Advances in Bioscience and Biotechnology
Vol.3 No.8(2012), Article ID:25338,8 pages DOI:10.4236/abb.2012.38134
Vanadium induces liver toxicity through reductive activation by glutathione and mitochondrial dysfunction
![]()
1Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran
2Department of Pharmacology and Toxicology, Faculty of Pharmacy, Zanjan University of Medical Sciences, Zanjan, Iran
3Department of Food Science and Engineering, Faculty of Biosystem Engineering, College of Agriculture, University of Tehran, Tehran, Iran
4Department of Pharmacology and Toxicology, Faculty of Pharmacy, Zabol University of Medical Sciences, Zabol, Iran
Email: *j.pourahmadjaktaji@utoronto.ca
Received 10 September 2012; revised 17 October 2012; accepted 29 November 2012
Keywords: Vanadium (V5+); Oxidative Stress; Glutathione Activation; Mitochondrial/Lysosomal Cross-Talk; Hepatotoxicity
ABSTRACT
Pentavalent vanadium (V5+) (metavanadate salt) toxicity is a challenging problem to the health professionals and has been recognized as an industrial hazard that adversely affects human and animal health, but its cytotoxic mechanisms have not yet been completely understood. In this study, we investigated the cytotoxic mechanisms of V5+ in freshly isolated rat hepatocytes. V5+ cytotoxicity was associated with reactive oxygen species (ROS) formation, collapse of mitochondrial membrane potential, lysosomal membrane rupture and cytochrome c release into the hepatocyte cytosol. All of the above mentioned V5+- induced cytotoxicity markers were significantly (p < 0.05) prevented by ROS scavengers, antioxidants and mitochondrial permeability transition (MPT) pore sealing agents. Hepatocyte glutathione (GSH) was also rapidly oxidized and GSH-depleted hepatocytes were more resistant to lithium-induced oxidative stress markers. This suggests that V5+ is activated by GSH. Our findings also showed that the lysosomotropic agents prevented V5+ induced mitochondrial membrane potential collapse. On the other hand, mitochondrial MPT pore sealing agents inhibited lysosomal membrane damage caused by V5+. It can therefore be suggested that there is probably a toxic interaction (cross-talk) between mitochondrial and lysosomal oxidative stress generating systems, which potentiates ROS formation and further damages both sub-or- ganelles in V5+-induced induced hepatotoxicity. In conclusion, V5+-induced cytotoxicity can be attributed to oxidative stress started from glutathione mediated metal reductive activation and continued by mitochondrial/lysosomal toxic interaction.
1. INTRODUCTION
There has been a growing concern about increase in toxic metals contamination i.e. V5+ that affect the environment, living organisms and human health [1]. In recent years, the toxicity of V5+ has been reviewed and suggested that V5+-induced lung tumors in mice and rats and caused chromosomal aberrations and aneuploidy in bone marrow cells [2]. In human fibroblasts, a low concentration of V5+ impaired the repair of DNA damage caused by UV irradiation or by bleomycin [3]. It showed that V5+ solutions induced DNA damage in specific organs, highest in liver, heart and kidney [4]. In addition, previous studies showed that liver is one of the most important target tissues for V5+ toxicity [1,5]. In a cell-free system containing rat liver microsomes, V5+ was reduced by NADH to V4+ and generated hydroxyl radicals [6]. On the other hand, V5+ reacted with thiols to produce V4+ and thiyl radicals [7]. Several studies have associated V5+-toxicity with its capacity to induce the formation of reactive oxygen species (ROS), probably by interacting with mitochondrial redox centers [8-10]. It is generally known that the GSH-related thiols participate in many important biological reactions, including the protection of cell membranes against oxidative damage. On the other hand, GSH could reduce V5+ to V4+ in most mammalian tissues [11].
Thus, the ability of V5+-to generate free radicals, for example, hydroxyl radicals in the Fenton-like reaction, and decrease concentration of biologically important cellular compounds, causes the weakness of antioxidant system making cells more vulnerable to oxidative damage [8,12]. The toxicity of V5+ is associated with the increase of oxidative processes and/or the decrease of the cell antioxidant potential. It seems the production of ROS and RNS by V5+ like other metals could leads to oxidative stress in cells [13].
Due to key role that oxidative stress plays in V5+ toxicity; we therefore planned to study molecular and cellular mechanisms of V5+ toxicity on isolated SpragueDawley hepatocytes. Although, rat hepatocyte metabolism is higher than human, but obtained results with rat hepatocytes can be extrapolated to human hepatocytes [14].
All intracellular pathways involved in V5+-induced liver toxicity have not yet been completely understood. So in this study we screened all the possible cellular toxicity mechanisms including ROS formation, Mitochondrial Membrane Potential collapse, increased caspase-3 activity, cytochrome c release and also lysosomal membrane disruption in isolated rat hepatocytes. Our results supported the hypothesis that V5+ cellular target is mitochondrion and V5+ causes mitochondrial dysfunction and release of cytochrome c that later signals cell death.
