Journal of Biophysical Chemistry
Vol.06 No.02(2015), Article ID:56112,9 pages
10.4236/jbpc.2015.62006
Gly→Ala Point Mutation and Conformation of Poly-Ala Stretch of PABPN1: A Molecular Dynamics Study
Mohd Shafique, Mohan Lal Garg, Fateh Singh Nandel
Department of Biophysics, Panjab University, Chandigarh, India
Email: fateh_nandel@yahoo.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 February 2015; accepted 30 April 2015; published 5 May 2015
ABSTRACT
Single nucleotide replacing mutations in genes cause a number of diseases, but sometimes these mutations mimic other genetic mutations such as trinucleotide repeats expansions. A mutation in codon GGG→GCG results in Gly→Ala at the N-terminal of PABPN1 protein that mimics the trinuc- leotide repeat expansion disease called Oculopharyngeal muscular dystrophy (OPMD). Molecular dynamics simulations in water with peptide models having sequence Ac-A10-GA2GG-NHme (pep- tide A) and Ac-A10A3GG-NHme (peptide B) reveal an increase in the length of helical segment in peptide B. The α-helical length is found to be stable in peptide B with starting geometry of a right handed helix, while in the case peptide A, the helical length is short. The interactions of water molecules at terminals, side chain-backbone interactions and hydrogen bonds provide stability to resultant conformation. The adopted helix by the poly-Ala stretch may lead to masking some other active parts of the PABPN1 that may trigger the aggregation, decrease in degradation and/or im- paired function of protein. Hence, further studies with N-terminal may be helpful to understand unclear disease mechanism.
Keywords:
Single Nucleotide Polymorphism, Gly→Ala Mutation, Poly-Ala, OPMD, PABPN1

1. Introduction
Many genetic diseases caused by expansions in trinucleotide repeats become more severe after each new genera- tion, a phenomenon known as-genetic anticipation [1] . These expansions can result in expansions of homopoly- amino acid repeats and cause a number of human neurodegenerative disorders like Huntigton’s, spinocellebelar ataxias, ocuopharyngeal muscular dystrophy (OPMD) and many others [2] . OPMD, an autosomal dominant, late onset disease with progressive ptosis and dysphagia, has been found to occur due to expansion of GCG10 repeat to GCG8-13. The GCG10 tract lies in the exon 1 of PABPN1 gene located on chromosome 14q11.2-13 [3] . The expanded pabpn1 protein forms aggregates (called intra nuclear inclusions) in the skeletal muscle nuclei and is the hallmark of OPMD [4] . Robinson et al. report a point mutation that occurs in the GGG codon that lies im- mediately after the TNR (trinucleotide repeat) for ten alanine residues [5] [6] . The mutation G→C in GGG co- don results in tandem thirteen GCG codons (see Figure 1), which mimics the expansion of trinucleotide re- peats in the GCG codons reported by Brais et al. in 1998 [3] .
This G→C substitution changes a glycine GGG codon, situated immediately to the trinucleotide repeats en- coding Ala10 residues, to an alanine GCG codon (Figure 1). Following the GGG codon, two alanine codons are present and after the point mutation (G→C) a stretch of a contiguous 13 codons for alanine is expressed (Figure 1). The GCG6 to GCG8-13 has been reported in OPMD cases but the patient with the Gly→Ala mutation does not have the amino acid sequence Gly-Ala-Ala immediately after the Ala10 stretch (normally found in OPMD pa- tients). Yet, patients having mutated Gly→Ala residue are found to have OPMD symptoms [5] .
Therefore, it is of interest to investigate the conformational change that may occur after point mutation (G→A) at the 12th position in the pabpn1 protein that may lead to OPMD. Here, in this study, we mainly emphasize to decipher the conformational changes that occur after this mutation in the N-terminal stretch (first 15 residues). This study may be fruitful to understand the conformational behavior of the stretch and may help to predict structural change after expansion by first N-terminal domain of pabpn1/in the whole protein. Here, we report the conformational behavior of the model peptides―Ac-A10GA2G2NHme (peptide A) and Ac-A10AA2G2NHme (peptide B) shown in Figure 1. These peptides with normal sequence of first sixteen residues except the first re- sidue (methionine not included) have been investigated for simulation study in water. It is also of our interest to know the stabilizing interactions in the conformations of these peptides sampled from the simulations.
2. Methodology
2.1. Choice of Starting Geometry
Alanine is simple amino acid with highest propensity for α-helix and have been reported by various studies, it can adopt structures that belongs to second quadrant of Ramachandran map [7] - [11] . Therefore, we have chosen starting geometries one from right hand side of second quadrant and another form the third quadrant corresponding to alpha helical region. The model peptides with protected groups (Ac- and -NHme) at the N and C terminals, respectively, were prepared using PyMOL software [12] for two starting geometries with distinct back- bone dihedral angles (φ, ψ) of −57˚, −47˚, and −51˚, 153˚ corresponding to alpha-helical and collagen-like structure, respectively.
