Journal of Environmental Protection
Vol.4 No.3(2013), Article ID:28638,12 pages DOI:10.4236/jep.2013.43028
The Role of Advanced Biological Data in the Rationality of Risk-Based Regulatory Decisions
![]()
Cambridge Centre for Climate Change Mitigation Research, University of Cambridge, Cambridge, UK.
Email: djc77@cam.ac.uk
Received January 7th, 2013; revised February 5th, 2013; accepted March 1st, 2013
Keywords: Health Risk; Environmental Policy; Risk Assessment
ABSTRACT
Advanced biological information such as computational biology, in vitro transformation assays, genome pathway analysis, genotoxicity assays, proteomics, gene expression, cell signaling disruption and hormone receptors offer the potential for significant improvements in the ability of regulatory agencies to consider the risks of the thousands of compounds—and mixtures of compounds—currently unexamined. While the science for performing the assays underlying such information is developing rapidly, there is significantly less understanding of the rationality of using these data in specific decision problems. This paper explores these issues of rationality, identifying the issues of rationality that remain to be developed for applications in regulatory risk assessment, .and providing a draft decision framework for these applications. The conclusion is that these rapid, high throughput methods hold the potential to significantly improve the protection of public health through better understanding of risks from compounds and mixtures, but incorporating them into existing risk assessment methodologies requires improvements in understanding the reliability and rates of Type I and Type II errors for specific applications.
1. Introduction
Human health regulatory risk assessments have relied historically on evidence from whole organism and human epidemiological or clinical studies. Such studies provide a legally well-tested basis for claims of human health impacts, in large measure because the health effects noted in these studies are directly observable as having an adverse impact on lifespan and/or quality of life. These same studies however have significant limitations in the levels of exposure to environmental toxins—and to mixtures of these compounds—for which effects can reliably be identified. The former limitation has been addressed in part through the application of policy defaults such as uncertainty factors to bring a level of precaution to decisions clouded by significant uncertainty as to the effects at environmental levels of exposure that are usually of regulatory interest. The latter limitation has been addressed both through building in margins of safety for each compound and through use of cumulative exposure indices such as the hazard index
(www.epa.gov/oswer/riskassessment/glossary.htm), in the event an individual is exposed to more than one risk agent simultaneously.
Recent advances in both the methods of molecular and cellular biology and in understanding the biochemical steps leading to clinical health effects is allowing development of a wider base of evidence on which risk assessments might rest [1]. This includes an array of methods from molecular systems biology such as computational biology, in vitro transformation assays, genome pathway analysis, genotoxicity assays, proteomics, gene expression, cell signaling disruption and hormone receptors [1]. These methods offer hope of being able to identify effects at stages earlier than clinical disease; to examine exposure-response relationships at much lower exposures than whole organism studies allow; to consider quantitatively the additive, synergistic or antagonistic effects of mixtures of chemicals; and to identify sensitive subpopulations requiring special consideration in risk assessment and risk management.
While potentially powerful tools for improving the scientific basis for risk assessment and beginning to deal with the backlog of tens of thousands of compounds that have not been examined to date because of the time and cost of whole organism studies, such methods have an ill-defined role in regulatory assessments. This lack of clarity stems from the fact that the observations from these methods are not of an adverse effect itself, but rather a precursor to, or perhaps correlate of, such clinically significant effects. This raises a question as to whether the advanced biological information might serve as a substitute for the whole organism data, in a supplementary role, or as “backing support” for claims of risk based primarily in whole organism data. In the first role they will truly increase the rate of production of regulatory risk assessments; in the latter two, it is not clear any increase will take place (although it may be the case that the evidential basis for traditional risk assessments will be improved, or will allow rapid focus on the most potentially hazardous compounds).