2. MATERIALS AND METHODS
2.1. Materials
Meta Vanadate sodium (NH4VO3) and other chemicals were purchased from Sigma-Aldrich Co. (Taufkrichen, Germany). All other chemicals were of the highest commercial grade available.
2.2. Animals
Male Sprague-Dawley rats (250 - 300 g) fed a standard chow diet and given water ad libitum, were used in all experiments. They were purchased from Institute Pasteur (Tehran, Iran) and were kept in individual cages in a controlled room temperature (20˚C - 25˚C), humidity (50% - 60%) and exposed to 12 h of daylight. The animals were fed with a normal standard chow diet and tap water ad libitum. All experiments were conducted according to the ethical standards and protocols approved by the Committee of Animal Experimentation of Shahid Beheshti University of Medical Sciences, Tehran, Iran. All efforts were made to minimize the number of animals used and their suffering.
2.3. Isolation and Incubation of Hepatocytes
Hepatocytes were obtained by collagenase perfusion of the liver and viability was assessed by plasma membrane disruption determined by trypan blue (0.2 w/v) exclusion test [15]. Cells were suspended at a density of 106 cells/mL in round-bottomed flasks rotating in a water bath maintained at 37˚C in Krebs-Henseleit buffer (pH = 7.4), supplemented with 12.5 mM HEPES under an atmosphere of 10% O2, 85% N2, and 5% CO2. Hepatocytes were pre-incubated for 30 min prior to addition of chemicals.
To avoid either non-toxic or very toxic conditions in this study, we used EC50 concentrations for V5+ (100 µM). The EC50 of a chemical in hepatocyte cytotoxicity assessment technique (with the total 3 hours incubation period) is defined as the concentration, which decreases the hepatocye viability down to 50% following the 2 hours of incubation [16]. In order to determine this value for V5+, dose-response curves were plotted and then EC50 was determined based on a regression plot of three different concentrations. Glutathione (GSH) depleted heaptocytes were prepared by pre-incubation of hepatocytes with 200 µM n-bromoheptane for 30 min as described by Khan and O’Brien [17].
2.4. Main Research Methods
To determine the cellular and molecular mechanisms involved V5+ liver toxicity, we used accelerated cytotoxicity mechanism screening (ACMS) methods. This technique determines the cytotoxic effectiveness of xenobiotics incubated for 3 h towards rat hepatocytes. A functionomic approach is used to understand the cytotoxic mechanism, e.g., the effects of different types of antagonist or substrates on the loss of cell viability, ROS formation, mitochondrial and lysosomal damages and cell death signaling induced by the xenobiotics (V5+) are investigated. The procedures used are as follows: 1) The concentration of xenobiotic (V5+) required inducing a 50% loss of membrane integrity (EC50) of freshly isolated rat hepatocytes in 2 h is determined by trypan blue exclusion and then this concentration will be applied to rat hepatocytes and all the above mentioned toxicity mechanisms will be assayed; 2) A major assumption with accelerated cytotoxicity mechanism screening (ACMS) is that high dose/short time (in vitro) simulates low dose/long time (in vivo) with relevance to human environmental exposure [18].
2.5. Cell Viability
The viability of isolated hepatocytes was assessed from the intactness of the plasma membrane as determined by the trypan blue (0.2% w/v) exclusion test [18]. Aliquots of the hepatocyte incubate were taken at different time points during the 3 hours incubation period.
2.6. Determination of Reactive Oxygen Species
To determine the rate of hepatocyte reactive oxygen species (ROS) generation induced by V5+, dichlorofluorescin diacetate (DCFH-DA) was added to the hepatocytes. It penetrates hepatocyte cells and is hydrolyzed to nonfluorescent dichlorofluorescin (DCFH). The latter then reacts with ROS to form the highly fluorescent dichlorofluorescein (DCF), which effluxes the cell. The fluorescence intensity of DCF was measured using a Shimadzu RF5000U fluorescence spectrophotometer. Excitation and emission wavelengths were 500 and 520 nm, respectively. The results were expressed as fluorescent intensity per 106 cells [19].
2.7. Mitochondrial Membrane Potential Assay
Mitochondrial uptake of the cationic fluorescent dye, rhodamine123, has been used for estimation of mitochondrial membrane potential [20]. The amount of rhodamine123 remaining in the incubation medium was measured fluorimeterically using a Shimadzu RF5000U fluorescence spectrophotometer set at 490 nm excitation and 520 nm emission wavelengths. The capacity of mitochondria to up take the rhodamine123 was calculated as the difference (between control and treated cells) in rhodamine123 fluorescence. Our data were shown as the percentage of mitochondrial membrane potential collapse (%ΔΨm) in all treated (test) hepatocyte groups [20].