2.2. Simulations
All molecular dynamics (MD) simulations were performed by using GROMACS (version 4.5.5) software package [13] in water model. The coordinate and topology for simulations were generated by pdb2gmx protocol of
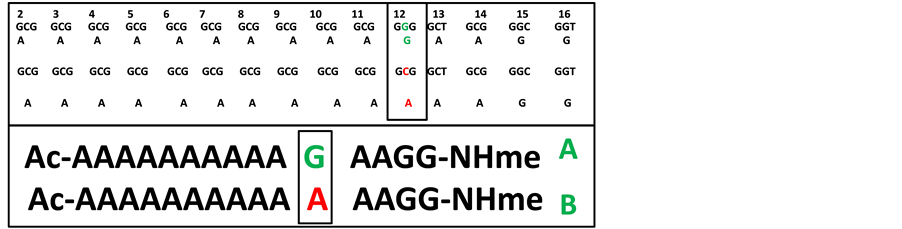
Figure 1. The trinucleotide codons and their encoded amino acid residues and the site of point mutation are given in upper panel; the complete sequence of the studied peptides A (native) and B (mutated) are shown in lower panel.
GROMACS using G43a1force field [14] and solvated using spc216 explicit water model and all atoms of the system were considered explicitly [15] . The steric conflicts between water and model peptides were removed by subjecting the system to energy minimization using steepest descent method with 500 maximum numbers of steps. Potential energy convergence has been checked by g_energy and analyzed for convergence. Following minimization, the system was equilibrated by using NVT ensemble at 300 K and the solvent molecules were allowed to equilibrate by keeping the peptide in fixed position for 100 ps using a v-rescale thermostat [16] . Then, isothermal-isobaric conditions were applied and again the system was equilibrated to maintain constant pressure and density for 100 ps. After removing the restraints, the molecular dynamics run was performed for 10 ns at 300 K in simple cubic periodic box, with a time step of 2 fs using the leapfrog integrator for all starting geometries [17] . Periodic boundary conditions have been applied in all the three directions of the cubic box. The LINCS algorithm was used to apply restraints on bond lengths [18] .The center of mass motion of the system was removed at every step to maintain T0 (reference temperature). Pressure was controlled using weak coupling with a time constant of 0.5 ps and a reference pressure of 1 bar [18] . Coulomb and van der Waals interactions were evaluated by using the cut-off of 1.0 nm and 1.2 nm, respectively and was updated every 10 fs. Particle Mesh- Ewald summation method was used to calculate long-range electrostatic interactions [19] . Initial velocities of all atoms were taken from a Maxwellian distribution at the desired initial temperature [18] .
3. Results
Polyalanine peptides have been reported to adopt a variety of conformations like right handed α-helix, β-sheet/ β-strand, PPII helix and random coil [20] - [24] . Alanine is a simple amino acid with methyl group as side chain and has highest propensity to form right handed helix [25] . PPII helix has also been a suggested conformation by polyalanine in water [26] . Therefore, the two starting geometries have been investigated one corresponding to the second quadrant and other belonging to the third quadrant of Ramachandran map.