Answering this question requires clarity on the role of such advanced biological data in the process of reasoning to a decision on regulations, clarity best brought by an understanding of the rationality of such decisions rooted in the concept of reasonable confidence that 1) a significant risk exists and 2) reduction of a particular compound in the environment will reduce this risk. This reasoning is in part related to scientific rationality (how evidence is used in science to support or detract from beliefs about risk), and in part to the rationality of public decisions (how evidence is used to justify public actions such as risk management). Especially in the case of public decisions, one must consider how reasoning incorporates consideration of the precautionary principle [2,3] to allow action in the face of uncertainty, while simultaneously avoiding unfair restrictions on the use of potential risk agents in economic and other activities when the evidence for a claim of risk does not rise above a reasonable threshold. This balancing of aims has been explored in traditional regulatory risk assessments, but is much less well explored in regard to use of the emerging advanced biological information.
The current paper explores this issue of the rationality of using advanced biological information in a framework of traditional risk assessment. Focus is not on the science itself (although examples are used), but rather on the process of reasoning from evidence to regulatory limits and risk management decisions, and how advanced biological information might play a role in such a process. The discussion is based on recognition that existing risk assessment methods are so resource intensive that they have led to only a few hundred potentially hazardous materials being assessed quantitatively for their risk, in contrast to the millions of compounds known to exist, and the tens of thousands used in daily industrial practice. Advanced biological methods offer the potential to greatly increase the number of compounds for which regulation can be considered, as well as improving the ability to determine risks in communities affected by multiple compounds simultaneously (a hallmark of environmental justice concerns). The trade-off is that these methods gain their power through observation of effects prior to clinical effects, and hence raise important scientific, statistical, philosophical and legal issues of their reliability in identifying significant risks before they are evident in observational studies of human populations exposed to environmental risk agents. The contention is that such advanced methods will take place in a regulatory setting that has already established a particular way of treating the related concepts of uncertainty, confidence and precaution, and that thought will need to be given as to how this new information can either fit reasonably within this framework, or lead to revisions of the framework.
2. Evidence and Regulation
Regulatory processes typically have several distinct stages, with varying requirements for evidence (that a risk exists) and varying degrees of conservatism built in to reflect precaution and a desire for a margin of safety. As an example, consider the process of setting regulatory limits on contaminants in drinking water in the US [4]. Under the Safe Drinking Water Act, the US Environmental Protection Agency (EPA) must develop a Contaminant Candidate List (CCL) through a process overseen by the National Drinking Water Advisory Committee (NDWAC). Those steps are:
1. Prepare a new list every few years (typically 5 years), of the contaminants in water that might be the focus of future regulatory decisions. The goal here is not to establish which contaminants will in fact require regulation, but simply to establish which of the currently unregulated contaminants warrant a further scientific look to better define the risks. There is no claim through the CCL process that the risks can be quantified at present; rather that the evidence to hand is more suggestive of risk for some contaminants than for others. As such, this stage is akin to a screening assessment and not a full risk assessment. The CCL process draws on methods that allow for rapid screening of candidates from amongst the “universe” of many tens of thousands of compounds.
2. Move some of the CCL candidates forward to Regulatory Determination. Here the process is to supplement the CCL screening material with more detailed data on actual levels of exposure, size of affected populations and severity of effect to determine which candidate compounds should be at least considered for environmental regulation. The decision at this stage is not the level at which a regulatory limit (standard) will be set, but rather whether it is worth the effort in time and resources to go through the full risk assessment process in the face of competing demands for those resources.
3. Finally, perform the regulatory risk assessment and take a regulatory decision on an exposure limit for those compounds passing step 2. Here the full process of risk assessment is engaged, including consideration of all available data in performing exposure-response extrapolation; defining health endpoints as adverse and/or critical effects; determining the treatment of uncertainty or perhaps reducing that uncertainty through further research; and defining sensitive subpopulations.
