2-Tc2 cytokine expression is also a factor that results in chronicity. Another potential mechanism of blocked type I cytokine production involves regulatory T cells. These cells can release IL-10 and TGF-β and inhibit the proliferation of T cells, either directly or through other cytokines. The presence of CD8 regulatory T cells has
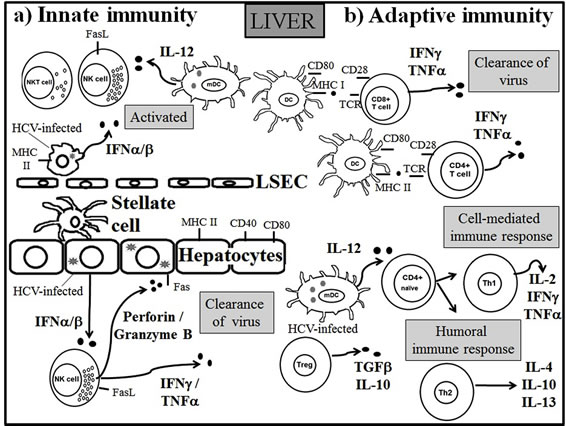
Figure 1. Cytokine-mediated immune response during HCV infection. As a result of HCV infection a) Innate immune response begins with the release of type I interferon which activates NK and NKT cells, which in turn release perforin, granzyme B, IFN-γ, TNF-α and finally by expressing FasL wich induces the death of infected hepatocytes; b) Adaptive immune response is mediated through the specific interactions between dendritic cells (DC) and CD4 T cells. These interactions include the release of IFN-γ, TNF-α, IL-2, IFN-γ, IL-4, IL-10, TGF-β and IL-13. HCV manage to escape immune response. This escape is mediated by the interference of various immune mechanisms including cytokine activity modulation [46-50].
been demonstrated in the liver of patients with chronic hepatitis C. These cells can inhibit HCV-specific CD8 T cells via IL-10 production. Furthermore, in vitro studies have shown that NK cells from HCV-infected patients, but not those from healthy control individuals, are impaired in their capacity to activate dendritic cells due to the overproduction of TGF-β and IL-10 [25]. In addition, IFN-γ production and cytotoxic activity in response to in vitro stimulation are reduced in HCV-specific T cells [26-30]. This early impairment might contribute to the lower probability of viral clearance. Immune cells with regulatory functions, such as peripheral CD4 + CD25 + FOXP3 + T regulatory cells and the presence of intrahepatic CD8 + CD25 + FOXP3 + T has been recently described in HCV-infected patients [11] (Figure 1).
4. THE EXQUISITE CONTROL OF CYTOKINE LEVELS DURING TYPE A VIRAL HEPATITIS INFECTION
Hepatitis A infection is typically acute in nature and is associated with extensive shedding of the virus in the feces during the 3- to 6-week incubation period. This shedding may explain the high prevalence of HAV infection in regions with low standards of sanitation, which in turns promote the transmission of the virus [31].
The adaptive immune response to HAV is robust and extremely effective in eliminating the virus. Neutralizing antibodies to the virus anti-HAV generally appear in the serum concurrent with the earliest evidence of hepatocellular injury and aminotransferase elevation. The mechanisms responsible for hepatocellular injury in individuals infected with hepatitis A are poorly characterized. However, HAV infection appears to result in a characteristic immune response different from that resulting from other hepatotropic viruses, including HCV. This difference is under the control of specific cytokine profiles and may explain why HAV infections do not cause significant liver damage and resolve during the acute stage. In general terms, during the innate immune response, HAV inhibits the expression of IFN α/β. This inhibition is mediated through the specific blocking of the IRF-3 signaling pathway by the virus and allows for an efficient infection [32]. The humoral immune response to HAV is characterized by the specific production of IgM antibodies against the VP1 viral protein and IgG antibodies against the VP1 and VP3 viral proteins [6,33]. In contrast to the responses to most viral infections, in which CD8 T cells play a predominant role, the responses to HAV infection seem to rely on CD4 T cells, which act during the first steps of the infection and produce numerous cytokines [34,35]. Our laboratory has recently shown that different cytokine profiles are associated with distinct clinical states induced by HAV. Minor HAVinduced hepatic injury is associated with the increased secretion of IL-8 and TGF-β, whereas intermediate liver injury is associated with increased serum levels of IL-1α, IL-6, IL-13, TNF-α and MCP-2 [36]. These changes in the cytokine profiles during HAV infection correlate with polymorphisms in the genes of cytokines, including IL-8, TNF-α and RANTES that are associated with the differential progression and severity of hepatic diseases.
