Journal of Biophysical Chemistry
Vol.07 No.01(2016), Article ID:63521,12 pages
10.4236/jbpc.2016.71003
Modified RNA with a Phosphate-Methylated Backbone. A Serious Omission in Our (Retracted) Study at HIV-1 RNA Loops and Integrated DNA. Specific Properties of the (Modified) RNA and DNA Dimers
Henk M. Buck
Kasteel Twikkelerf 94, Tilburg, The Netherlands

Copyright © 2016 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 1 December 2015; accepted 15 February 2016; published 18 February 2016
ABSTRACT
After the recent publication in the Journal of Biophysical Chemistry entitled “Retracted HIV Study Provides New Information about the Status of the in Vitro Inhibition of DNA Replication by Back- bone Methylation”, it is of importance to review the results of Buck’s group on the synthesis and conformation analyses of phosphate-methylated RNAs in order to afford information on the absence of a further investigation with regard to this de facto acceptable approach. In fact these compounds belong to the very first group of RNAs with a modified neutral backbone by phospha- temethylation. In contrast to the corresponding phosphate-methylated DNAs with a frozen B- conformation, the phosphate-methylated RNAs show an A-conformation. The latter is a prerequi- site for duplex formation with (complementary) (natural) RNA. A number of experiments support this fundamental statement. After the HIV study was retracted, the overall results concerning the phosphate-methylated RNAs were published without mentioning Buck’s initial proof of concept and his contributions. Generally, the (modified) dimer RNAs and DNAs possess a number of specific biophysical properties. A novel explanation is given for conflicting structural determinations.
Keywords:
Phosphate-Methylated RNA and DNA, Conformational Study, Replication and Transcription Inhibition, Chemistry of DNA Dimers during Crystallization, 5-Substituted Cytosines in DNA, Conflicting Structural Determinations, Duchenne

1. Introduction
The phosphate-methylated RNAs synthesized by Buck’s group demonstrated the formation of a parallel right- handed non-Watson and Crick base-pairing and the traditional right-handed antiparallel duplex formation. The dimer duplex formation was strongly influenced by the configuration of phosphorus. There is correspondence with the phosphate-methylated DNA. The fundamental difference is found in the ring puckering between deoxy- ribose and ribose rings. A unique example is given for the phosphate-methylated RNA dimer with the bases C and U. In the modified RNA the 2ꞌ-OH group is replaced by 2ꞌ-OCH3 in order to preclude intramolecular attack of 2ꞌ-OH on the phosphorus, a process favored by methylation of the phosphate group [1] . An illustration of the dimer is given in Figure 1.
For the S-configuration of phosphorus (SP) a parallel duplex was found, based on a C-C base pair with two equivalent N(4)-H…N(3) hydrogen bonds and a U-U pair with two equivalent O(4)…H-N(3) hydrogen bonds. In the case of the RP-configuration no duplex is formed. A similar observation was done for the corresponding phosphate-methylated DNA dimer consisting of the bases C and T. This difference in configuration results in an outward (SP) and inward orientation (RP) of the backbone methyl group.
An antiparallel orientation may be expected for the self-complementary dimer r(2ꞌ-OMe)APU. For r(2ꞌ-OMe)CPG a right-handed antiparallel duplex has been established for both phosphorus configurations [2] [3] . In the corresponding phosphate-methylated DNA dimer d(CPG) a left-handed duplex with CD spectroscopy has been established [4] . The structural properties were investigated in aqueous solution with high-field NMR (1H 400 and 600 MHz, and 31P 120 MHz) and UV hyperchromicity.
An interesting procedure to inhibit the HIV-1 RNA loops (TAR, PBS, NEF, and VIF) as shown in the retracted Science article [5] -[7] , see Figure 3(B), was suggested for the preparation of a 20-nucleotide RNA consisting of short 2ꞌ-O-methyl-phosphate-methylated RNAs in combination with relatively long 2ꞌ-O-methyl natural RNAs. However, after the retracted Science article was published, our further research on these modified nucleic acids was definitively terminated.
Later, related experiments were carried out successfully with regard to antiviral activity of locked nucleic acids (LNA with preferred A-conformation [8] ) targeting RNA stem-loops by Brown et al., and Jakobsen et al. [9] [10] . The latter group concluded that “the neutralization of HIV-1 expression declined in the following order: antisense LNA > LNAzymes > DNAzymes and antisense DNA. The LNA modifications strongly enhanced the in vivo inhibitory activity of all the antisense constructs and some of the DNAzymes”. So the conclusion is justified that the biophysical data of the different phosphate-methylated RNAs gave rise to place this modified RNA in the list of RNA backbone modifications causing inhibition of gene expression, indicated as short interfering RNA (siRNA). It has been generally accepted that a careful design of the RNA sequence and structure can impart target affinity and nuclease stability while reducing immune response [11] . The systems discussed in that
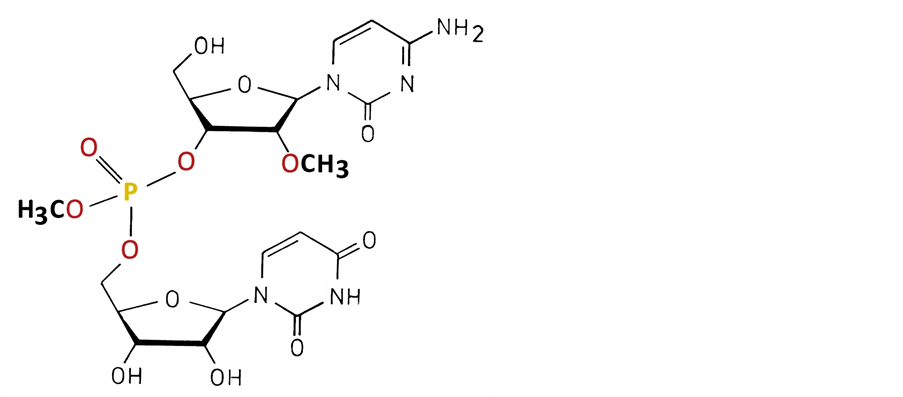
Figure 1. The RNA dimer 2ꞌ-O-methyl cytidyl 3ꞌ→5ꞌ uridine O-methyl phosphate indicated as r(2ꞌ-OMe)CPU.
paper are focused on experimental data based on effective and clinically advanced siRNA conjugates. Using this approach, the methyl group of the phosphate triester can be considered as the smallest conjugating delivery fragment to the siRNA. In any case the phosphate-methylated RNA promotes the lipophilicity and the stereo- specificity, par excellence an important aspect in intra- and inter-molecular recognition. In addition, the effective shielding of the phosphate anionic backbones via methylation makes the encapsulation with cationic additives or non-specific conjugates not directly necessary. The specificity of conjugate delivery systems will be found in the recognition of the target in combination with its biochemical environment. Additional properties of these modified RNAs as the effect of (selective) aggregation will be discussed. The synthesis of the phosphate-methylated RNAs corresponds with the concept of the preparation of the phosphate-methylated DNAs. Generally, this concept can be considered as a gradual non-automated synthesis, indicated as solution-phase chemistry. The validity of this chemistry is in sharp contrast with the interrupted solid-phase triester procedure [12] .