The CCL process offers an object lesson in the connections between the nature of decisions being taken and the strength of evidence required to underpin and justify those decisions. The evidential basis required for decisions—both legally and rationally—becomes stronger as one moves from the first to the third step of the process above because the implications of each of these steps increases. This raises in turn significant questions about the potential role in risk assessment of advanced biological methods that allow for more rapid development of information via high-throughput assays, but are also increasingly removed from direct observation of adverse effects in whole organisms, observation that has formed the legal foundation of regulatory decisions.
So long as one is simply defining compounds deserving a “further look”, the regulatory, regulated and affected communities tend to be comfortable with methods that have the advantage of low cost, high-throughput and some degree of conservatism in the sense of errors being more likely to lead to over-identification rather than under-identification of potential risk agents. These advanced methods are already being used in programs such as REACH in the EU [5], and are at least hinted at in the discussions surrounding re-authorisation of the Toxic Substances Control Act (TSCA) in the US [6]. But as one gets closer to actual regulatory decisions in the third stage, the simplicity, low cost and high volume of these advanced methods become less compelling reasons for their use as a significant basis for decisions, and the focus turns more to the ability of such information to produce reliable estimates of risk, or to enhance the ability of traditional risk assessments to do so.
Existing approaches to risk assessment based largely on whole-animal or human epidemiological studies, however slow and resource intensive they may be, have withstood tests in the court. They have been formalized in methods that explicitly and often quantitatively address uncertainties in risk estimates, how those uncertainties should be treated in a framework of the precautionary principle, and how uncertainty is related to the confidence underlying risk assessment and risk management claims. If advanced biological methods are to play a significant role in risk-based assessments and decisions, there will be a high evidential barrier to cross in demonstrating that they are reliable and lead to reasonable protection of public health (meaning neither insufficient nor excessive levels of precaution).
The problem here is that the attempt to avoid coming into conflict with traditional and legally tested approaches to regulation may cause the advanced biological data to be treated in the same way as current whole organism information used in regulatory decisions. The tendency will be to simply replace direct observation of clinical effects from traditional risk assessment (“critical effects” in regulatory risk terminology) with the observation of the precursor effects of Figure 1, and then to use the methods of uncertainty factors, precaution, etc. that developed historically to overcome the limitations in whole organism data. The question that arises is then: under what conditions might this be acceptable as a reasonable basis for establishing risks requiring regulation, and how might the process of regulatory risk assessment be adjusted to allow use of these potentially powerful insights into the process of disease development?
At present, most regulatory decisions on at least noncancer effects are based on a process [7] that 1) examines effects in humans or whole animals to identify a No Observed Effects Level (NOEL) or Lowest Observed Effects Level (LOEL); 2) determines which effects are to count as truly adverse rather than merely present, therefore leading to identification of a No Observed Adverse Effects Level (NOAEL) or Lowest Observed Adverse Effects Level (LOAEL); (3) establishes some form of Point of Departure based on the NOAEL or LOAEL; and 4) applies uncertainty factors to account for uncertainty in the exposure at which the adverse effect either ceases to be adverse (e.g. dropping in severity so it is no longer considered adverse) or ceases to be present in a signifi-
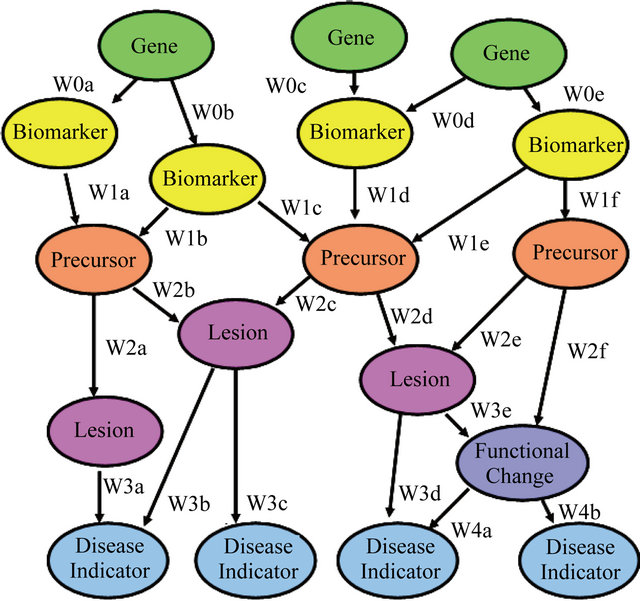
Figure 1. Advanced biological information, as used in this paper, refers to information on effects subsequent to, but part of the causal chain leading to, a clinically observable effect shown here as a disease indicator. Such information can be collected at much lower exposures than the clinical effects and with much lower costs and higher volumes.