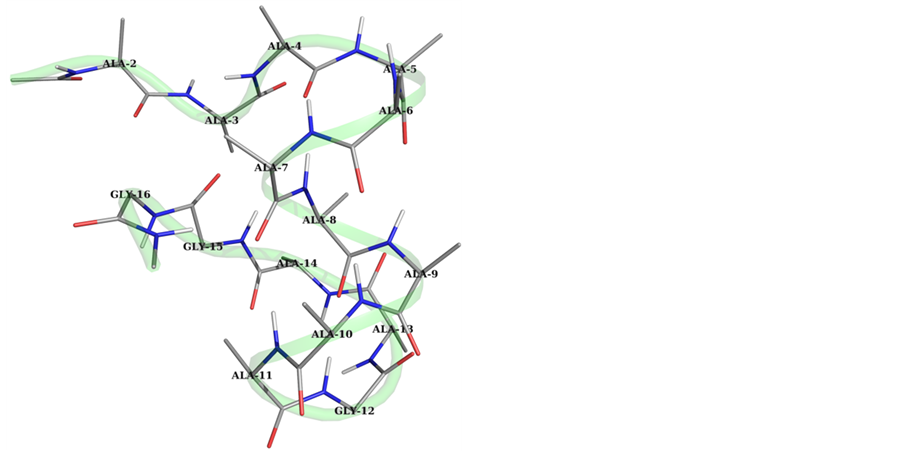
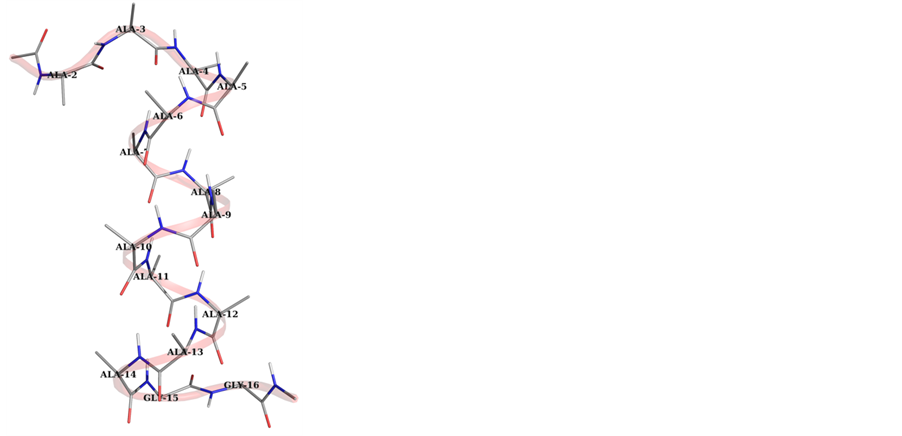
The results after 10 ns simulations in water for studied peptide A (Gly at the 11th position) and peptide B (Ala residue at the 11th position) are shown in Table 1 and clearly depicts that the peptides with the starting geometry of α-helix remain in helical conformation for native as well as the peptide with single point mutation i.e., peptide A and B. The (φ, ψ) values in Table 1 show that the residues from the 3rdto 10th adopt (φ, ψ) values in the right handed helical region of the Ramachandran map in Peptide A (Figure 2), while in case of peptide with the single point mutation (peptide B) have also helical content. The helical segment increased in the latter due to mutation of the Gly with Ala residue (Figure 2). The (φ, ψ) values of the peptide B given in Table 1 clearly indicate
 Peptide A Peptide B
Peptide A Peptide B
Figure 2. Graphical view of peptides within 3 Å of the peptide surface after 10 ns simulation in water clearly depicts the increase in the helical segment with starting geometry with (φ, ψ) values (−57˚, −47˚).

Table 1. The MD results in terms of φ, ψ and ω values for model peptides A and B after 10 ns in water.
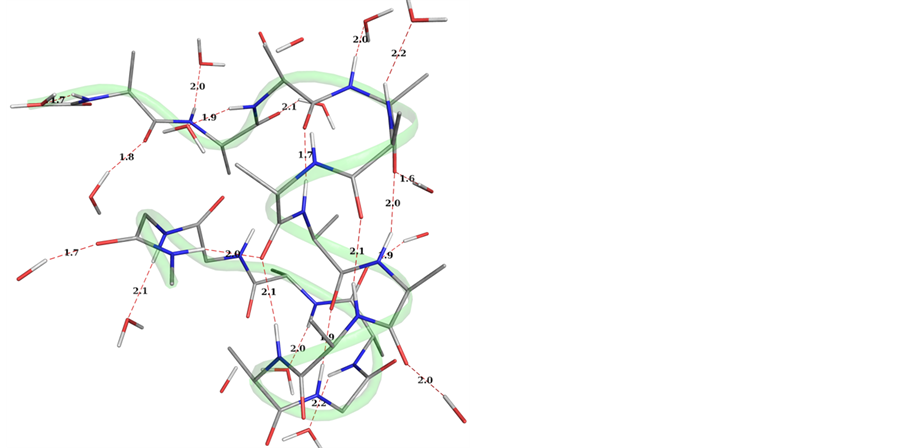
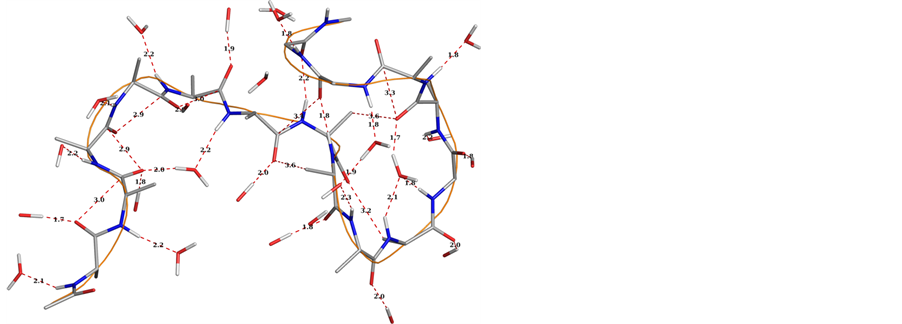
that it has an extended α-helix region (from 3rd to 13th residues) and can also be seen in Ramachandran map (Supporting Information SF1) and the residues in the helical region increase as compared to peptide A. Therefore, the placement of Gly→Ala has increased the helix in peptide B. The stabilizing interactions for the both peptides are carbonyl-carbonyl interactions, CH of the methyl group of the alanine side chain interacts with the oxygen atoms of the carbonyl group, Carbonyl oxygens interact with water molecules, and NH of the peptide backbone interacts with the water oxygens as shown in the Figure 3. The hydrogen bonds are abundant in the peptide B compared to the peptide A (Figure 3(a) and Figure 3(b)), likely providing the basis for the former containing more helical segment than the latter. Analysis of the trajectories for both the peptides with starting geometry (−57˚, −47˚) revealed that the helices remain stable and slightly open from termini but middle residues remain in the right handed helical conformation.