5. INTEGRATING CYTOKINE PRODUCTION DURING HEPATITIS A VIRUS INFECTION IN FOUR STEPS
Cytokine production during HAV infection may be analyzed during four steps. The first step is the incubation period of the virus once it has been internalized through its specific receptor HAVCR1 in hepatocytes [37-39]. Interestingly, same receptor is expressed on T cell surface, where it can act as a co-stimulatory molecule called TIM-1 [40-42]. TIM-1 may be then implicated with the specific production of cytokines by T cells in response to HAV infection. When HAV begins to replicate in hepatocytes and reaches the maximum viral titer, the second step begins. During, this step the number of IgM antibodies and cytokines such as IL-22 reach their maximum levels. Th17 cells may be essential players during this step by secreting IL-22. Moreover, during this step, a reduction in the level of TGF-β secreted by T regulatory cells is observed. This reduction suggests that during this step, T effector cells are active, whereas T regulatory cells are inactive. The third step is characterized by the increased production of IgG antibodies. This increase of IgG production correlates with a reduction in the viral titer. The TGF-β levels gradually increase until the fourth step. During this last step, the virus is undetectable, and minimal levels of IL-22 are found. Thus, it is possible that during this step, T regulatory cells are active, resulting in the down-regulation of T effector functions. Finally, cytokine control during HAV infection results in viral clearance within 108 days [43] (Figure 2).
6. CYTOKINE-BASED THERAPIES
IFN-α is the only cytokine currently used for the treatment of chronic viral hepatitis. Pegylated IFN-α combined with ribavirin leads to sustained viral clearance in 50% of patients with chronic hepatitis C [22]. Ribavirin is a broad-spectrum antiviral agent used as part of combination therapies for hepatitis C. This drug has immunomodulating effects that induce type I cytokine production. Sustained viral load reductions with antiviral agents have also been observed to facilitate the recovery of specific T responses involving type I cytokine production in patients with hepatitis C. Moreover, genetic polymorphisms in cytokine genes, particularly IL-28 [44,45] gene have
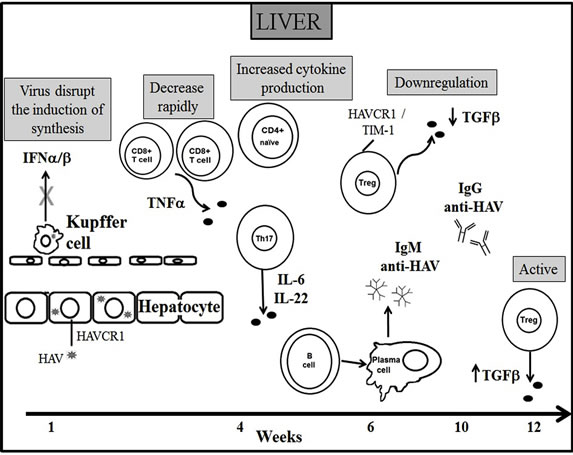
Figure 2. The cytokine production during the course of HAV infection. Once HAV is internalized through its specific receptor HAVCR1 in hepatocytes, a robust and effective immune response is induced. Neutralizing antibodies to the virus antiHAV appear in the serum concurrent with the earliest evidence of hepatocellular injury. HAV inhibits the expression of IFN α/β and CD4 T cells which act during the first steps of the infection produce numerous cytokines. The direct interaction between HAV and its receptor on T cells surface (HAVCR1/TIM-1) transitorily inhibits T regulatory function during viremia by reducing the production of IL-10 and TGF-β and increasing IL-6 and IL-22. Reactivation of T regulatory function occurs once HAV is cleared. This results in a limited liver damage and illness resolution [6,31,32,35,43].