cant fraction of the population, and applies other modifying factors to account for inter-species and intraspecies differences (e.g. dose modification due to pharmacokinetic or pharamcodynamic differences). Much of this treatment of uncertainty comes from the facts that one often cannot measure effects in humans directly, therefore requiring use of experimental animals, and from statistical noise in whole animal or human epidemiological/clinical studies becoming significant and disabling at exposures approaching those found in the environment. The uncertainty factors are a means of producing high levels of confidence that a regulatory limit will still be protective of public health even in the face of this uncertainty that prevents reliable observation of effects at levels and conditions of exposure of regulatory interest.
Much of the emerging advanced biological data, by contrast, can be extended to significantly lower exposures than is the case for traditional whole organism studies. This will drive down—perhaps by orders of magnitude—the NOELs and LOELs. Uncertainty factors were applied to these Points of Departure in the past based on the argument that regulators can’t “see” down into the low exposures of regulatory interest, and so the uncertainty factors produced increased confidence that the proposed regulatory limit is protective of public health. The introduction of advanced biological methods, however, allows regulators to “see” down into this region of direct environmental interest, and so it would be inappropriate to continue to apply these same uncertainty factors. Nevertheless, there will be strong pressure to do precisely that, if for no other reason than that the courts, regulators and the affected communities have become accustomed to seeing these uncertainty factors applied, and have come to expect them as the sign that precaution is being exercised. The solution to this problem is to either change the magnitude of uncertainty factors applied when advanced biological information is the basis for a Point of Departure so the same degree of confidence in protection of health is achieved, or to apply the advanced biological information as supplement to, but not surrogate for, the observations of clinical effects.
Additionally, the application of uncertainty factors to Points of Departure based on whole organism data has several decades of research underlying understanding of the degree of conservatism and margin of safety built in through their application. Research by Dourson et al. [8] and others allows approximate identification of the percentiles of both uncertainty and inter-subject variability distributions generated through use of specific uncertainty factors. This allows regulators to ensure regulatory limits are neither insufficiently nor excessively protective in the face of uncertainty. These same probabilistic interpretations and understandings are not yet available for advanced biological information, and so it is not clear how their application will be justified probabilistically in assessing resulting margins of safety.
To frame this issue, the current paper turns now to the reasoning process underlying regulatory risk assessments, and explores how advanced biological information might find either supplementary or new roles in that process.
3. Evidence and Risk
An advantage of advanced biological information and methods is that they hold the potential to harmonise cancer and non-cancer risk assessments, placing these both on a probabilistic basis. There has always been a rather arbitrary distinction between these categories of effect, with the former treated probabilistically and the latter as a threshold phenomenon [9]. Protection against noncancer effects arose during a time when industrial hygiene was rooted firmly in the idea of a distinct threshold for effect; the resulting regulatory framework was one rooted in the idea of reducing exposures below this threshold so the clinical effect would not appear. Cancer risk assessment, on the other hand, is a more recent process, and was affected significantly by the health physics community for which radiation effects are considered largely stochastic at low doses. Hence the introduction of probabilistic concepts of risk in the latter case but not the former, with regulation of carcinogens being treated as a problem of reducing the probability of cancer below an acceptable level.