Hydrogen bonds stabilizing the helices are interesting in the Figure 3(b); the same carboxyl oxygen interacts with the HN groups of the 5th and 6th alanine residues (2.3 Å, 2.2 Å) and forms weak hydrogen bonds, but it results in the deviation of amide bond the 2nd residue up to 30˚. Interestingly, the capped peptide (with protecting groups) with (Supporting Information SF2) Gly at the 11th position (peptide A) with starting geometry collagen (−51˚, 153˚) results in unordered structure stabilized by hydrogen bonds and between the NH and C=O moieties of the backbone, CH-O, C=O-C=O, NH-O of water etc. Similar results have been obtained for peptide with Ala at 11th position (peptide B) but larger number of residues fell in the right handed helical region of the Ramachandran map (Supporting Information SF2) and Cα RMSD also shows the overall stability (equilibrium) of the both peptides (A and B) during simulations for 10 ns (Supporting Information SF3). Additional 10 ns simulations for both the peptide A and B again revealed more population of residues in helical region for peptide B as compared to peptide A (Supporting Information SF4).
Amide Bond Geometry
Trajectory analysis for all the studied conformations shows that no conversion of trans amide bond to cis form, however, the deviations in ω (amide bond) have been observed. Representative plots for deviation in ω (∆ω, +ve for ω value >180 and ?ve for ω value <180) from trans amide bond geometry as a function of residue number are shown in Figure 4 with the starting geometries (−57˚, −47˚) and (−51˚, 153˚) for peptides A and B after 10 ns simulations in water. The plots clearly indicate that the deviations in ω are less in peptide B for starting geometry (−57˚, −47˚) but the residue with (φ, ψ) values of −80˚, 135˚ have deviation ~30˚. The deviations in ω are supported by spectroscopy, computational results on model systems and analysis of proteins crystal structure [27] - [29] .
 (a) (b)
(a) (b)
Figure 3. Graphical views of the peptides within 3 Å of the peptide surfaces, with starting geometry (−57˚, −47˚) for peptides A and B (3a and 3b, respectively) depicting water―HN and water―O=C of the peptide backbone interactions as well as the hydrogen bond stabilizing the helices.
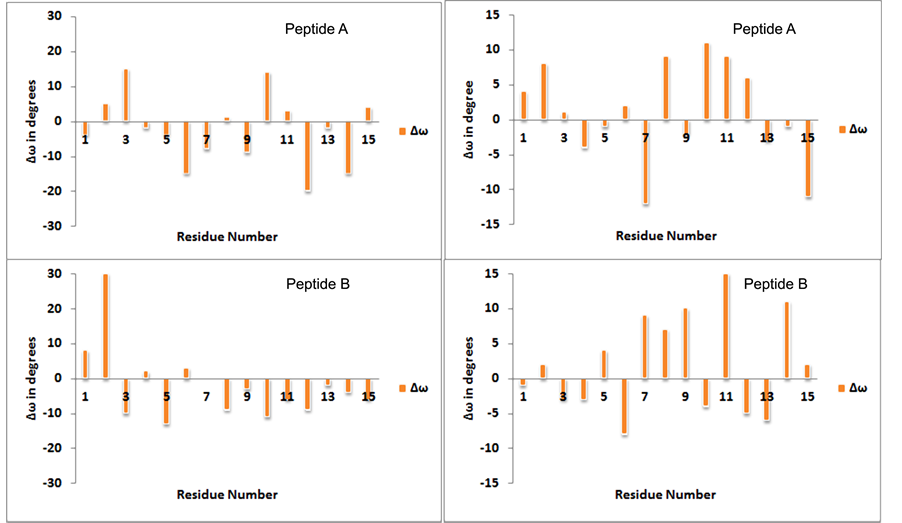
Figure 4. Plots for deviation in omega (∆ω) in degrees for peptide A and peptide B with starting geometries corresponding to collagen (right) and right handed helix (left respectively).
4. Discussion
The single point mutation in codon sequence of PABPN1 has been shown to mimics the polyalanine expansion that results in Oculopharyngeal muscular dystrophy [5] . Here, we study the behavior of the expanded alanine stretch (due to mutation G→A) and its preference for helical conformation. Alanine is usual amino acid and has high propensity to form α-helical structure, stabilized by hydrogen bonds. This conformational study on the peptides reveals that the helix is the preferred conformation for the initial geometries with (φ, ψ) values corresponding to helical region of the Ramachandran map and increased after G→A substitution as consistent with the literature [30] [31] . Glycine residues are poor helix former and called as helix breakers [32] . Such differences between Ala and Gly are also related with the solubility of glycine as compared with the alanine residue as in the latter the potential hydrogen binding sites are not easily available for interaction due to methyl side chain. Energetically alanine forms more stable (0.4 - 2 kcalmol−1) helix relative to glycine [30] [33] . Therefore, increase in hydrophobic character or helicity may be of pathological relevance in mutant PABPN1. The stabilizing interactions and deviation in ω may play a role in conformational preferences adopted by homopolymeric peptides. Tryptophan fluorescence and real time NMR studies found to be in favor of the current simulation study that adoption of insoluble folded character may cause aggregation [34] . Interactions of the poly-Ala stretch with functional domain of the protein may also trigger pathological behavior of PABPN1. But before concluding this type of remarks, further conformational study with the whole N-terminal domain and protein PABPN1 in native as well as mutant form should be considered as Winter et al. report poly-Ala independent aggregation of PABPN1 protein [35] . The helix adopting character of poly-Ala stretch and its relation to pathological mechanism by mutant PABPN1 will be published in our forthcoming publication. Therefore, the present work throws some light on the possible conformational behavior of polyalanine stretch in isolation with two Gly residues at C terminal (i.e., first 15 residues of PABPN1 except met at first position).