been associated with viral clearance and response to the treatment. The exogenous administration of Th1 inducing cytokines such as IL-12 or antiinflammatory cytokines such as IL-10 has also been attempted as a method to reduce the severity of the intrahepatic inflammation [6]. However, such therapies remain experimental, and their effectiveness is unclear.
Because acute hepatitis A is a self-limiting disease and in most cases resolves spontaneously without residual damage or sequelae and because no specific therapy is available, treatment is based on monitoring. However, severe cases require the use of therapeutic strategies that in turn will help improve health. The use of cytokines represents a potential tool for the management of acute viral infections.
7. REMARKS
Hepatitis A and C viruses are the major causes of virusrelated liver damage. These viruses interact differentially with the host immune system. Because the immune responses to HAV and HCV determine the amount of liver damage that results from infection, studies should be undertaken to evaluate the use of HAVand HCV-related cytokines as susceptibility and/or resistance markers for the development of advanced liver damage. The identification of such markers could lead to early intervention with antiviral therapy for those individuals determined to be at the highest risk of developing advanced liver damage. HAV and its interactions with the immune response represent a field for future investigation. In particular, exploring the nature of these interactions may provide clues as to why this virus does not persist in the infected host, whereas HCV does.
8. ACKNOWLEDGEMENTS
This work was funded by grants from the Consejo Nacional de Ciencia y Tecnologia (CONACYT) #127229, #188240 and the Consejo Estatal de Ciencia y Tecnologia de Jalisco (COECYTJAL) #849 to NAF and the Programa de Mejora al Profesorado (PROMEP) to AP. FPCG was supported by PhD scholarship from the CONACYT.
![]()
![]()
REFERENCES
- Panduro, A., Escobedo Melendez, G., Fierro, N.A., Ruiz Madrigal, B., Zepeda-Carrillo, E.A. and Roman, S. (2011) Epidemiology of viral hepatitis in Mexico. Salud Publica de Mexico, 53, S37-S45.
- Costa-Mattioli, M., Cristina, J., Romero, H., Perez-Bercof, R., Casane, D., Colina, R., Garcia, L., Vega, I., Glikman, G., Romanowsky, V., et al. (2002) Molecular evolution of hepatitis A virus: A new classification based on the complete VP1 protein. Journal of Virology, 76, 9516- 9525. doi:10.1128/JVI.76.18.9516-9525.2002
- Escobedo-Melendez, G., Fierro, N.A., Roman, S., Maldonado-Gonzalez, M., Zepeda-Carrillo, E. and Panduro, A. (2012) Prevalence of hepatitis A, B and C serological markers in children from western Mexico. Annals of Hepatology, 11, 194-201.