2.8. Lysosomal Membrane Integrity Assay
Hepatocyte lysosomal membrane stability was determined from the redistribution of the fluorescent dye, acridine orange [20]. Aliquots of the cell suspension (0.5 mL) that were previously stained with acridine orange (5 μM) were separated from the incubation medium by 1 min centrifugation at 1000 rpm. The cell pellet was then resuspended in 2 mL of fresh incubation medium. This washing process was carried out for two times to remove the fluorescent dye from the media. Acridine orange redistribution in the cell suspension was then measured fluorimetrically using a Shimadzu RF5000U fluorescence spectrophotometer set at 495 nm excitation and 530 nm emission wavelengths.
2.9. Determination of Cytochrome c Release
Release of cytochrome c from mitochondria in to cytosol is a key initiating step in both apoptotic and necrotic cell death processes in intact cells [21]. Detection of cytochrome c release was determined by cytochrome c ELISA Kit (Quantikine M., R&D Systems, Abingdon, UK) according to the manufacturer’s instructions. This assay employs the quantitative sandwich enzyme immunoassay technique and is designed to accurately quantify natural rat or mouse cytochrome c in cell lysates and subcellular fractions.
2.10. Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s HSD as the post hoc test. Levene’s test was used to check the homogeneity of variances. Results were presented as mean ± SD of triplicate samples. The triplicate samples were representative of 3 separate rats (n = 3). The minimal level of significance chosen was p < 0.05.
3. RESULTS
At least 80% - 90% of the control cells were viable following 3 hours of incubation. The EC502h found for V5+ was 100 µM. EC50 was determined based on a regression plot of four different concentrations (25, 50, 100 and 200 μM) in isolated hepatocyte .The EC502h of V5+ in heaptocytes (with total 3-h incubation period) is defined as the concentration, which decreases the viability down to 50% following the 2 h of incubation. As shown in Table 1, V5+ (100 µM) significantly increased cytotoxicity compared to control hepatocytes (p < 0.05). In addition, when isolated hepatocytes were incubated with V5+ (100 µM), ROS formation was significantly (p < 0.05) increased in time-dependent manner (Table 2). On the other hand, these factors (cytotoxicity and ROS Formation) were prevented by antioxidants and ROS scavengers (BHT, DMSO and mannitol), MPT pore sealing agent (carnitine) and lysosomotropic agent (chloroquine) (Tables 1 and 2). However, depleting hepatocyte glutathione (GSH) beforehand decreased cytotoxicity and ROS formation (Tables 1 and 2). All of the reagents used in cytotoxicity and ROS study including antioxidants and ROS scavengers, MPT pore sealing agent and 1-bromoheptan (used for GSH depleting) did not significantly (p < 0.05) increase hepatocyte membrane lysis and ROS formation at concentrations used while incubated alone in isolated hepatocytes (data not shown).
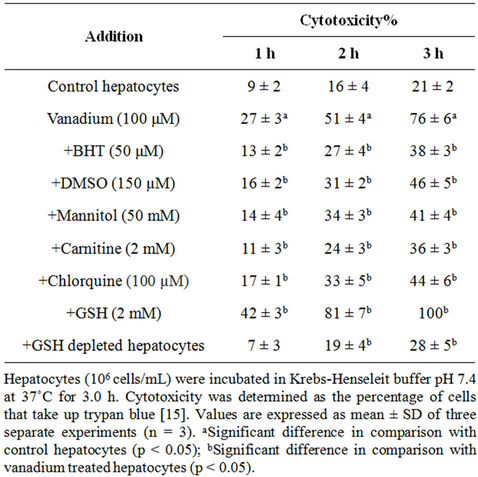
Table 1. Effect of antioxidant, ROS scavengers, MPT pore sealing agents, lysosomotropic agents and GSH depletion on V5+-induced hepatotoxicity.
As shown in Table 3, V5+ (100 µM) induced mitochondrial membrane potential collapse during 1 hours of incubation which was prevented by antioxidant and ROS scavengers (BHT, DMSO and mannitol) suggesting that V5+-induced mitochondrial membrane potential decrease was subsequent of ROS formation. All of these mentioned inhibitors did not show any significant effect (p < 0.05) on hepatocytes mitochondrial membrane potential at concentrations used while incubated alone in hepatocytes (data not shown).
When hepatocyte lysosomes were loaded with acridine orange (a lysosomotropic agent), a significant release of acridine orange into the cytosolic fraction ensued within 60 min of incubation with V5+ indicating a severe damage to lysosomal membrane (Table 4).