Recently, the group of Caruthers (Paul et al., [13] ) describes a new route for the synthesis of post-syntheti- cally modified DNAs. With the preparation of boranephosphonate dimers, substitution reactions are activated by iodine. In fact, it involves the formation of iodophosphate as intermediate. This intermediate (characterized as shown in Figure 4 of Ref. [13] ) undergoes nucleophilic substitution with nucleophiles as methanol under formation of a phosphate-methylated dimer with a defined phosphorus configuration. Such a conversion proceeds via a trigonal bipyramidal geometry with methanol (incoming) and iodine (leaving) in the apical positions. In a similar way corresponding modified RNAs may be synthesized. It may be of importance to compare the different biophysical and chemical results of the pre- and post-synthetically modified DNAs based on the solid-phase procedure.
2. Methods and Results
2.1. Synthesis of Phosphate-Methylated RNA Dimers and Their Ability for Duplex Formation
The general procedure for these modified RNAs is given in Scheme 1. In fact the synthesis is closely related to the preparation of the phosphate-methylated DNAs. Extension to longer phosphate-methylated RNAs will certainly improve specificity, facilitated by conjugates selected by their ability to afford additional support for target selectivity. It is necessary to mention that duplex formation with its natural (charge) complement for a selected composition of sequences may have the possibility for an allosteric-like interaction between the phosphate-methylated strand and its natural complement. This property has been exclusively demonstrated for phosphate-methylated DNAs with their natural counterpart [14] . Accepting a fixed A-geometry for the phosphate- methylated RNA, duplex formation will occur with natural RNA as well as with natural DNA. This means that at different levels of genetic transmission there is a blockade for this information. These phosphate-methylated RNAs may play this specific role in the genetic inhibition of retroviruses that replicate through double helical DNA intermediates.
Generally speaking, not so much attention was given to the various backbone modified DNAs and RNAs concerning their conformational stereochemistry. A serious exception was the locked nucleic acid (LNA) with preferred A-conformation, vide supra. These compounds are still operative with anionic phosphate backbones. It is to be expected that phosphate methylation will add an additional improvement on their inhibition selectivity in combination with nuclease resistance.
2.2. Conformational Analysis of Self-Complementary Phosphate-Methylated RNA Dimers. Parallel and Antiparallel Duplex Formation
It was established that one of the diastereoisomers of r(2ꞌ-OMe)CPU and r(2ꞌ-OMe)APU [1] shows a NOE contact between the methyl group of phosphorus and the H(3ꞌ) ribose of the bases C and A. This effect was assigned to the RP-configuration of both dimers, corresponding with lower mobility in reversed-phase HPLC and a lower 31P NMR chemical shift in comparison with the SP-configuration.
The conformation of the backbone and the ribose ring was derived from vicinal proton-proton and proton- phosphorus coupling constants. In both dimers there is a dominant C(3ꞌ)-endo-conformation, corresponding with the A-geometry of natural RNA.
Duplex formation was found for r(2ꞌ-OMe)CPU with the S-configuration of phosphorus, representing a

Scheme 1. The RNA bases need transient protection of the NH2-groups with the exception of U. The reaction proceeds in the following way: (i) Trimethylchlorosilane, 9-fluorenyl methoxycarbonyl chloride (Fmoc-Cl), H2O (Koole et al., [15] ); (ii) 4-Monomethoxytrityl chloride; (iii) Bis-(N,N-diisopropylamino)-methoxy phosphine/0.5eq. 1H-tetrazole (Marugg et al., [16] ); (iv) Coupling with 2ꞌ,3ꞌ-di-O-levulinyl base; (v) Oxidation with tert-butylhydroperox- ide; (vi) Removal of Fmoc and levulinyl (Lev) groups by mild base treatment (Kuijpers et al., [17] ); (vii) Detritylation under acidic conditions; (viii) Separation of the RP- and SP-diastereo- isomers with reversed-phase HPLC.
parallel duplex composed of a C-C base pair with two equivalent N(4)-H…N(3) hydrogen bonds and a U-U pair with two equivalent O(4)…H-N(3) hydrogen bonds, vide supra. For the other configuration no duplex is formed. With UV hyperchromicity a sigmoidal curve corresponding with the S-configuration was found at 11.2˚C for a concentration of 8.55 µM. This observation was confirmed with a sigmoidal chemical shift for the protons H(1ꞌ) related to C and H(5) of U. An antiparallel duplex with Watson and Crick pairing for the other dimer r(2ꞌ-OMe)APU was not observable under the corresponding temperature conditions.
As to be expected, for the dimer (2ꞌ-OMe)CPG [2] [3] with RP- and SP-configuration an antiparallel duplex has been observed based on corresponding NMR results, resulting in a preferred C(3ꞌ)-endo-conformation of the ribose ring. The tight conformational fixation is clearly demonstrated with the observation that both phosphorus configurations are able to accommodate a self-complementary right-handed duplex, in contrast with the corres- ponding phosphate-methylated DNA dimers which demonstrate for both configurations a left-handed self-com- plementary duplex [3] [4] . Generally, the melting-transitions for the duplex formation of the phosphate-methy- lated RNAs in comparison with the corresponding DNAs occur at substantial higher concentrations.
2.3. Comparison with a Self-Complementary Natural RNA and Its 2ꞌ-OMe Derivative. Left- and Right-Handed Duplex Formation
The work of Davis et al., [18] and Popenda et al., [19] on the high salt structure of a left-handed RNA double helix is well-known. It was found that for the natural ribohexanucleotide r(CPGPCPGPCPG) at 1M concentration a duplex is formed with an A-conformation. At higher concentrations in 6M a left-handed duplex is formed with a Z-conformation, similar to Z DNA. In the turn-turn model of Z RNA, the base pairs are much closer to the axis than in Z DNA. It appears that the location of the 2ꞌ-OH groups plays a crucial role, explaining the position of 2ꞌ-O-methyl for the realization of the conformational change from A to Z in the hexanucleotide.
From the different compositions it can be deduced that the conversion C(3ꞌ-endo) into C(2ꞌ-endo) with cytosine in the anti-conformation is the decisive step in the A to Z transition.