There is no fundamental scientific reason for these two classes of effect to be treated differently. Each has an underlying exposure-response curve (perhaps different ones for different groups of individuals). At some point along the exposure axis, the probability and/or severity of the effect in a population goes above a de minimus level and regulation is warranted to protect public health. The curve may not be monotonic in the sense of always increasing as exposure is increased, and may be quite complex, but it is a curve nonetheless. The sole difference is ambiguity as to whether the probabilities calculated are truly stochastic (i.e. the probability that a given individual develops the effect) or refer instead to fractions of a population with an effect (which would be a property of the population, not of any given individual).
Through harmonisation, it is possible for both categories of effects to establish a target probability and/or severity of effect to be protected against by regulation, and to set the regulatory limit accordingly. Any form of treatment of uncertainty can then be applied to this result to increase the confidence that a regulatory limit remains protective even if the risk estimate is proven to have been incorrect. This latter concern has been addressed in traditional regulatory risk assessment by uncertainty factors that make it much more likely that errors introduced by inadequate data cause a regulatory standard to be overly protective than underprotective of human health.
Under this harmonization of cancer and non-cancer risk assessment, advanced biological information and methods could play any of three roles in assessments and decisions:
1. They could be used to determine the shape of the exposure-response function below the levels at which clinical effects can be seen in the whole organism data, as well as to determine which sub-populations might have distinct curves representing the more sensitive members of a population. The advanced information would establish the model form for extrapolation (e.g. establishing whether it is linear, quadratic…), but not otherwise provide parameters for the models. Numerical values for parameters would instead be obtained from the whole organism data by fitting the exposure-response function discerned from the advanced data to the results of the whole organism data. For example, the mathematical shape of exposure-response curves from cell transformation studies on radiation have been used to provide the functional form for extrapolation equations in whole organism carcinogenicity, but all parameter values in the equation have been obtained by fitting to the whole organism data [10].
This approach has been termed formal relevance [11], where there is reasonable confidence that the mathematical form is the same at both the clinical and subclinical biological levels of Figure 1, but where there is not reasonable confidence that the numerical values of parameters apply at all levels. Such use of advanced biological information requires reasonable demonstration that the mathematical form of the exposure-response relationship is the same for both the advanced biological effect (e.g. lesions in Figure 1) and the clinical effect of regulatory interest. The primary problem is that there are intervening steps in the process of disease between the advanced biological effect and the clinical disease. If any of these intervening steps are non-linear and/or nonmonotonic, such as would be the case if repair mechanisms are operating in these intervening steps [12], then the mathematical form of the exposure-response relationship in the advanced biological data will be less complex that that in the whole organism data. There is at present inadequate testing and demonstration of this assumption, and so the rationality of using the advanced biological information in this way has not yet been established.
2. They could be used as in item 1, but in addition at least some of the parameters within the extrapolation model—parameters that relate directly to processes measured by other advanced biological information—would be determined from those advanced biological studies, although others within the model would be determined by calibrating the extrapolation function to the whole animal data as in the first option. For example, extrapolation models for radiation based on cell transformation studies have been given prior estimates of cell killing influences based on in vitro cytotoxicity studies, with remaining parameters discerned from fitting to whole organism data [12]. This approach has been termed numerical relevance [11], where there is reasonable confidence both that the mathematical form is the same at both the clinical and sub-clinical biological levels of Figure 1, and that at least some of the numerical values of parameters apply at all levels. This requires evidence that numerical relevance holds across the various contexts (whole organisms, in vitro, blood samples, etc.) in which assays are performed. At present, evidence is inadequate to establish the reliability of this assumption.
3. They would be used as the primary basis for estimates of probability and/or severity of health effect, generating the exposure-response curves directly without recourse to the whole organism data. This approach is a full expression of both formal and numerical relevance, applicable where there is reasonable confidence that both the mathematical form and the parameter values in the extrapolation equation are the same at both the clinical and sub-clinical biological levels of Figure 1.