V5+-induced acridine orange release was again prevented by antioxidant (BTH) and radical scavengers (mannitol and DMSO) (Table 4). Again depleting heaptocyte GSH beforehand protected the acridine orange loaded hepatocytes against V5+-induced acridine orange release.
All of these inhibitors did not show any significant effect (p < 0.05) on acridine orange redistribution from lysosomes into cytosol at concentrations used when incubated alone in isolated hepatocytes (Data not shown). The interesting finding was that vanadium (V) induced hepatocyte mitochondrial membrane potential collapse prevented by lysosomotropic agent (chloroquine) and also lysosomal membrane leakiness induced by V5+ were also prevented by mitochondrial MPT pore sealing agents (CsA) (Tables 3 and 4).

Table 2. Effect of antioxidant, ROS scavengers, MPT pore sealing agents, lysosomotropic agents and GSH depletion on vanadium induced ROS formation.
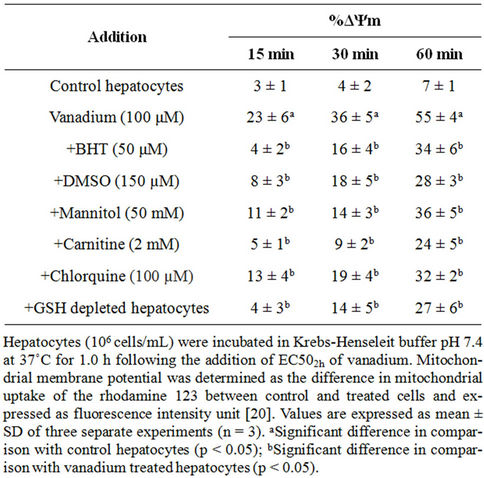
Table 3. Preventing of mitochondrial membrane potential decline induced by vanadium by antioxidant, ROS scavengers, MPT pore sealing agents, lysosomotropic agents and GSH depleting agent.

Table 4. Preventing of lysosomal membrane damage induced by vanadium by antioxidant, ROS scavengers, MPT pore sealing agents, lysosomotropic agents and GSH depleting agent.
Activity of caspase-3 enzyme (final mediator of apoptosis) was significantly increased compared to that of control hepatocytes following 2 h of exposure with V5+-increased caspase-3 activity was prevented by antioxidant (BHT), hydroxyl radical scavengers (DMSO and mannitol), MPT pore sealing agents (CsA), lysosomotropic agent (chloroquine). Depleting hepatocyte GSH beforehand prevented hepatocytes against V5+-induced caspase-3 activity. All of these mentioned agents did not change hepatocytes caspase-3 activity at concentrations used (Figure 1). Induction of mitochondrial permeability transition (MPT) is supposed to be resulted from opening of the related pores in mitochondrial membrane leading to the release of cytochrome c [22].
In order to confirm the occurrence of this intra-mitochondrial protein release in to the cytosol, cytochrome c release in V5+-treated rat hepatocyte was determined spectrophotometrically at wavelength of 450 nm with an ELIZA reader.
Our results showed that V5+ (100 µM) induced release of cytochrome c from mitochondria in to the cytosol following 1 hours of incubation in isolated rat hepatocyte before cytotoxicity ensued. V5+-induced cytochrome c release was prevented by antioxidant (BHT), hydroxyl radical scavengers (DMSO and mannitol), and MPT pore sealing agents (carnitine), lysosomotropic agent (chloroquine) suggesting that V5+-induced cytochrome c expulsion was subsequent of MPT pore opening due to ROS formation. All of these mentioned inhibitors did not show any significant effect (p > 0.05) on the hepatocyte mitochondrial cytochrome c release at concentrations used (Figure 2).
4. DISCUSION
This study contribute to a better understanding of responsible mechanisms involved in V5+-induced oxidative stress and cell death in the liver. Our findings revealed
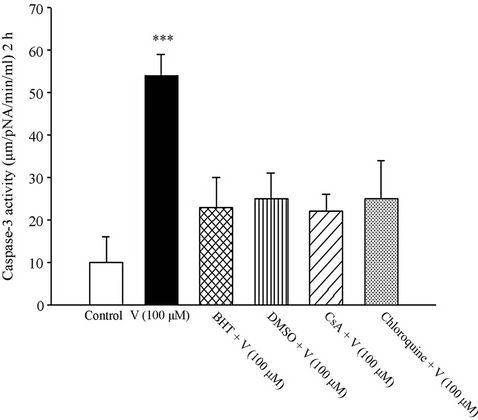
Figure 1. Prevention of vanadium-induced caspase-3 activation by antioxidant (BHT) and MPT pore sealing agent (CsA). Caspase-3 activity was measured by sigma colorimetric assay kit. Each histogram represents the mean ± SD of three different determinations (*p < 0.05; **p < 0.01; ***p < 0.001).