The crystal structure of the duplex of 2ꞌ-OMe(CPGPCPGPCPG) clearly shows that the 2ꞌ-OMe substituent points towards the minor groove [20] . Besides narrowing, there is also an increase in hydrophobicity. Interaction with 2-methyl-2,4-pentanediol (R,S-MPD) as diastereoisomeric mixture resulted in the S-enantiomer bound in the minor groove during crystallography experiments. It has been suggested that both methyl groups of the tertiary alcohol function of MPD point towards the 2ꞌ-O-methyl groups of the duplex promoting hydrogen bonds between the secondary hydroxyl group and the exo-amino hydrogens of guanosine. This coordination model reflects the preference for the S-enantiomer.
2.4. Bio-Physical Characteristics of Phosphate-Methylated DNA. Comparison with Phosphate-Methylated RNA for Self-Base-Pairing in Parallel Duplexes
In an article dealing with the formation of the phosphate-methylated DNA dimers d(CPC) and d(TPC) and their ability to form parallel mini-duplexes for the S-configuration only, an unusual mode of base-base stacking was derived [21] .
The results were obtained with UV hyperchromicity and 1H NMR chemical shift vs temperature profiles. With the thermodynamic equation of Marky and Breslauer [22] , two equations are derived:
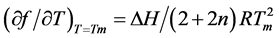 (1)
(1)
in which 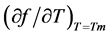 is the slope of the dissociation curve at the midpoint of the transition, f is the fraction of single strands, Tm the melting transition, n the number of strands that form an n-mer complex, and ∆H the van’t Hoff dissociation enthalpy.
is the slope of the dissociation curve at the midpoint of the transition, f is the fraction of single strands, Tm the melting transition, n the number of strands that form an n-mer complex, and ∆H the van’t Hoff dissociation enthalpy.
In addition, a (linear) plot of 1/Tm vs ln CTot (total strand concentration):
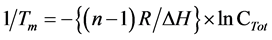 (2)
(2)
The SP-configuration of d(CPC) and d(TPC) resulted for n and ∆H in 1.94, 198 kcal/mol and 2.23, 189 kcal/mol, respectively. The ∆H values are extremely high. Equation (2) is necessary because the duplexes are absent under “natural” conditions.
For the phosphate-methylated parallel duplex with T-T base pairs molecular mechanics calculations were performed [23] . Taking into account all specific interactions, we calculated for the T-T base pairs in the mini- duplex of the dimer an (average) binding energy of 10.0 kcal/mol, an intra-strand stacking energy of 6.3 kcal/mol, and 2.9 kcal/mol for inter-strand, demonstrating no specific effects compared with the unmethylated duplex shielded with counter-ions. On the other hand there is significant reduction of the inter-strand phosphate-phosphate repulsion after phosphate methylation. Apparently, the experimental dissociation enthalpies in the related parallel duplexes seem overvalued in contrast with the expectation values of n = 2.
In order to afford an explanation, we must separate direct binding within the duplex from effects determined through specific physical-chemical properties corresponding with their lipophilicity, vide supra, or other factors based on specific inter-molecular crystal orientations. Apparently, from the overall enthalpy only a part is used for the hydrogen bridge dissociation between the bases. Therefore it seems realistic to accept that under the absence of charge the lipophilicity of phosphate-methylated duplexes may result in various aggregates with a high degree of compactness showing attractive interactions for the remarkably slim parallel pyrimidine-pyrimidine duplexes via van der Waals-London dispersion forces. An appreciation of this type of interactions is a prerequisite for a full understanding of bonding and physicochemical behavior.
The same procedure for a corresponding phosphate-methylated RNA dimer, the SP right-handed parallel duplex, was carried out. For demonstrating the correct modus operandi, the plot of 1/Tm vs ln CTot (Equation (2), vide supra) is demonstrated for r(2ꞌ-OMe)CPU in Figure 2. The slope of the linear plot equals .
.
Determination of (∂f/∂T) vs temperature plot at the Tm-value with a slope equals 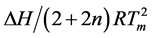 (Equation (1)). From the Equations (1) and (2) the values for n and ∆H can be abstracted. For r(2ꞌ-OMe)CPU with SP-configuration n = 2.04 with a relatively high ∆H value of 73 kcal/mol, although considerably smaller in comparison with the corresponding phosphate-methylated DNA, d(TPC) [1] [21] . We hypothesize, taking into account the fundamental geometric differences between the A- and B-geometry, that the spatial alignment of both type of duplexes causes the difference in ∆H focused on the intermolecular interaction in the formation of aggregates. To the best of our knowledge calculations for such model studies at the ab-initio level are unknown.
(Equation (1)). From the Equations (1) and (2) the values for n and ∆H can be abstracted. For r(2ꞌ-OMe)CPU with SP-configuration n = 2.04 with a relatively high ∆H value of 73 kcal/mol, although considerably smaller in comparison with the corresponding phosphate-methylated DNA, d(TPC) [1] [21] . We hypothesize, taking into account the fundamental geometric differences between the A- and B-geometry, that the spatial alignment of both type of duplexes causes the difference in ∆H focused on the intermolecular interaction in the formation of aggregates. To the best of our knowledge calculations for such model studies at the ab-initio level are unknown.
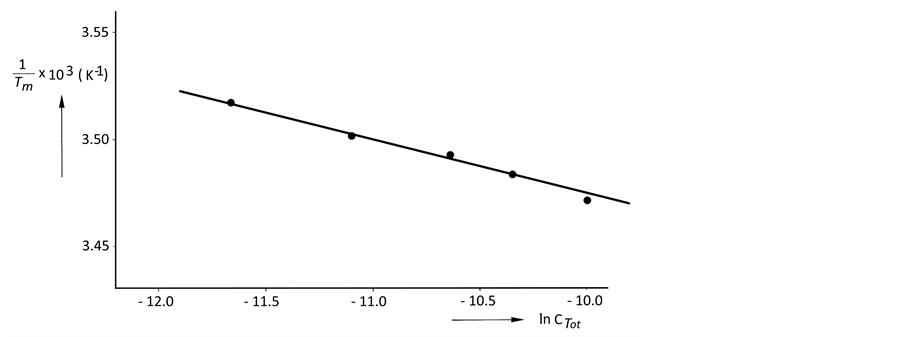
Figure 2. A linear plot of 1/Tm vs ln CTot (Equation (2), slope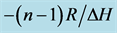 ) as measured for the parallel duplex of r(2ꞌ-OMe)CPU with SP-configuration. Tm-values were determined by computer fitting of the experimental melting profiles into sigmoidal curves. The CTot-values were obtained from UV extinction measurements.