What might be the evidential requirements to allow the advanced biological data to play any of these three roles? The key lies in understanding the nature of evidence and rationality in regulatory decisions, and how that rationality provides a reasonably compelling basis for the belief that a risk exists and that a particular regulatory limit is protective of health. Rationality of regulatory risk assessment can be founded in any of five categories of evidential reasoning [13,14], examined below. Throughout the discussion, consider the modes by which a decisionmaker might form a claim about whether tropospheric ozone produces an adverse effect at 20 ppm:
1. Direct Empirical: In this case, there are data directly displaying whether there is (or is not) an effect at 20 ppm in the organism of interest (humans will be assumed here), and in a subpopulation of interest (e.g. asthmatics). The quality of the claim concerning the level of risk of asthmatic attacks at 20 ppm then rests on the quality of the methods used to produce the data (sample size, control for confounding, exposure estimation, etc.). For ozone, these data traditionally have been human epidemiological studies. For advanced data—such as measures of oxidative stress in cells—to play a similar role here, they would need to form the basis for this direct observation of adverse effect, replacing the traditional data on prevalence or incidence of asthma in a population with the measure of oxidative stress. This would in turn require asserting that the advanced data are measuring an effect (oxidative stress) that is itself to be considered adverse in the regulatory sense. Within the existing framework of risk assessment, this is unlikely to occur, and the advanced biological information would at best play the roles noted in items 3 or 4 below. An exception is noted in Section 4.
2. Semi-Empirical Extrapolation: In this case, there might be whole organism data on incidence or severity of asthmatic attacks at perhaps 50, 100 and 200 ppm, but not below 50 ppm. There is, however, a clear exposureresponse relationship observed in the data, and that relationship could be followed down to 20 ppm. The evidence of a risk at 20 ppm is not then directly empirical, but there is nonetheless a strongly empirical foundation due to the data at higher exposures. The epistemic quality of the claim of a risk at 20 ppm then rests on the quality of the methods used to produce the data as in the Direct Empirical mode of reasoning, and the clarity of the exposure-response curve seen in those data.
For advanced data to play a role here, they either would need to form the basis for the observations at higher exposures, which would in turn require asserting that the effect (e.g. oxidative stress) measured is itself adverse and can form the sole basis of the semi-empirical extrapolation, or would need to provide a more reliable basis for developing the mathematical form of extrapolation functions below the lowest observed exposure in the whole organism studies. The latter approach requires evidence that the form of the exposure-response function is the same for the advanced effect and the adverse effect in the whole organism; i.e. it requires a demonstration of at least formal relevance as defined previously.
3. Empirical Correlation: In this case, the adverse effect of interest (e.g. an increase in prevalence of asthma) is not observed, but some other advanced biological effect (e.g. a biochemical marker of oxidative stress) is observed at 20 ppm. Further, that precursor effect has been shown to be correlated with, or prognostic of, the adverse effect itself. The epistemic quality of the claim of a risk of clinical effect at 20 ppm based on the advanced biological information then rests on the quality of the methods used to produce the advanced biological data (sample size, control of confounding, etc.), and the quality of the correlation (strength and specificity) between appearance of the precursor effect and the clinical effect. For advanced data to play a role here, there would be a need to characterize the strength and specificity of the correlations used, which is the same as characterizing the prognostic reliability of the advanced data for the adverse effect of interest. We return later in this section to the issue of prognostic reliability 4. Theory-Based Inference: In this case, the adverse effect of interest (e.g. an increase in the prevalence of asthma) would not be observed, but some other advanced biological effect (e.g. a biochemical marker of oxidative stress) is observed at 20 ppm and that effect is established scientifically to play a causal role in the adverse effect. The epistemic quality of the claim of a risk of clinical effect at 20 ppm then rests on the quality of the methods used to produce the advanced biological data (sample size, control of confounding, etc.) and the degree of rational support for the causal theory connecting the precursor and clinical effects. For advanced biological data to play a role here, there would be a need to characterise the evidential support for this causal theory across a wide variety of compounds, including an assessment of whether the measured effect was a necessary and/or sufficient condition to trigger the cascade of events leading to the adverse effect of regulatory interest.