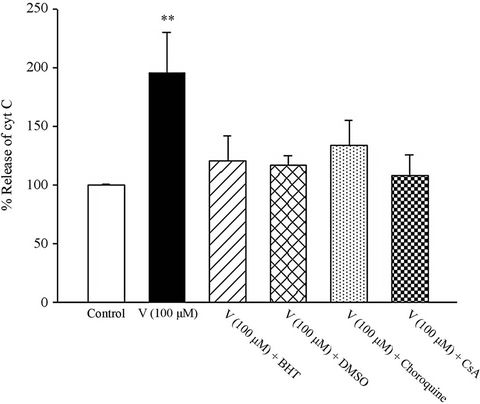
Figure 2. Effect of vanadium-induced cytochrome c release in isolated rat hepatocyte. Cytochrome c release was measured with an ELISA kit. Each histogram represents the mean ± SEM of three different determinations. (*p < 0.05; **p < 0.01; ***p < 0.001).
that at first vanadium needs a glutathione mediated reductive activation for ROS formation. Then, the oxidative stress induced in hepatocytes targets mitochondrial and lysosomal pathways. Vanadium (V) is a trace element known to be essential for a number of species that is widely distributed on earth. But, its role as a micronutrient in humans has yet to be established [23,24]. Therefore, V5+ toxicity is a challenging problem to the health. V5+ enters the organism by inhalation, the gastrointestinal tract and the skin and it is specifically stored in certain organs mainly in the liver, kidney and bones and to a lesser in lungs and tests [23,25]. Variety of toxic effects reported for V5+ compounds such as gastrointestinal Disturbance, anorexia, weight loss, abdominal pain and most importantly histopathological alterations of liver and kidneys [26,27].
It was suggested that production hydrogen peroxide and other ROS by V5+ compounds in the cytosol or in the mitochondria, results in mitochondrial damage and the release of cytochrome c which activates the caspases. Activated caspases such as caspase-3 and caspease-8, provoke further mitochondrial damage and activate cellular substrates such as poly(ADP-ribose)-polymerase (PARP), leading to apoptosis [28]. Our results showed that V5+ induced sharp rise in hepatocyte membrane lysis (cytotoxicity), ROS formation and glutathione depletion. Our data also supported the hypothesis that V5+-induced cytotoxicity is directly related to ROS formation which could be a key player in the mechanism underlying V5+- induced liver injury. Overproduction of reactive oxygen species (ROS) or significant depletion of antioxidant defense molecules (e.g. GSH) causes oxidative stress in different cells [29].
Some recent studies have highlighted the ability of GSH to promote oxidative processes by participating in metal ion mediated reactions eventually leading to the formation of ROS and free radicals. A crucial role in these phenomena is played by membrane bound γ-glutamyltransferase (GGT) activity [30]. It was already reported that some metals such as Li+ could significantly increase GPx and GGT activities, and decrease GSSG reductase activity in protein-deficient rats [31]. On the other hand V5+ also promoted ROS formation and cell death, when incubated with GSH. Also, depleting heaptocyte GSH beforehand prevented the hepatocytes against V5+-induced cytotoxicity, ROS generation, mitochondrial membrane damage which reminds toxic reactions of GSH with some other metals including copper, chromium, uranium, thallium and lithium [18,32-36]. Thiol compounds especially when dissociated to their thiolate anion (R-S-) forms can in fact affect the reduction of metal cations, for example, iron and copper. Electrons can be then transferred in turn from metal ions to molecular oxygen, thus generating superoxide anions, which will easily dismutate to the strong prooxidant hydrogen peroxide [36].
GSH, a tripeptide consisting of glycine (Gly), cysteine (Cys), and glutamic acid (Glu), itself is rather less efficient than other thiols, an aspect which can be explained by the vicinity of the SH group of cysteine to the a-carboxyl group of glutamic acid [37]. The removal of glutamic acid in fact produces a marked increase in the metal-reducing ability, resulting in a promotion of oxidative processes [38,39]. It is therefore suggested that V5+ is reduced by a one-electron transfer reaction, which in turn reduce O2 to , the superoxide anion radical. Thus, these reactions result in the generation of further reactive oxygen species (ROS) and further toxicity.
, the superoxide anion radical. Thus, these reactions result in the generation of further reactive oxygen species (ROS) and further toxicity.