) as measured for the parallel duplex of r(2ꞌ-OMe)CPU with SP-configuration. Tm-values were determined by computer fitting of the experimental melting profiles into sigmoidal curves. The CTot-values were obtained from UV extinction measurements.
More recently this type of calculations has been carried out by the group of Schreiner et al. They prepared highly rigid structures known as diamondoids and calculated that the London dispersion interactions are responsible for the extraordinary stability of the coupling products between the intramolecular H…H contact regions [24] . Another interesting example of Schreiner’s group more focused on biological systems has been given for dispersion interactions playing a key role in membranes. They demonstrated that the spatial alignment of strained all-trans-fused cyclobutane moieties as a natural part of membranes, leads to an increase of attractive dispersion interactions as compared to alkyl chains of the same length [25] .
Generally, this kind of interactions demonstrates clearly that besides the size of the systems, an important factor is stored in its molecular structure. Therefore we think that the triester-phosphate-backbone, with an exo- POCH3-group, perfectly orientated by the base-pairing of the duplex strands delivers a reservoir for dispersion interactions. It is superfluous to say that the contribution of these type of interactions will be strongly reduced between natural duplexes and modified-natural duplexes due to the presence of negatively charged phosphate ions in both or one of the backbones. Phosphate-methylated antiparallel duplex DNAs with purine-pyrimidine bases are not studied with regard to their dissociation enthalpy, vide infra.
For the same combination of duplex formation with one phosphate-methylated strand, it was established that ∆H shows no particular anomaly as reported for the duplex of the phosphate-methylated trimer d(APAPA) and poly(dT) in comparison with its natural trimer [26] . For this combination, the ∆H-value has been calculated with:
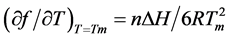 (3)
(3)
as derived from Equation (1) with n the number of base pairs.
The melting curves are equally sharp with a difference of 21.0˚C in favor of the phosphate-methylated one. The four diastereoisomers correspond with identical Tm-values of 41.0˚C (for the natural counterpart Tm = 20.0˚C) as has been observed with UV-hyperchromicity and the 1H NMR chemical shift of the imino protons vs temperature. The slopes of the dissociation curves at the midpoint of both transitions ((∂f/∂T)T=Tm) are equal. From these observations it could be abstracted that the ∆H of the phosphate-methylated d(APAPA).poly(dT) is 23.4 kcal/mol (∆Hmod) and for the natural duplex 20.4 kcal/mol (∆Hnat), using Equation (3), presented as:
 (4)
(4)
Substituting for the Tm-values in ˚K, the ratio is 1.15. With ∆Hnat = 6.8 kcal/mol per base pair [27] , the corresponding ∆H value of the phosphate-methylated trimer is obtained. This value in comparison with the phosphate-methylated parallel pyrimidine-pyrimidine duplexes indicates that formation of aggregates is absent as caused by the negatively charged phosphates, vide supra. The phosphate-methylated antiparallel DNAs amongst themselves can be described with this thermodynamic approach in a proper way. We compare the combinations of the interrelated phosphate-methylated trimers in the duplex formations with their natural complement, namely phosphate-methylated d(APAPA).poly(dT) and phosphate-methylated d(CPCPC).poly(dG) with Tm-values of 41.0˚ and 55.0˚C, respectively, as has been observed with UV and NMR [26] [28] . In order to calculate the corresponding ∆H’s a factor ~1.5 must be introduced (three hydrogen bridges in C-G and two in A-T base-pairs). This ratio can be abstracted from Figure 3 [14] .
From Figure 3 the ratio is as to be expected between 1.4 - 1.5. This figure also demonstrates a distinct and sharp sigmoidal transition for n = 3 to n = 4. Increasing the k-value gives a linear relationship.
The ratio of the ∆H’s of the phosphate-methylated duplexes d(CPCPC).poly(dG) and d(APAPA).poly(dT), using the latter equation with 1.09 for equal (∂f/∂T)T=Tm values and the factor 1.4 - 1.5 as derived from Figure 3, is then equal to 1.526 - 1.635. With ∆H for the latter combination we obtain for d(CPCPC).poly(dG) 35.7 - 38.3 kcal/mol. Via a study based on the thermodynamics of polymolecular duplexes between phosphate-methylated DNA and natural DNA the corresponding value of phosphate-methylated d(CPCPC).poly(dG) is 33.0 ± 2.0 kcal/mol. In the concluding remarks, Moody et al. [29] focus the attention on work in progress: “Further, it is of significance to investigate the thermodynamic properties of phosphate-methylated DNA-natural RNA duplexes because of possible interactions with mRNA leading to inhibition or stimulation of translation”. Anyway, even in the Science article we afford information on the relatively small level of natural RNA inhibition with phosphate-methylated DNA [5] . The optimal values were obtained for the phosphate-methylated DNA dimers: d(APOCH3A).poly(U) is 13˚C, d(APOCH3APOCH3A).poly(U) < 10˚C and d(CPOCH3C).poly(rG) is 28˚C, d(CPOCH3CPOCH3C).poly(rG) is 12˚C. For comparison, the much higher corresponding values with poly(dT) and poly(dG) are 30 and 41˚C, and 45 and 55˚C, respectively (see also Figure 3) [28] [30] . The fact that still longer DNAs were able to lock selected RNA sequences, at a level of 18 - 20 nucleotide oligomers, was based on an NMR hybridization experiment of phosphate-methylated DNA 18-mer with yeast phenylalanine transfer RNA (tRNAPhe) for the bases 59 to 76. Unfortunately, this fraudulent NMR-spectrum in Science [5] : see text under Figure 2 and Figure 2(B1) and Figure 2(B2) of that article, escaped my accuracy. This specific aspect with a thorough analysis of this spectrum was mentioned to the Editor of Science. At a first glance it looked as though the figures and the corresponding text are not manipulated. However, all information in Figure 2B(1) concerning the upfield part < 12.5 ppm has been omitted in order to show the appearance of the new A-T hybridization in Figure 2(B2). For a complete analysis see Ref. [31] on p. 1841, under b. All information needed for this analysis, that became available soon after the retraction, was accepted for publication in the journal of the Proc. Kon. Ned. Akad. v. Wetensch., 1993 (communicated by Buck at the meetings of Arts and Sciences of the Royal Netherlands Academy of January 27, and February 24, 1992). Shortly thereafter the entire issue was removed because
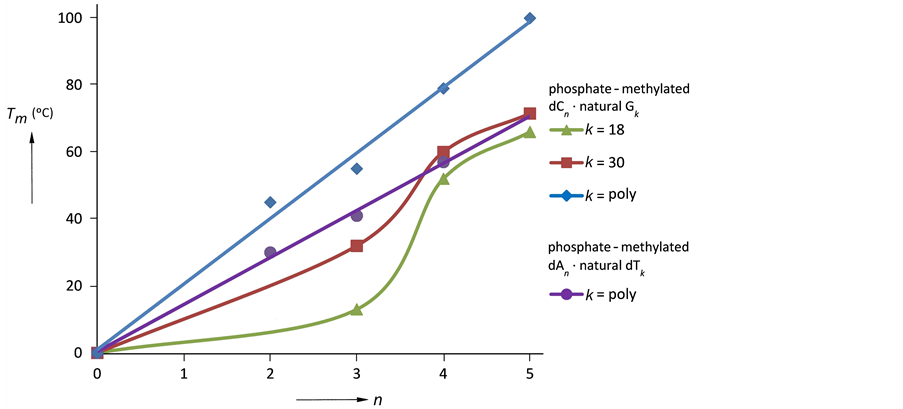
Figure 3.Melting temperature (Tm) of phosphate-methylated Cn for n = 2 - 5 bases hybridized with its natural complement Gk for different k values (the numbers are given in the column) and of phosphate-methylated An for n = 2 - 4 bases with poly (dT).