5. Expert Insight: In this case, the advanced biological data are not used to calculate a risk at all. Instead, they are used to support a claim (informed, but subjective) by a reflective expert that the adverse effect is or is not expected to appear at 20 ppm. This mode of reasoning is not considered further here as it is judged inadequate to provide a reasonable evidential basis for risk estimates.
These uses of the advanced biological data in any of the modes of evidential reasoning however raise issues of coherence of findings across compounds, across levels of exposure and across species.
To see what is meant by this issue of coherence, consider the case of a single compound (ozone) and species (humans). Turning first to Empirical Correlation, imagine that there are no direct measurements of the adverse effect (increased prevalence of asthma) in humans due to ozone exposures at 20 ppm. Instead, there are measurements of markers of oxidative stress at a variety of levels of exposure. Developing the necessary correlation between these two biological levels of effect (asthma and oxidative stress) requires data from an array of compounds, and a demonstration that the strength and specificity of the correlation are high enough to pass standards of minimal epistemic status [13] that can then withstand legal challenges. No amount of collection of data on ozone itself will be sufficient to establish the reliability of the correlation, since strength and specificity are properties of defined sets of compounds and not of any compound individually.
The use of advanced biological data in Empirical Correlation rests, therefore, on the prognostic ability of these data in supporting the claim that a regulator might reasonably expect the truly adverse effect (asthma) to be present even if only the precursor effect (marker of oxidative stress) has been observed. A suite of such markers will need to be examined, since lack of response to one marker does not imply the compound produces no risk of the clinical effect. Recent research makes it clear that there can be many pathways, mechanisms and modes of action leading to the same disease state, and that any one stage of a pathway may be neither necessary nor sufficient to cause the next stage [15,16]. Instead of thinking of a particular event (such as oxidative stress) as uniquely causing the next stage (such as inflammation, and from there to clinical effect), that precursor event will have only a probabilistic, prognostic ability to foresee any of the more overtly clinical stages taking place later in a causal sequence of events leading to disease.
An example can be seen in Figure 2, produced here for perchlorate. In this figure, there are four stages of the clinical disease: 1) inhibition of iodine uptake into the thyroid; 2) down-regulation of T3 or T4 (or up-regulation of TSH); 3) altered development or thyroid hyperplasia and 4) birth defects or tumours as clinical, adverse effects to be regulated. Advanced biological data can provide information on the first three stages. The question is how well appearance of any or all of these stages is prognostic of the appearance of the fourth, clinical stage of the disease pathway.
But such one-to-one correlations (between an individual advanced biological effect and the disease) as predictors of clinical effects would fail to make use of the full prognostic power of suites of advanced biological assays, whose high throughput nature allows collection of data throughout the chain of events in Figure 1 or 2. This prognostic power can be improved significantly by considering the combined impact of data on several of the precursors simultaneously. By setting prognostic ability within a framework such as Bayesian analysis [17,18], it is possible to combine information at many stages of action in a pathway to calculate the overall confidence in the clinical, adverse effect at the end of that pathway. The result is a methodology that reflects quantitatively and probabilistically the coherence of findings on a given compound across levels of effect, as well as providing uncertainty distributions on the risk of disease. This is where the utility of the advanced biological data becomes most evident, although the databases required to develop the joint probabilities and/or the Bayesian network capabilities has yet to be developed.
The situation concerning the rather poor state of development of the rational base is similar for TheoryBased inference. Theories are generalised statements about how specific classes of events (oxidative stress, inflammation, asthma) are related causally. If the theory is not generalisable, how is one to know whether it is reliable to invoke it when using advanced biological data for the case of assessing risks from ozone? Confidence in invoking a particular theory justifying use of the advanced biological data will require testing the theory across a wide class of compounds and exposure conditions, to determine whether a consistent theory emerges that accounts for all of these tests simultaneously, or explains why some compounds and conditions might violate the causal scheme of the theory. Again, these are early days for such tests, and so the rationality of employing such theories is not yet established.