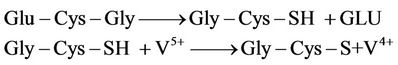
In the following another suggested mechanism could be explained by interactions of generated thiolate anion in the first reaction with the thiyl radical in the second reaction:

We can therefore conclude that toxic GSH and V5+ reactions leads to more ROS production and oxidative stress culminated in higher V5+ liver toxicity. We demonstrated that addition of antioxidant (BHT), hydroxyl radical scavengers (DMSO and mannitol), MPT pore sealing agent (carnitine) and lysosomotropic agent (chloroquine), which significantly reduced V5+-induced ROS formation and cell death. Our finding would thus argue for a pivotal role of lysosomal damage and mitochondrial dysfunction in V5+-induced oxidative stress and liver toxicity. In our study V5+ caused lysosomal membrane damage that was again prevented by antioxidants and radical scavengers suggesting that lysosoms are one of the ROS production sites in V5+ toxicity. Our other interesting results were that the lysosomotropic agents prevented V5+-induced mitochondrial membrane potential collapse and mitochondrial MPT pore sealing agents inhibited lysosomal membrane damage caused by V5+. It can therefore be suggested that there is probably a toxic interaction between mitochondrial and lysosomal oxidative stress generating systems, which potentiates each organelle damage and ROS formation in V5+ model of hepatotoxicity.
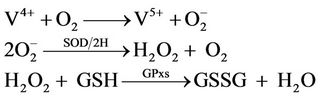
Reductive activation of V5+ through glutathione interaction leads to considerable free radical formation. Increased ROS formation could directly damage hepatocyte mitochondria via MPT pore opening, cytochrome c expulsion and disruption of electron transfer chain. Hydrogen peroxide (H2O2) originated either from V5+ metabolic activation or damaged mitochondria diffuses into lysosomes (due to lipophilic nature) and a Fenton type reaction (Haber-weiss) catalyzed by intra-lysosomal redox-active Fe2+/Fe3+ occurs. This leads to highly reactive hydroxyl radical (HO.) generation. Hydroxyl radicals could destabilize the lysosomal membrane integrity and release of digestive proteases (i.e. cathepsins). These released proteases and hydroxyl radicals could either open the mitochondrial MPT pore via oxidation of surrounding thiol groups or through activation of Bid or Bax pro-apoptotic proteins and other lytic enzymes including phospholipase A2 (PLA2). Disruption of electron transfer chain potentiates mitochondrial H2O2 generation and continues the cycle of mitochondrial/lysosomal oxidative stress toxic cross-talk that potentiates V5+ hepatocyte toxicity.
5. CONCLUSIONS
Our results confirmed that V5+-induced liver toxicity is resulted from glutathione mediated reductive activation which increased ROS formation and subsequent changes in mitochondrial membrane conformation and cytochrome c release in to the cytosol which causes mitochondrial collapse of Ψm. Cytochrome c release is the regulating step for cell death signaling which could lead to both apoptosis and necrosis. At the core of the apoptotic pathways, caspases exist. Caspase-3 is the most notable among the executioner caspases. In view of that, we measured the caspase-3 activity that was increased in hepatocytes when incubated with V5+.
To our knowledge this is the first report which declares that V5+-induced liver toxicity is the result of its reductive activation by hepatocytes glutathione which is the obvious cause of metal induced ROS formation and lipid peroxidation. And metal-induced liver cytotoxicity is further potentiated by mitochondrial/lysosomal toxic cross-talk.
6. ACKNOWLEDGEMENTS
The investigation described in this paper was extracted from Ph.D. thesis of Dr. Mir-Jamal Hosseini (graduated from Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences in 2012). The thesis conducted under supervision of Prof. Jalal Pourahmad. This study was supported by a grant from Shahid Beheshti University of Medical Sciences (90/01/94/7902, 2011).
REFERENCES
- Soares, S.S., Martins, H., Gutiérrez-Merino, C. and Aureliano, M. (2008) Vanadium and cadmium in vivo effects in teleost cardiac muscle: Metal accumulation and oxidative stress markers. Comparative Biochemistry and Physiology. Part C, Pharmacology, Toxicology & Endocrinology, 147, 168-178. doi:10.1016/j.cbpc.2007.09.003
- International Agency for Research on Cancer (IARC) (2006) Cobalt in hard metals and cobalt sulfate, gallium arsenide, indium phosphide and vanadium pentoxide. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 86, 119-237.