of my critical view on the fraudulent NMR-spectrum of one of the postdocs and on the different interpretations as given by a (faculty) commission, vide supra [31] .
2.5. Information on the Intra- and Inter-Molecular Interactions with an X-Ray Study of a Natural Self-Complementary d(CPG) Duplex and of the Corresponding Methylphosphonate Duplex, a Substitute for the Phosphate-Methylated Dimer
For the role of dispersion interactions it is necessary to have at one’s disposal a representative model for the solely duplex- and filling-space structures of the aforementioned (modified) DNA and RNA. For the antiparallel phosphate-methylated DNA there is a self-complementary left-handed dimer d(CPG) [4] , its (right-handed) tetramer d(CPGPCPG) [14] , and the right-handed r(2ꞌ-OMe)CPG [19] available. The individual duplex structures were obtained from CD and NMR data, vide supra. Unfortunately, no X-ray structures are available for the investigated phosphate-methylated nucleotides. For comparison with these relatively simple model compounds, the X-ray of the natural dimer d(CPG) and the corresponding methylphosphonate RP d(CPG) is given [32] [33] . The natural ammonium salt of the left-handed d(CPG) duplex was obtained at −20˚C containing two d(CPG) molecules in the asymmetric unit [32] . An interesting point is that the stacking in this duplex is related by the 21 screw symmetry in such a way that the cytosine base stacks on cytosine and the guanine base on guanine. This means that the dimer duplex does not have the three-dimensional structure i.e., a C-G-C-G-C-G…・ sequence necessary for a continuous Z-DNA helix as visualized in the well-known hexamer [34] . Additionally, the cytosine-cytosine stacking is far more extensive than the guanine-guanine stacking.
The methylphosphonate RP d(CPG) demonstrates in contrast with the left-handed phosphate-methylated d(CPG) a right-handed DNA conformation. Generally, the backbone conformation of the methylphosphonates differs strongly from their phosphate-methylated ones [4] [33] . Using the helical parameters of the dimer duplex of the methylphosphonate RP d(CPG), the development of the growing chain results in a more stretched B DNA.
The crystalline state of the natural pyrimidine dimers is of special interest because they demonstrate a duplex, closely related to the corresponding phosphate-methylated dimers as parallel duplexes, vide supra. This was demonstrated with a fiber diffraction pattern of d(TPT) [35] [36] . A double helical structure was established with about seven units per turn and a rise per base-pair of 3.8Å. The backbone strands are alternately interrupted by the absence of opposite phosphates, resulting in the elimination of the phosphate-phosphate charge repulsion as demonstrated in Figure 4.
A parallel right-handed phosphate-methylated hexamer d(TPTPTPTPTPT) in solution has been experimentally established by Koole et al., and supported by molecular mechanics calculations [36] . We expect similar observations for d(CPC) in the crystalline state based on the parallel duplex formation of the corresponding phosphate-methylated SP d(CPC). The life time of this unusual duplex formation, as illustrated in Figure 4, in solution compared to the crystalline state will be short. The suggested system as illustrated deserves more interest because it belongs to a unique duplex formation with well-defined sites structured by the “clefts” in a strict geometrical orientation. In fact, it resembles one of the fundamental aspects of enzyme-substrate modeling. It is suggested that the polar water molecules (and probably other nucleophiles) enter the opposite phosphate site because the phosphate-methylated hexamer d(TPTPTPTPTPT) exists as a duplex with exceptional stability in aque- ous solution. The molecular dynamics of the process as given in Figure 4 most likely proceeds with “inversion” of the attacking phosphorus under disruption of the intermolecular T-T base-pairing. On the other hand the stability in the crystal state of the left-handed antiparallel duplex d(CPG) differs besides the specific cationic inter- actions in the crystal state of the right-handed parallel duplex d(TPT) through its intermolecular pronounced cytosine-cytosine stacking in both strands of the “continuous” left-handed helix in comparison with the inter-

Figure 4. The fiber diffraction pattern of d(TPT). A parallel duplex alternately interrupted. A proposed intercalation of water molecules in the present vacatures as indicated with the blue squares.
molecular thymine-thymine base-pairing between both strands.
Self-base-pairing has been observed also for natural d(CPG) under acidic conditions with a pH = 4 [37] . Under this condition there are three hydrogen bonds between cytosine and its protonated form with two equivalent N(4)-H…O(2) hydrogen bonds and the N(3)-H…N(3) hydrogen bond. Thus we are dealing with two different modes of parallel C-C base-pairing. The guanine base-pairing accommodates this parallel orientation by two equivalent N(2)-H…N(3) hydrogen bonds. The latter hydrogen bonding resembles a similar bonding as demonstrated in the X-ray crystal structure of 2ꞌ-deoxycytidine [38] . The stacking of the bases within the duplex are weak. This accommodation is absent in d(CPTPCPTPCPT) at pH = 3. The parallel-pairs of the cytosines is accompanied with thymine-thymine bulges [39] .
The significance of short phosphate-methylated DNA in the suppression of genetic information as replication, has been demonstrated in various experiments [40] . Since during the replication one strand of DNA is duplicated in short fragments, known as Okazaki fragments, phosphate-methylated DNA is an excellent inhibitor, see Figure 3. The transcribed mRNA showed no hybridization with (short) phosphate-methylated DNA in its translation into protein [40] . Unfortunately, the significance of (short) phosphate-methylated RNA in the suppression of transcription and translation remained stuck in the results with the modelsystems.