Acknowledgements
We are thankful to IUAC, New Delhi for providing the HPC facility.
References
- Christopher, E.P., Edamura, K.N. and Cleary, J.D. (2005) Repeat Instability: Mechanisms of Dynamic Mutations. Nature Reviews Genetics, 6, 729-742. http://dx.doi.org/10.1038/nrg1689
- Mirkin, S.M. (2007) Expandable DNA Repeats and Human Diseases. Nature, 447, 932-940. http://dx.doi.org/10.1038/nature05977
- Brais, B., Bouchard, J.P., Xie, Y.G., Rochefort, D.L., Chretien, N., Tome, F.M., Lafrenier, R.G., Rommens, J.M., Uyama, E. and Nohira, O. (1998) Short GCG Expansions In The PABP2 Gene Cause Oculopharyngeal Muscular Dystrophy. Nature Genetics, 18, 164-166. http://dx.doi.org/10.1038/ng0298-164
- Bao, Y., Cook, L.J., O’Donovan, D., Uyama, E. and Rubinsztein, D.C. (2002) Mammalian, Yeast, Bacterial and Hemical Chaperones Reduce Aggregate Formation and Death in a Cell Model of Oculopharyngeal Muscular Dystrophy. Jour- nal of Biological Chemistry, 277, 12263-12269. http://dx.doi.org/10.1074/jbc.M109633200
- Robinson, D.O., Wills, A.J., Hammans, S.R., Read, S.P. and Sillibourne, J. (2006) Oculopharyngeal Muscular Dystrophy: A Point Mutation Which Mimics the Effect of the Pabpn1 Gene Triplet Repeat Expansion Mutation. Journal of Medical Genetics, 43, e23-e24. http://dx.doi.org/10.1136/jmg.2005.037598
- Robinson, D.O., Hilton-Jones, D., Mansfield, D., Hildebrand, R.D., Marks, S., Mechan, D. and Ramsay, J. (2011) Two Cases of Oculopharyngeal Muscular Dystrophy (OPMD) with the Rare PABPN1 C.35g > C; P.Gly12ala Point Mutation. Neuromuscular Disorders, 21, 809-811. http://dx.doi.org/10.1016/j.nmd.2011.06.003
- Asakura, T., Kumiko, Y.K., Furitsu, H., Yusuke, S., Katsuyuki, N. and Kaji, N.H. (2014) Difference in the Structures of Alanine Tri- and Tetra-Peptides with Antiparallel β-Sheet Assessed by X-Ray Diffraction, Solid-State NMR and Chemical Shift Calculations by GIPAW. Biopolymers, 101, 13-20. http://dx.doi.org/10.1002/bip.22241
- Palenčár, P. and Bleha, T. (2011) Molecular Dynamics Simulations of the Folding of Poly (Alanine) Peptides. Journal of molecular Modeling, 17, 2367-2374. http://dx.doi.org/10.1007/s00894-011-0997-4
- Mezei, M., Fleming P.J., Srinivasan, R. and Rose, G.D. (2004) Polyproline II Helix Is the Preferred Conformation for Unfolded Polyalanine in Water. Proteins: Structure Function and Bioinformatics, 55, 502-507. http://dx.doi.org/10.1002/prot.20050
- Kentsis, A., Mezei, M., Gindin, T. and Osman, R. (2004) Unfolded State of Polyalanine Is a Segmented Polyproline II Helix. Proteins: Structure Function and Bioinformatics, 55, 493-501. http://dx.doi.org/10.1002/prot.20051
- Soto, P., Baumketner, A. and Shea, J.E. (2006) Aggregation of Polyalanine in Hydrophobic Environment. Journal of Chemical Physics, 124, 134904-134907. http://dx.doi.org/10.1063/1.2179803
- The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC, New York.
- Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A.E. and Berendsen, H.J.C. (2005) GROMACS: Fast, Flexible, and Free. Journal of Computational Chemistry, 26, 1701-1718. http://dx.doi.org/10.1002/jcc.20291
- Van Gunsteren, W.F., et al. (1996) Biomolecular Simulation: The GROMOS 96 Manual and User Guide. VDF Hochs- chulverlag AG an der ETH Zürich, Zürich, 1-1042.
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F. and Hermans, J. (1981) Interaction Models for Water in Relation to Protein Hydration. In: Pullman, B., Ed., Intermolecular Forces, Springer, Berlin, 331-342.