- Ivancsits, S., Pilger, A., Diem, E., Scha Ver, A. and Rüdiger, H.W. (2002) Vanadate induces DNA strand breaks in cultured human fibroblasts at doses relevant to occupational exposure. Mutation Research, 519, 25-35. doi:10.1016/S1383-5718(02)00138-9
- Altamirano-Lozano, M., Valverde, M., Alvarez-Barrera, L., Molina, B. and Rojas, E. (1999) Genotoxic studies of vanadium pentoxide (V2O5) in male mice. II. Effects in several mouse tissues. Teratogenesis, Carcinogenesis, and Mutagenesis, 19, 243-255. doi:10.1002/(SICI)1520-6866(1999)19:4<243::AID-TCM1>3.0.CO;2-J
- Scibior, A., Zaporowska, H., Ostrowski, J. and Banach, A. (2006) Combined effect of vanadium(V) and chromium (III) on lipid peroxidation in liver and kidney of rats. Chemico-Biological Interactions, 159, 213-222. doi:10.1016/j.cbi.2005.11.008
- Shi, X. and Dalal, N.S. (1992) Hydroxyl radical generation in the NADH/microsomal reduction of vanadate. Free Radical Research Communications, 17, 369-376. doi:10.3109/10715769209083141
- Shi, X., Jiang, H., Mao, Y., Ye, J. and Sayotti, U. (1996) Vanadium(IV)-mediated free radical generation and related 2-deoxyguanosine hydroxylation and DNA damage. Toxicology, 106, 27-38. doi:10.1016/0300-483X(95)03151-5
- Valko, M., Morris, H. and Cronin, M.T. (2005) Metals, toxicity and oxidative stress. Current Medicinal Chemistry, 12, 1161-1208. doi:10.2174/0929867053764635
- Yang, X.G., Yang, X.D., Yuan, L., Wang, K. and Crans, D.C. (2004) The permeability and cytotoxicity of insulin-mimetic vanadium compounds. Pharmaceutical Research, 21, 1026-1033. doi:10.1023/B:PHAM.0000029293.89113.d5
- Soares, S.S., Gutie´rrez-Merino, C. and Aureliano, M. (2007) Decavanadate induces mitochondrial membrane depolarization and inhibits oxygen consumption. Journal of Inorganic Biochemistry, 101, 789-796. doi:10.1016/j.jinorgbio.2007.01.012
- Macara, I.G., Kustin, K. and Cantley Jr. (1980) Glutathione reduces cytoplasmic vanadate. Mechanism and physiological implications. Biochimica et Biophysica Acta, 629, 95-106. doi:10.1016/0304-4165(80)90268-8
- Leonard, S.S., Harris, G.K. and Shi, X. (2004) Metalinduced oxidative stress and signal transduction. Free Radical Biology & Medicine, 37, 1921-1942. doi:10.1016/j.freeradbiomed.2004.09.010
- Shi, H., Hudson, L.G. and Liu, K.J. (2004) Oxidative stress and apoptosis in metal ion-induced carcinogenesis, Free Radical Biology & Medicine, 37, 582-593. doi:10.1016/j.freeradbiomed.2004.03.012
- Pourahmad, J., Hosseini, M.J., Eskandari, M.R. and Rahmani, F. (2012) Involvement of four different intracellular sites in chloroacetaldehyde-induced oxidative stress cytotoxicity. Iranian Journal of Pharmaceutical Research, 11, 265-276.
- Pourahmad, J., Hosseini, M.J., Bakan, S. and GhaziKhansari, M. (2011) Hepatoprotective activity of angiotensin-converting enzyme (ACE) inhibitors, captopril and enalapril, against paraquat toxicity. Pesticide Biochemistry and Physiology, 99, 105-110. doi:10.1016/j.pestbp.2010.11.006
- Galati, G., Teng, S., Moridani, M.Y., Chan, T.S. and O’Brien, P.J. (2000) Cancer chemoprevention and apoptosis mechanisms induced by dietary polyphenolics. Drug Metabolism and Drug Interactions, 17, 311-349. doi:10.1515/DMDI.2000.17.1-4.311
- Khan, S. and O’Brien, P.J. (1991) 1-Bromoalkanes as new potent nontoxic glutathione depletors in isolated rat hepatocytes. Biochemical and Biophysical Research Communications, 179, 436-441. doi:10.1016/0006-291X(91)91389-T
- Pourahmad, J., Eskandari, M.R., Alavian, G. and Shaki, F. (2010) Lysosomal membrane leakiness and metabolic biomethylation play key roles in methyl tertiary butyl ether-induced toxicity and detoxification. Toxicological Environental Chemistry, 94, 281-293. doi:10.1080/02772248.2011.643566
- Daraei, B., Pourahmad, J., Hamidi-Pour, N., Hosseini, M.J., Shaki, F. and Suleiman, M. (2012) Uranyl acetate induces oxidative stress and mitochondrial membrane potential collapse in the human dermal fibroblast primary cells. Iranian Journal of Pharmaceutical Research, 11, 495-501.
- Andersson, B.S., Aw, T.Y. and Jones, D.P. (1987) Mitochondrial transmembrane potential and pH gradient during anoxia. The American Journal of Physiology, 252, 349-355.