- Hussain, Z., Husain, S.A., Almajhdi, F.N. and Kar, P. (2011) Immunological and molecular epidemiological characteristics of acute and fulminant viral hepatitis A. Virology Journal, 8, 254. doi:10.1186/1743-422X-8-254
- Jacobsen, K.H. and Wiersma, S.T. (2010) Hepatitis A virus seroprevalence by age and world region, 1990 and 2005. Vaccine, 28, 6653-6657. doi:10.1016/j.vaccine.2010.08.037
- Lanford, R.E., Feng, Z., Chavez, D., Guerra, B., Brasky, K.M., Zhou, Y., Yamane, D., Perelson, A.S., Walker, C.M. and Lemon, S.M. (2011) Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proceedings of the National Academy of Sciences of the United States of America, 108, 11223-11228. doi:10.1073/pnas.1101939108
- Rouse, B.T. and Sehrawat, S. (2010) Immunity and immunopathology to viruses: What decides the outcome? Nature reviews. Immunology, 10, 514-526. doi:10.1038/nri2802
- Charo, I.F. and Ransohoff, R.M. (2006) The many roles of chemokines and chemokine receptors in inflammation. The New England Journal of Medicine, 354, 610-621. doi:10.1056/NEJMra052723
- Rehermann, B. and Nascimbeni, M. (2005) Immunology of hepatitis B virus and hepatitis C virus infection. Nature reviews. Immunology, 5, 215-229. doi:10.1038/nri1573
- Buonaguro, L., Petrizzo, A., Tornesello, M.L. and Buonaguro, F.M. (2012) Innate immunity and hepatitis C virus infection: A microarray’s view. Infectious Agents and Cancer, 7, 7. doi:10.1186/1750-9378-7-7
- Billerbeck, E., Bottler, T. and Thimme, R. (2007) Regulatory T cells in viral hepatitis. World Journal of Gastroenterology, 13, 4858-4864.
- Lauer, G.M., Barnes, E., Lucas, M., Timm, J., Ouchi, K., Kim, A.Y., Day, C.L., Robbins, G.K., Casson, D.R., Reiser, M., et al. (2004) High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology, 127, 924-936. doi:10.1053/j.gastro.2004.06.015
- Soghoian, D.Z. and Streeck, H. (2010) Cytolytic CD4(+) T cells in viral immunity. Expert Review of Vaccines, 9, 1453-1463. doi:10.1586/erv.10.132
- Dustin, L.B. and Charles, E.D. (2012) Primary, postprimary and non-specific immunoglobulin M responses in HCV infection. Antiviral Therapy, 17, 1449-1452. doi:10.3851/IMP2222
- He, X.S. (2006) Regulation of adaptive immunity by HCV. In Tan, S.L., Ed., Hepatitis C viruses: Genomes and Molecular Biology, Norfolk, UK.
- Hiroishi, K., Eguchi, J., Ishii, S., Hiraide, A., Sakaki, M., Doi, H., Omori, R. and Imawari, M. (2010) Immune response of cytotoxic T lymphocytes and possibility of vaccine development for hepatitis C virus infection. Journal of Biomedicine & Biotechnology, 2010, 263810. doi:10.1155/2010/263810
- Giron-Gonzalez, J.A., Martinez-Sierra, C., Rodriguez-Ramos, C., Macias, M.A., Rendon, P., Diaz, F., FernandezGutierrez, C. and Martin-Herrera, L. (2004) Implication of inflammation-related cytokines in the natural history of liver cirrhosis. Liver International: Official Journal of the International Association for the Study of the Liver, 24, 437-445. doi:10.1111/j.1478-3231.2004.0951.x
- Racanelli, V. and Rehermann, B. (2006) The liver as an immunological organ. Hepatology, 43, S54-S62. doi:10.1002/hep.21060
- Larrubia, J.R., Benito-Martinez, S., Calvino, M., Sanzde-Villalobos, E. and Parra-Cid, T. (2008) Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World Journal of Gastroenterology, 14, 7149-7159. doi:10.3748/wjg.14.7149
- Nattermann, J., Nischalke, H.D., Feldmann, G., Ahlenstiel, G., Sauerbruch, T. and Spengler, U. (2004) Binding of HCV E2 to CD81 induces RANTES secretion and internalization of CC chemokine receptor 5. Journal of Viral Hepatitis, 11, 519-526. doi:10.1111/j.1365-2893.2004.00545.x
- Edlich, B., Ahlenstiel, G., Zabaleta Azpiroz, A., Stoltzfus, J., Noureddin, M., Serti, E., Feld, J.J., Liang, T.J., Rotman, Y. and Rehermann, B. (2012) Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology, 55, 39-48. doi:10.1002/hep.24628
- Heim, M.H. (2012) Interferons and hepatitis C virus. Swiss Medical Weekly, 142, w13586.