- Bussi, G., Donadio, D. and Parrinello, M. (2007) Canonical Sampling through Velocity Rescaling. Journal of Chemical Physics, 126, Article ID: 014101. http://dx.doi.org/10.1063/1.2408420
- Cuendet, M.A. and van Gunsteren, W.F. (2007) On the Calculation of Velocity-Dependent Properties in Molecular Dynamics Simulations Using the Leapfrog Integration Algorithm. Journal of Chemical Physics, 127, Article ID: 184102. http://dx.doi.org/10.1063/1.2779878
- Hess, B., Bekker, H., Berendsen, H.J.C. and Fraaije, J.G.E.M. (1997) LINCS: A Linear Constraint Solver for Molecular Simulations. Journal of Computational Chemistry, 18, 1463-1472. http://dx.doi.org/10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
- Essmann, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H. and Pedersen, L.G. (1995) A Smooth Particle Mesh Ewald Method. Journal of Chemical Physics, 103, 8577-8592. http://dx.doi.org/10.1063/1.470117
- Marqusee, S., Robbins, V.H. and Baldwin, R.L. (1989) Unusually Stable Helix Formation in Short Alanine-Based Pep- tides. Proceedings of the National Academy of Sciences of the United States of America, 86, 5286-5290. http://dx.doi.org/10.1073/pnas.86.14.5286
- Nguyen, H.D. and Hall, C.K. (2004) Molecular Dynamics Simulations of Spontaneous Fibril Formation by Random- Coil Peptides. Proceedings of the National Academy of Sciences of the United States of America, 101, 16180-16185. http://dx.doi.org/10.1073/pnas.0407273101
- Nguyen, H.D., Marchut, A.J. and Hall, C.K. (2004) Solvent Effects on the Conformational Transition of a Model Po- lyalanine Peptide. Protein Science, 13, 2909-2924. http://dx.doi.org/10.1110/ps.04701304
- Graf, J., Nguyen, P.H., Stock, G. and Schwalbe, H. (2007) Structure and Dynamics of the Homologous Series of Ala- nine Peptides: A Joint Molecular Dynamics/NMR Study. Journal of the American Chemical Society, 129, 1179-1189. http://dx.doi.org/10.1021/ja0660406
- Yamauchi, K., Okonogi, M., Kurosu, H., Tansho, M., Shimizu, T., Gullion, T. and Asakura, T. (2008) High Field 17O Solid-State NMR Study of Alanine Tripeptides. Journal of Magnetic Resonance, 190, 327-332. http://dx.doi.org/10.1016/j.jmr.2007.11.006
- Scott, K.A., Alonso, D.O.V., Sato, S., Fersht, A.R. and Daggett, V. (2007) Conformational Entropy of Alanine versus Glycine in Protein Denatured States. Proceedings of the National Academy of Sciences of the United States of America, 104, 2661-2666. http://dx.doi.org/10.1073/pnas.0611182104
- Mezei, M., Fleming, P.J., Srinivasan, R. and Rose, G.D. (2004) Polyproline II Helix Is the Preferred Conformation for Unfolded Polyalanine in Water. Proteins: Structure, Function, and Bioinformatics, 55, 502-507. http://dx.doi.org/10.1002/prot.20050
- Bednárová, L., Malo?, P. and Bouř, P. (2007) Spectroscopic Properties of the Nonplanar Amide Group: A Computa- tional Study. Chirality, 19, 775-786. http://dx.doi.org/10.1002/chir.20462