- Kamada, S., Kusano, H., Fujita, H., Ohtsu, M., Koya, R.C., Kuzumaki, N. and Tsujimoto, Y. (1998) A cloning method for caspase substrates that uses the yeast twohybrid system: Cloning of the antiapoptotic gene gelsolin. Proceedings of the National Academy of Sciences of the United States of America, 95, 8532-8537. doi:10.1073/pnas.95.15.8532
- Gogvadze, V., Orrenius, S. and Zhivotovsky, B. (2006) Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochimica et Biophysica Acta, 1757, 639-647. doi:10.1016/j.bbabio.2006.03.016
- Evangelou, A.M. (2002) Vanadium in cancer treatment. Critical Reviews in Oncology/Hematology, 42, 249-265. doi:10.1016/S1040-8428(01)00221-9
- Scibior, A., Zaporowska, H., Ostrowski, J. and Banach, A. (2006) Combined effect of vanadium(V) and chromium (III) on lipid peroxidation in liver and kidney of rats. Chemico-Biological Interactions, 159, 213-222. doi:10.1016/j.cbi.2005.11.008
- Cortizo, A.M., Bruzzone, L., Molinuevo, S. and Etcheverry, S.B. (2000) A possible role of oxidative stress in the vanadium-induced cytotoxicity in the MC3T3E1 osteoblast and UMR106 osteosarcoma cell lines. Toxicology, 8, 89- 99. doi:10.1016/S0300-483X(00)00181-5
- Goldfine, A.B., Simonson, D.C., Folli, F., Patti, M.E. and Kahn, C.R. (1995) In vivo and in vitro studies of vanadate in human and rodent diabetes mellitus. Molecular and Cellular Biochemistry, 3, 217-231. doi:10.1007/BF01075941
- Merritt, K. and Brown, S.A. (1995) Distribution of titanium and vanadium following repeated injection of highdose salts. Journal of Biomedical Materials Research, 29, 1175-1178. doi:10.1002/jbm.820291003
- Green, D.R. and Reed, J.C. (1998) Mitochondria and apoptosis. Science, 281, 1309-1312. doi:10.1126/science.281.5381.1309
- Valko, M., Leibfritz, D., Moncol, J., Cronin, M.T., Mazur, M. and Telser, J. (2007) Free radicals and antioxidants in normal physiological functions and human disease. The International Journal of Biochemistry & Cell Biology, 39, 44-84. doi:10.1016/j.biocel.2006.07.001
- Pompella, A., Visvikis, A., Paolicchi, A., De Tata, V. and Casini, A.F. (2003) The changing faces of glutathione, a cellular protagonist. Biochemical Pharmacology, 66, 1499- 1503. doi:10.1016/S0006-2952(03)00504-5
- Pourahmad, J. and O’Brien, P.J. (2000) A comparison of hepatocyte cytotoxic mechanisms for Cu2+ and Cd2+ Toxicology, 143, 263-273.
- Chadha, V.D., Bhalla, P. and Dhawan, D.K. (2008) Zinc modulates lithium-induced hepatotoxicity in rats. Liver International, 28, 558-565. doi:10.1111/j.1478-3231.2008.01674.x
- Pourahmad, J. and O’Brien, P.J. (2001) Biological reactive intermediates that mediate chromium (VI) toxicity. Advances in Experimental Medicine and Biology, 500, 203-207. doi:10.1007/978-1-4615-0667-6_27
- Pourahmad, J., Ghashang, M., Ettehadi, H. and Ghalandari, A. (2006) A search for cellular and molecular mechanisms involved in depleted uranium toxicity. Environmental Toxicology, 21, 349-354. doi:10.1002/tox.20196
- Pourahmad, J., Eskandari, M.R. and Daraei, B. (2010) A comparison of hepatocyte cytotoxic mechanisms for thallium (I) and thallium (III). Environtal Toxicology, 25, 456- 467. doi:10.1002/tox.20590
- Riadh, N., Allagui, M.S., Bourogaa, E., Vincent, C., Croute, F. and Elfeki, A. (2011) Neuroprotective and neurotrophic effects of long term lithium treatment in mouse brain. Biometals, 24, 747-757. doi:10.1007/s10534-011-9433-6
- Kappus, H. and Sies, H. (1981) Toxic drug effects associated with oxygen metabolism: Redox cycling and lipid peroxidation. Cellular and Molecular Life Sciences, 37, 1233-1241. doi:10.1007/BF01948335
- Paolicchi, A., Minotti, G., Tonarelli, P., Tongiani, R., De Cesare, D., Mezzetti, A., Dominici, S., Comporti, M. and Pompella, A. (1999) γ-Glutamyl transpeptidase-dependent iron reduction and low density lipoprotein oxidation—A potential mechanism in atherosclerosis. Journal of Investigative Medicine, 47, 151-160.
- Tien, M., Bucher, J.R. and Aust, S.D. (1982) Thiol-dependent lipid peroxidation. Biochemical and Biophysical Research Communications, 107, 279-285. doi:10.1016/0006-291X(82)91701-6
NOTES
*Corresponding author.