- Li, K., Foy, E., Ferreon, J.C., Nakamura, M., Ferreon, A.C., Ikeda, M., Ray, S.C., Gale Jr., M. and Lemon, S.M. (2005) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proceedings of the National Academy of Sciences of the United States of America, 102, 2992- 2997. doi:10.1073/pnas.0408824102
- Maheshwari, A., Ray, S. and Thuluvath, P.J. (2008) Acute hepatitis C. Lancet, 372, 321-332. doi:10.1016/S0140-6736(08)61116-2
- Jinushi, M., Takehara, T., Tatsumi, T., Kanto, T., Miyagi, T., Suzuki, T., Kanazawa, Y., Hiramatsu, N. and Hayashi, N. (2004) Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cellinduced modulation of dendritic cell functions in chronic hepatitis C virus infection. Journal of Immunology, 173, 6072-6081.
- Ferrari, C., Penna, A., Bertoletti, A., Valli, A., Antoni, A.D., Giuberti, T., Cavalli, A., Petit, M.A. and Fiaccadori, F. (1990) Cellular immune response to hepatitis B virusencoded antigens in acute and chronic hepatitis B virus infection. Journal of Immunology, 145, 3442-3449.
- Thimme, R., Oldach, D., Chang, K.M., Steiger, C., Ray, S.C. and Chisari, F.V. (2001) Determinants of viral clearance and persistence during acute hepatitis C virus infection. The Journal of Experimental Medicine, 194, 1395- 1406. doi:10.1084/jem.194.10.1395
- Urbani, S., Boni, C., Missale, G., Elia, G., Cavallo, C., Massari, M., Raimondo, G. and Ferrari, C. (2002) Virusspecific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. Journal of Virology, 76, 12423- 12434. doi:10.1128/JVI.76.24.12423-12434.2002
- Accapezzato, D., Francavilla, V., Paroli, M., Casciaro, M., Chircu, L.V., Cividini, A., Abrignani, S., Mondelli, M.U. and Barnaba, V. (2004) Hepatic expansion of a virusspecific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. The Journal of Clinical Investigation, 113, 963-972.
- Sugimoto, K., Ikeda, F., Stadanlick, J., Nunes, F.A., Alter, H.J. and Chang, K.M. (2003) Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology, 38, 1437-1448.
- Cuthbert, J.A. (2001) Hepatitis A: Old and new. Clinical Microbiology Reviews, 14, 38-58. doi:10.1128/CMR.14.1.38-58.2001
- Fensterl, V., Grotheer, D., Berk, I., Schlemminger, S., Vallbracht, A. and Dotzauer, A. (2005). Hepatitis A virus suppresses RIG-I-mediated IRF-3 activation to block induction of beta interferon. Journal of Virology, 79, 10968- 10977. doi:10.1128/JVI.79.17.10968-10977.2005
- Martin, A. and Lemon, S.M. (2006) Hepatitis A virus: From discovery to vaccines. Hepatology, 43, S164-S172. doi:10.1002/hep.21052
- Knosp, C.A. and Johnston, J.A. (2012) Regulation of CD4+ T-cell polarization by suppressor of cytokine signalling proteins. Journal of Immunology, 135, 101-111. doi:10.1111/j.1365-2567.2011.03520.x
- Zhou, Y., Callendret, B., Xu, D., Brasky, K.M., Feng, Z., Hensley, L.L., Guedj, J., Perelson, A.S., Lemon, S.M., Lanford, R.E., et al. (2012) Dominance of the CD4(+) T helper cell response during acute resolving hepatitis A virus infection. The Journal of Experimental Medicine, 209, 1481-1492. doi:10.1084/jem.20111906
- Fierro, N.A., Escobedo-Melendez, G., De Paz, L., Realpe, M., Roman, S. and Panduro, A. (2012) Cytokine expression profiles associated with distinct clinical courses in hepatitis A virus-infected children. The Pediatric Infectious Disease Journal, 31, 870-871. doi:10.1097/INF.0b013e318258e808
- Feigelstock, D., Thompson, P., Mattoo, P., Zhang, Y. and Kaplan, G.G. (1998) The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. Journal of Virology, 72, 6621-6628.