- Improta, R., Vitagliano, L. and Esposito, L. (2011) Peptide Bond Distortions from Planarity: New Insights from Quan- tum Mechanical Calculations and Peptide/Protein Crystal Structures. PLoS ONE, 6, e24533. http://dx.doi.org/10.1371/journal.pone.0024533
- Platts, J.A., Maarof, H., Harris, K.D.M., Lim, G.K. and Willock, D.J. (2012) The Effect of Intermolecular Hydrogen Bonding on the Planarity of Amides. Physical Chemistry Chemical Physics, 14, 11944-11952. http://dx.doi.org/10.1039/c2cp41716b
- López-Llano, J., Campos, L.A. and Sancho, J. (2006) α-Helix Stabilization by Alanine Relative to Glycine: Roles of Polar and Apolar Solvent Exposures and of Backbone Entropy. Proteins: Structure, Function, and Bioinformatics, 64, 769-778. http://dx.doi.org/10.1002/prot.21041
- Chakarbourty, A., Schellman, J.A. and Baldwin, R.L. (1991) Large Differences in the Helix Propensities of Alanine and Glycine. Nature, 351, 586-588. http://dx.doi.org/10.1038/351586a0
- Levitt, M. (1978) Conformational Preferences of Amino Acids in Globular Proteins? Biochemistry, 17, 4277-4285. http://dx.doi.org/10.1021/bi00613a026
- Rohl, C.A., Chakrabartty, A. and Baldwin, R.L. (1996) Helix Propagation and N-Cap Propensities of the Amino Acids Measured in Alanine-Based Peptides in 40 Volume Percent Trifluoroethanol. Protein Science, 5, 2623-2637. http://dx.doi.org/10.1002/pro.5560051225
- Rohrberg, J., Sachs, R., Lodderstedt, G., Sackewitz, M., Balbach, J. and Schwarz, E. (2008) Monitoring Fibril Forma- tion of the N-Terminal Domain of PABPN1 Carrying an Alanine Repeat by Tryptophan Fluorescence and Real-Time NMR. FEBS Letters, 582, 1587-1592. http://dx.doi.org/10.1016/j.febslet.2008.04.002
- Winter, R., Kühn, U., Hause, G. and Schwarz, E. (2012) Polyalanine-Independent Conformational Conversion of Nuc- lear Poly(A)-Binding Protein 1 (PABPN1). Journal of Biological Chemistry, 287, 22662-22671. http://dx.doi.org/10.1074/jbc.M112.362327
Supporting Information
 (a)
(a)
 (b)
(b)
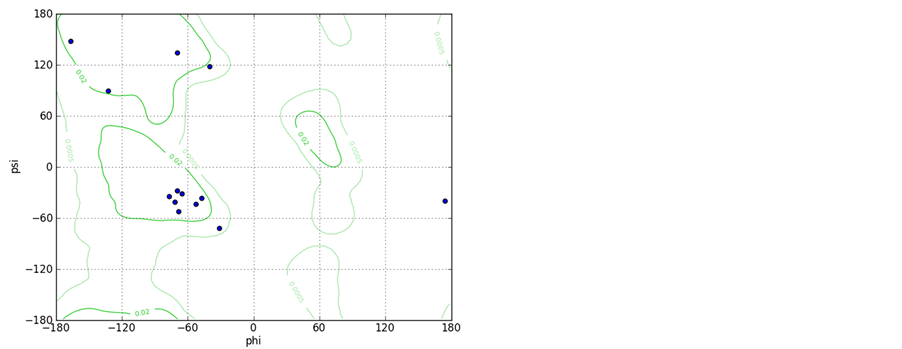
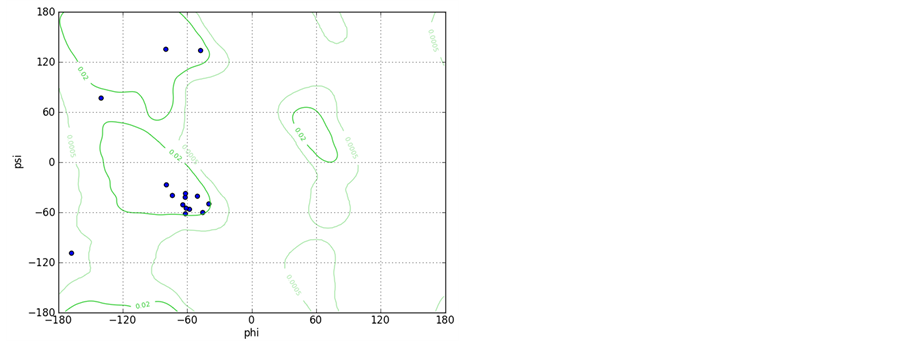
Supporting Information 1. The Ramachandran plots for peptide A and B with starting geometry of the right handed helix shows that the most of the residues fall in helical region of the Ramachandran map.
 Peptide A Peptide B
Peptide A Peptide B
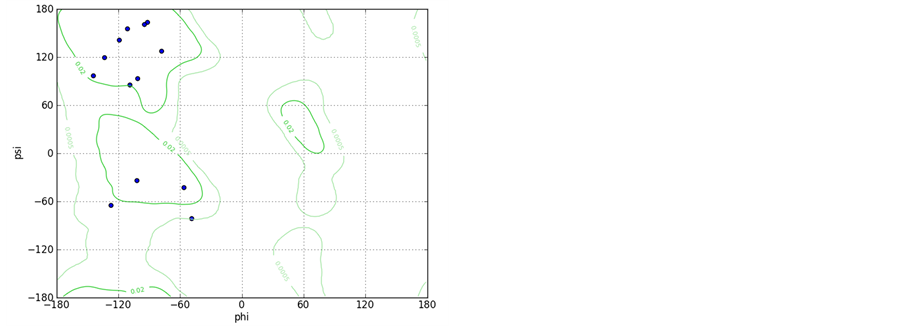
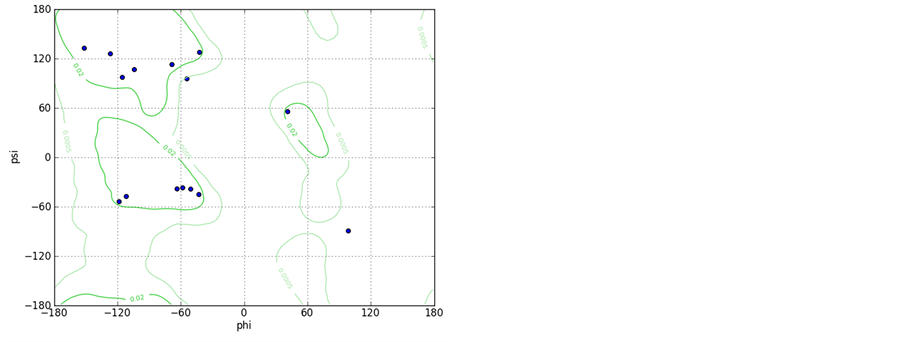
Supporting Information 2. Graphical views of the peptides A (upper left) and B (upper right) after 10 ns with water molecules within 3 Å of peptide surface depicts different types of interaction; Ramachandran plots for both the peptides A (lower left) and peptide B (lower Right) shows increase in the number of the residues falling in right handed helical region for peptide B.