2.6. Future Perspectives in the Understanding and Evaluation of the Significance of Partial and Complete Shielding of the Phosphate Backbone in DNA and RNA
It is of general interest to focus the attention on the base manipulation in recent studies of the DNA conformation. Especially at DNA systems for B-Z transition and their significance as epigenetic model [41] . Closely related is a study with regard to the effect on the B-Z transition of the three different cytosine modifications i.e. 5-hydroxymethyl, 5-formyl, and 5-carboxyl as derived from 5-methyl cytosine and mediated by the TET family of enzymes, known as new DNA bases on CPG dinucleotide sequences [42] . It was demonstrated that 5-hy- droxymethylcytosine inhibits the B-Z transition, whereas the other bases facilitate this conversion. At the same time a study showed the significance of 5-formylcytosine in altering the structure of a DNA double helix [43] . With X-ray, the crystal structure of a DNA dodecamer with three 5-formyl CPG units shows a clear change in the conformation of the grooves resulting in helical underwinding. Hydrogen reduction results in 5-hydroxy- methylcytosine with formation of a B DNA structure. An interesting aspect of this study is that increase of concentrations of spermine results in conversion of the 5-formylcytosine containing oligomers into B DNA. Apparently shielding of regio-specific phosphate anions plays an important role in this conformational change. Therefore it may be suggested to introduce 5-substituted cytosines with phosphate-methylated backbones in these studies, as compared with the Z-conformation of the phosphate-methylated d(CPG). Even on this dimer level fundamental information will become available to predict the conformational behavior of corresponding long oligomers. This information may be helpful, in part, to scan the possible parameters which might be responsible for the different conformational behavior of DNA and RNA with respect to the backbone- and base-manipulation. Through methylation of the phosphate backbone the coordination via hydrogen networks with phosphate anions will be absent. From this approach it is probable that the surplus value of geometric information of these molecular structures is most likely the result of chosen experimental conditions. In fact this may be an answer on the apparent contradiction in the outcome of the molecular arrangement in the study of the aforementioned groups [42] [43] . From the output of the abovementioned results it is evident that the cooperativity of DNA is based on its potency to accommodate, via various conformations, a fascinating template for interacting molecular systems.
At the end we will focus the attention on the antisense method, based on the inhibition of (pre)-mRNA with complementary modified RNAs, vide supra, specifically for the treatment of Duchenne muscular dystrophy. The treatment is directed on exon skipping. Based on the various modified RNAs (ionic and non-ionic) there are differences in skipping specific exons and sequence specificity. As an example we mention 2ꞌ-OMe RNA with a phosphorothioate backbone with 17 - 21 nucleotides in the key study of Aartsma-Rus and van Ommen [44] . Unfortunately, there are no explicit physical data available in comparison with model- and other modified systems. In fact this type of modified RNA differs from 2ꞌ-OMe RNA with a non-ionic phosphate-methylated backbone, vide supra.
In order to obtain knowledge of the use of a specific modified RNA oligonucleotide, it is of importance to scan its spatial arrangement. Most of the study of Buck’s group was based on this aspect demonstrating unique differences caused by backbone- and base manipulations as shown in various publications. Furthermore the role of a specific conjugate delivery system for an effective transport of the (modified) RNA to the site of action is of great importance [11] . Finally it is of significance to note that there is a risk in the preparation of artificial oligo- nucleotides via an interrupted automated solid-phase triester procedure. These results differ from the corresponding results obtained by a gradual non-automated synthesis [12] .
3. Conclusion
The significance of small nucleotide dimers in biology has been neglected to some extent even for the natural ones in their ability to form different duplexes on the DNA and RNA level. The same limited description is given for the modified nucleotides focused on the backbone-modified phosphate-methylated triesters. This article summarizes the biophysical and biological results up to a level of five phosphate-methylated DNA nucleotides and a two-level of modified RNA. Longer phosphate-methylated DNAs were synthesized following a different route [15] [45] . From the conformation studies it is concluded or expected that the A-conformation of the phosphate-methylated RNAs results in duplex formation with natural RNA (A-conformation) as well as with natural DNA (A- and B-conformation). This means that at different levels of genetic transmission there is a blockade for this information. The corresponding phosphate-methylated DNAs with a frozen B-conformation give a duplex with natural DNA whereas its duplex formation with natural RNA is of much less significance. An evaluation of the significance of partial and complete shielding of the phosphate backbone in DNA and RNA has been demonstrated for different conformational transitions.
Cite this paper
Henk M.Buck, (2016) Modified RNA with a Phosphate-Methylated Backbone. A Serious Omission in Our (Retracted) Study at HIV-1 RNA Loops and Integrated DNA. Specific Properties of the (Modified) RNA and DNA Dimers. Journal of Biophysical Chemistry,07,30-41. doi: 10.4236/jbpc.2016.71003
References
- 1. Quaedflieg, P.J.L.M., van der Heiden, A.P., Koole, L.H., van Genderen, M.H.P., Coenen, A.J.J.M., van der Wal, S. and Buck, H.M. (1990) Synthesis and Conformation of Phosphate-Methylated r(CPU) and r(APU). Formation of a Parallel Right-Handed Duplex for SP r(CPU). Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 93, 33-38.
- 2. Quaedflieg, P.J.L.M., van der Heiden, A.P., Koole, L.H., Coenen, A.J.J.M., van der Wal, S. and Meijer, E.M. (1991) Synthesis and Conformational Analysis of Phosphate-Methylated RNA Dinucleotides. Journal of Organic Chemistry, 56, 5846-5859.
http://dx.doi.org/10.1021/jo00020a028 - 3. Quaedflieg, P.J.L.M. (1993) Synthesis and Structural Aspects of Backbone-Modified Nucleic Acids. Thesis University of Leiden.
- 4. Quaedflieg, P.J.L.M., Koole, L.H., van Genderen, M.H.P. and Buck, H.M. (1989) A Structural Study of Phosphate-Methylated d(CPG)n and d(GPC)n DNA Oligomers. Implications of Phosphate Shielding for the Isomerization of B-DNA into Z-DNA. Recueil Travaux Chimiques des Pays-Bas, 108, 421-423.
http://dx.doi.org/10.1002/recl.19891081107 - 5. Buck, H.M., Koole, L.H., van Genderen, M.H.P., Smit, L., Geelen, J.L.M.C., Jurriaans, S. and Goudsmit, J. (1990) Phosphate-Methylated DNA Aimed at HIV-1 RNA Loops and Integrated DNA Inhibits Viral Infectivity. Science, 248, 208-211.
http://dx.doi.org/10.1126/science.2326635 - 6. Moody, H.M., Quaedflieg, P.J.L.M., Koole, L.H., van Genderen, M.H.P., Buck, H.M., Smit, L., Jurriaans, S., Geelen, J.L.M.C. and Goudsmit, J. (1990) Retraction: Inhibition of HIV-1 Infectivity by Phosphate-Methylated DNA. Science, 250, 125.
http://dx.doi.org/10.1126/science.2218505 - 7. Buck, H.M. (1996) Phosphate-Methylated DNA: A Unique Oligodeoxynucleotide as Compared with Other Modified DNAs. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 99, 145-153.