- Nakajima, T., Wooding, S., Satta, Y., Jinnai, N., Goto, S., Hayasaka, I., Saitou, N., Guan-Jun, J., Tokunaga, K., Jorde, L.B., et al. (2005) Evidence for natural selection in the HAVCR1 gene: High degree of amino-acid variability in the mucin domain of human HAVCR1 protein. Genes and Immunity, 6, 398-406. doi:10.1038/sj.gene.6364215
- Silberstein, E., Konduru, K. and Kaplan, G.G. (2009) The interaction of hepatitis A virus (HAV) with soluble forms of its cellular receptor 1 (HAVCR1) share the physiological requirements of infectivity in cell culture. Virology Journal, 6, 175. doi:10.1186/1743-422X-6-175
- Freeman, G.J., Casasnovas, J.M., Umetsu, D.T. and DeKruyff, R.H. (2010) TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunological Reviews, 235, 172-189.
- Lee, J., Phong, B., Egloff, A.M. and Kane, L.P. (2011) TIM polymorphisms—Genetics and function. Genes and Immunity, 12, 595-604. doi:10.1038/gene.2011.75
- Rodriguez-Manzanet, R., DeKruyff, R., Kuchroo, V.K. and Umetsu, D.T. (2009). The costimulatory role of TIM molecules. Immunological Reviews, 229, 259-270. doi:10.1111/j.1600-065X.2009.00772.x
- Manangeeswaran, M., Jacques, J., Tami, C., Konduru, K., Amharref, N., Perrella, O., Casasnovas, J.M., Umetsu, D.T., Dekruyff, R.H., Freeman, G.J., et al. (2012) Binding of hepatitis A virus to its cellular receptor 1 inhibits T-regulatory cell functions in humans. Gastroenterology, 142, 1516-1525. doi:10.1053/j.gastro.2012.02.039
- Thomas, D.L., Thio, C.L., Marin, M.P., Qi, Y., Ge, D., O’Huigin, C., Kidd, J., Kidd, K., Khakoo, S.I., Alexander, G., et al. (2009) Genetic variation in IL28B and spontaneous clearence of hepatitis C virus. Nature, 461, 798- 801.
- Hsu, C.S., Hsu, S.J., Chen, H.C., Liu, C.H., Jeng, J., Liu, C.J., Chen, P.J., Chen, D.S. and Kao, J.H. (2012) Association of IL28B genotypes with metabolic profiles and viral clearance rate in chronic hepatitis C patients. Hepatology International, 7, 1-9.
- Bowen, D.G. and Walker, C.M. (2005) Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature, 436, 946-952. doi:10.1038/nature04079
- Charles, E.D. and Dustin, L.B. (2011) Chemokine antagonism in chronic hepatitis C virus infection. The Journal of Clinical Investigation, 121, 25-27. doi:10.1172/JCI45610
- Golden-Mason, L., Castelblanco, N., O'Farrelly, C. and Rosen, H.R. (2007) Phenotypic and functional changes of cytotoxic CD56pos natural T cells determine outcome of acute hepatitis C virus infection. Journal of Virology, 81, 9292-9298. doi:10.1128/JVI.00834-07
- Rehermann, B. (2009) Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. The Journal of Clinical Investigation, 119, 1745-1754. doi:10.1172/JCI39133
- Thimme, R., Bukh, J., Spangenberg, H.C., Wieland, S., Pemberton, J., Steiger, C., Govindarajan, S., Purcell, R.H. and Chisari, F.V. (2002) Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 15661-15668. doi:10.1073/pnas.202608299
NOTES
*Corresponding author.