 Peptide A Peptide B
Peptide A Peptide B
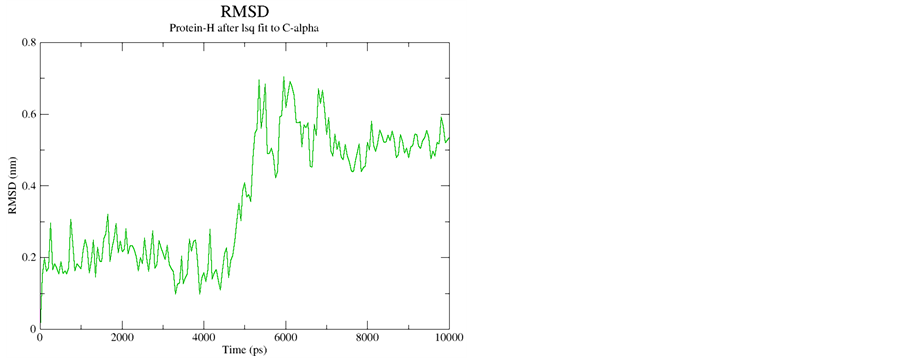
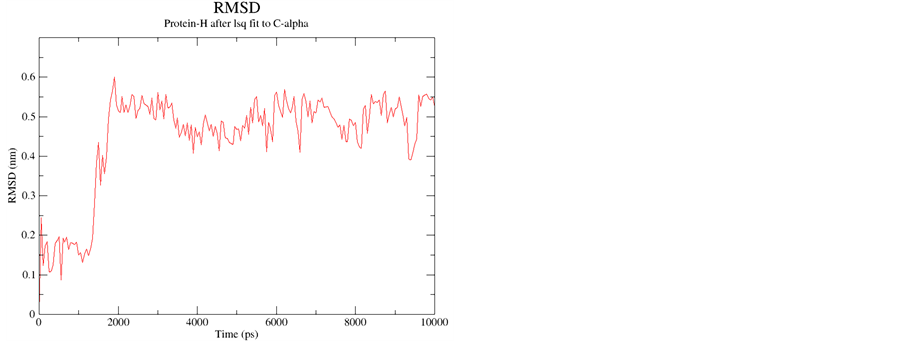
Supporting Information 3. Cα RMSD for Peptide A and Peptide B with starting geometry right handed α-helix depicting equilibrium during 10 ns simulations.
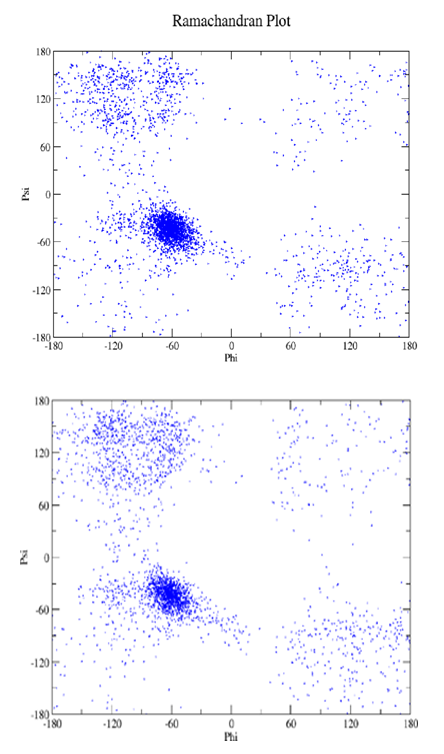
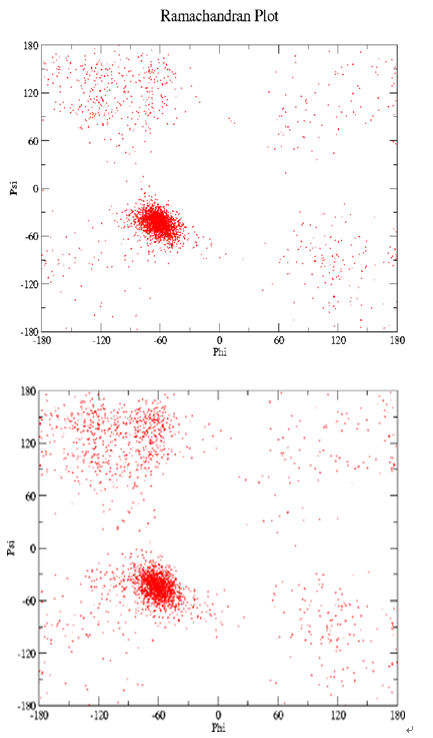 Peptide A Peptide B
Peptide A Peptide B
Supporting Information 4. The distributions of φ, ψ values as a function of time after 10 ns (upper panel) and 20 ns (lower panel) simulations in water for peptide A and B depicting denser helical region for peptide B.