- 8. Vester, B. and Wengel, J. (2004) LNA (Locked Nucleic Acid): High-Affinity Targeting of Complementary RNA and DNA. Biochemistry, 43, 13233-13241.
http://dx.doi.org/10.1021/bi0485732 - 9. Brown, D., Arzumanov, A.A., Turner, J.J., Stetsenko, D.A., Lever, A.M.L. and Gait, M.J. (2005) Antiviral Activity Steric-Block Oligonucleotides Targeting the HIV-1 Trans-Activation Response and Packaging Signal Stem-Loop RNAs. Nucleosides, Nucleotides and Nucleic Acids, 24, 393-396.
http://dx.doi.org/10.1081/NCN-200059813 - 10. Jakobsen, M.R., Haasnoot, J., Wengel, J., Berkhout, B. and Kjems, J. (2007) Efficient Inhibition by LNA Modified Antisense Oligonucleotides and DNAzymes Targeted to Functionally Selected Binding Sites. Retrovirology, 4, 29.
http://dx.doi.org/10.1186/1742-4690-4-29 - 11. Kanasty, R., Dorkin, J.R., Vegas, A. and Anderson, D. (2013) Delivery Materials for siRNA Therapeutics. Nature Materials, 12, 967-977.
http://dx.doi.org/10.1038/nmat3765 - 12. Buck, H.M. (2015) The Risk of the Preparation of Artificial DNAs via an Interrupted Automated Solid-Phase Triester Method. Nucleosides, Nucleotides and Nucleic Acids, 34, 400-415.
http://dx.doi.org/10.1080/15257770.2015.1006774 - 13. Paul, S., Roy, S., Monfregola, L., Shang, S., Shoemaker, R. and Caruthers, M.H. (2015) Oxidative Substitution of Borane Phosphonate Diesters as a Route to Post-Synthetically Modified DNA. Journal of the American Chemical Society, 137, 3253-3264.
http://dx.doi.org/10.1021/ja511145h - 14. Buck, H.M. (2013) A Conformational B-Z Study Monitored with Phosphatemethylated DNA as a Model for Epigenetic Dynamics Focused on 5-(Hydroxy)methylcytosine. Journal of Biophysical Chemistry, 4, 37-46.
http://dx.doi.org/10.4236/jbpc.2013.42005 - 15. Koole, L.H., Moody, H.M., Broeders, N.L.H.L., Quaedflieg, P.J.L.M., Kuijpers, W.H.A., van Genderen, M.H.P., Coenen, A.J.J.M., van der Wal, S. and Buck, H.M. (1989) Synthesis of Phosphate-Methylated DNA Fragments Using 9-Fluorenylmethoxycarbonyl as Transient Base Protecting Group. Journal of Organic Chemistry, 54, 1657-1664.
http://dx.doi.org/10.1021/jo00268a030 - 16. Marugg, J.E., Nielsen, J., Dahl, O., Burik, A., van der Marel, G.A. and van Boom, J.H. (1987) (2-Cyano-1,1-dimethyl-ethoxy)bis(diethylamino)phosphine: A Convenient Reagent for the Synthesis of DNA Fragments. Recueil des Travaux Chimiques des Pays-Bas, 106, 72-76.
http://dx.doi.org/10.1002/recl.19871060302 - 17. Kuijpers, W.H., Huskens, J., Koole, L.H. and van Boeckel, C.C.A. (1990) Synthesis of Well-Defined Phosphate-Methylated DNA Fragments in Methanol as Deprotecting Agent: The Application of Potassium Carbonate in Methanol as Deprotecting Agent. Nucleic Acids Research, 18, 5197-5205.
http://dx.doi.org/10.1093/nar/18.17.5197 - 18. Davis, P.W., Adamiak, R.W. and Tinoco Jr., I. (2004) Z-RNA: The Solution NMR Structure of r(CGCGCG). Biopoymers, 29, 109-122.
http://dx.doi.org/10.1002/bip.360290116 - 19. Popenda, M., Milecki, J. and Adamiak, R.W. (2004) High Salt Solution of a Left-Handed RNA Double Helix. Nucleic Acids Research, 32, 4044-4054.
http://dx.doi.org/10.1093/nar/gkh736 - 20. Adamiak, D.A., Rypniewski, W.R., Milecki, J. and Adamiak, R.W. (2001) The 1.19Å X-Ray Structure of 2’-O-Me (CGCGCG)2 Duplex Shows Dehydrated RNA with 2-Methyl-2,4-pentanediol in the Minor Groove. Nucleic Acids Research, 29, 4144-4153.
http://dx.doi.org/10.1093/nar/29.20.4144 - 21. Quaedflieg, P.J.L.M., Broeders, N.L.H.L., Koole, L.H., van Genderen, M.H.P. and Buck, H.M. (1990) Conformation of the Phosphate-Methylated DNA Dinucleotides d(CPC) and d(TPC). Formation of a Parallel Mini-Duplex Exclusively for the S Configuration at Phosphorus. Journal of Organic Chemistry, 55, 122-127.
http://dx.doi.org/10.1021/jo00288a025 - 22. Marky, L.A. and Breslauer, J. (1987) Calculating Thermodynamic Data for Transitions of Any Molecularity from Equilibrium Melting Curves. Biopolymers, 26, 1601-1620.
http://dx.doi.org/10.1002/bip.360260911 - 23. Van Genderen, M.H.P., Koole, L.H., Aagaard, O.M., van Lare, C.E.J. and Buck, H.M. (1987) Molecular Mechanics Studies of Parallel and Antiparallel Phosphate-Methylated DNA. Biopolymers, 26, 1447-1461.
http://dx.doi.org/10.1002/bip.360260902 - 24. Schreiner, P.R., Chernish, L.V., Gunchenko, P.A., Tikhonchuk, E.Y., Hausman, H., Serafin, M., Schlecht, S., Dahl, J.E.P., Carlson, R.M.K. and Fokin, A.A. (2011) Overcoming Lability of Extremely Long Alkane Carbon-Carbon Bonds through Dispersion Forces. Nature, 477, 308-311.
http://dx.doi.org/10.1038/nature10367 - 25. Wagner, J.P. and Schreiner, P.R. (2014) Nature Utilizes Unusual High London Dispersion Interactions for Compact Membranes of Molecular Ladders. Journal of Chemical Theory and Computation, 10, 1353-1358.
http://dx.doi.org/10.1021/ct5000499 - 26. Koole, L.H., van Genderen, M.H.P., Reiniers, R.G. and Buck, H.M. (1987) Enhanced Stability of a Watson & Crick DNA Duplex Structure by Methylation of the Phosphate Group. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 90, 41-46.
- 27. Pless, R.C. and Ts’o, P.O.P. (1977) Duplex Formation of Nonionic Oligo(deoxythymidylate) Analogue [Heptadeoxythymidilyl-(3’-5’)-deoxythymidine heptaethyl ester d[d(TP(Et)]7T] with Poly-(Deoxyadenylate). Evaluation of the Electrostatic Interaction. Biochemistry, 16, 1239-1250.
http://dx.doi.org/10.1021/bi00625a033 - 28. Van Genderen, M.H.P., Koole, L.H. and Buck, H.M. (1989) Hybrization of Phosphate-Methylated DNA and Natural Oligonucleotides. Implications for Protein-Induced DNA Duplex Destabilization. Recueil des Travaux Chimiques des Pays-Bas, 108, 28-35.
http://dx.doi.org/10.1002/recl.19891080106 - 29. Moody, M.R., van Genderen, M.H.P. and Buck, H.M. (1990) Thermodynamics of Polymolecular Duplexes between Phosphate-Methylated DNA and Natural DNA. Biopolymers, 30, 609-618.
http://dx.doi.org/10.1002/bip.360300513 - 30. Van Genderen, M.H.P. (1989) Structure and Stability of Phosphate-Methylated DNA Duplexes. Master’s Thesis, Eindhoven University of Technology, Eindhoven.
- 31. Buck, H.M. (2004) The Chemical and Biochemical Properties of Methyl Phosphotriester DNA. Nucleosides, Nucleotides and Nucleic Acids, 23, 1833-1847.
http://dx.doi.org/10.1081/NCN-200040620 - 32. Ramakrishnan, B. and Viswamitra, M.A. (1988) Crystal and Molecular Structure of the Ammonium Salt of the Dinucleoside Monophosphate d(CPG). Journal of Biomolecular Structure and Dynamics, 6, 511-523.
http://dx.doi.org/10.1080/07391102.1988.10506504 - 33. Han, F., Watt, W., Duchamp, D., Callahan, L., Kézdy, F.J. and Agarwal, K. (1990) Molecular Structure of Deoxycytidyl-3’-methylphosphonate (RP) 5’-Deoxyguanidine, d[CP(CH3)G]. A Neutral Dinucleotide with Watson-Crick Base Pairing and a Right Handed Helical Twist. Nucleic Acids Research, 18, 2759-2767.
http://dx.doi.org/10.1093/nar/18.9.2759 - 34. Wang, A.H.J., Quigley, G.J., Kolpak, F.J., Crawford, J.L., van Boom, J.H., van der Marel, G. and Rich, A. (1979) Molecular Structure of a Left-Handed Double Helical DNA Fragment at Atomic Resolution. Nature, 282, 680-686.
http://dx.doi.org/10.1038/282680a0 - 35. Tollin, P., Walker, R.T. and Wilson, H.R. (1984) Is There Sometimes T-T Wobble Pairing in Thymidylyl-3’,5’? Nucleic Acids Research, 12, 8345-8347.
http://dx.doi.org/10.1093/nar/12.22.8345 - 36. Koole, L.H., van Genderen, M.H.P. and Buck, H.M. (1987) A Parallel Right-Handed Duplex of the Hexamer d(TPTPTPTPTPT) with Phosphate Triester Linkages. Journal of the American Chemical Society, 109, 3916-3921.
http://dx.doi.org/10.1021/ja00247a015 - 37. Cruse, W.B.T., Egert, E., Kennard, O., Sala, G.B., Salisbury, S.A. and Viswamitra, M.A. (1983) Self Base Pairing in a Complementary Deoxydinucleoside Monophosphate Duplex Crystal and Molecular Structure of Deoxycytidylyl-(3’-5’)-deoxyguanosine. Biochemistry, 22, 1833-1839.
http://dx.doi.org/10.1021/bi00277a014 - 38. Young, D.W. and Wilson, H.R. (1975) The Crystal and Molecular Structure of 2’-Deoxycytidine. Acta Crystallographica Section B, 31, 961-965.
http://dx.doi.org/10.1107/S0567740875004281 - 39. Sarma, M.H., Gupta, G. and Sharma, R.H. (1986) A Cytosine-Cytosine Base Paired Parallel DNA Double Helix with Thymine-Thymine Bulges. FEBS Letters, 205, 223-229.
http://dx.doi.org/10.1016/0014-5793(86)80902-4 - 40. Van Genderen, M.H.P., Koole, L.H. and Buck, H.M. (1988) Duplex Stability of Hybrids between Phosphate-Methylated DNA and Natural RNA. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 91, 53-57.
- 41. Buck, H.M. (2011) DNA Systems for B-Z Transition and Their Significance as Epigenetic Model: The Fundamental Role of the Methyl Group. Nucleosides, Nucleotides and Nucleic Acids, 30, 918-944.
http://dx.doi.org/10.1080/15257770.2011.620580 - 42. Wang, S., Long, Y., Wang, J., Ge, Y., Guo, P., Liu, Y., Tian, T. and Zhou, X. (2014) Systematic Investigations of Different Cytosine Modifications on CPG Dinucleotide Sequences: The Effect on the B-Z Transition. Journal of the American Chemical Society, 136, 56-59.
http://dx.doi.org/10.1021/ja4107012 - 43. Raiber, E.-A., Murat, P., Chirgadze, D.Y., Beraldi, D., Luisi, B.F. and Balasubramanian, S. (2015) 5-Formylcytosine Alters the Structure of the DNA Double Helix. Nature Structural & Molecular Biology, 22, 44-49.
http://dx.doi.org/10.1038/nsmb.2936 - 44. Aartsma-Rus, A. and van Ommen, G.J.B. (2007) Antisense-Mediated Exon Skipping: A Versatile Tool with Therapeutic and Research Applications. RNA, 13, 1609-1624.
http://dx.doi.org/10.1261/rna.653607 - 45. Buck, H.M. (2015) Retracted HIV Study Provides New Information about the Status of the in Vitro Inhibition of DNA Replication by Backbone Methylation. Journal of Biophysical Chemistry, 6, 29-34.
http://dx.doi.org/10.4236/jbpc.2015.61003