The use of advanced biological data in semi-empirical extrapolation requires a further consideration of rationality: the “meaning” of the data within any extrapolation function (the desideratum of clarity of conception in rationality [11]). Consider that exposure-response extrapolation equations usually employ linear, quadratic or multi-stage models. In such models, the coefficients of
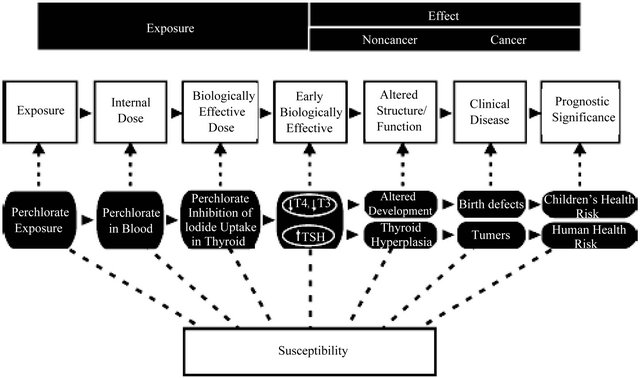
Figure 2. The stages of disease generally (white boxes) and in the specific case of perchlorate (black boxes in the row below).
the terms have no essential scientific “meaning”. For example in the multi-stage model often used in cancer risk assessment, the α, β, etc. coefficients don’t “mean” anything; they are simply fitting coefficients to the whole organism data. As a result, it becomes impossible to use the advanced biological data to gain a better estimate of these coefficients such as might be used in putting Bayesian priors on the coefficients before fitting to the whole organism data, since it is not clear which data in the pathway prior to appearance of the full clinical effect relates to any particular term or coefficient in the model.
Why is this issue important? Consider the case of a multi-stage model and Semi-Empirical Extrapolation. This model is fit to whole organism data (clinical disease in Figure 1 or 2) at higher levels of exposure than those of interest in regulation. Rationality requires some test of the model to establish its reliability as guide to estimates of risk at lower levels of exposure. If the multi-stage model has many terms, and if there are no prior constraints on reasonable values for the coefficients in these terms, then the model has large degrees of flexibility. This means it can fit almost any set of data. This in turn means that its ability to fit any particular set of data (such as those for the whole animal effects) is not a test of the truthfulness or reliability of the model. It provides no confidence that the model is in some sense correct and therefore can be followed reliably downwards in exposure into the region of regulatory interest.
Getting around this problem requires constraining the coefficients of the model based on advanced biological data from the stages or processes in the pathway to full adverse effect (the stages to the left in Figure 2, or above in Figure 1). This requires, however, that the terms in the extrapolation model have “meaning” in the sense described earlier, so the assessor can determine which specific set of advanced biological data apply in constraining any particular coefficient prior to fitting to the whole organism data. This in turn calls for greater development and testing of biologically-based models of exposureresponse [19].
A core feature of these biologically-based models is the role of dosimetry. Figure 2 shows a stage of assessment related to the biologically effective dose, which is the amount of the active form of a compound that reaches a target cell (here, the cells of the thyroid). A key difference between advanced biological data and whole organism data is that the latter are collected in a biological context (a body) that is significantly more complex in terms of the relationship between application of the original compound (here, perchlorate) and delivery of the active form of that compound to the target cells. This difference can affect not only the mode and mechanism of action at the two levels of biological organization (affecting the strength and specificity of correlations), but also parameters in an exposure-response relationship (reducing the reliability of any attempt at employing numerical relevance of the advanced biological data). In traditional risk assessment, this difference is accounted for through application of modifying factors. In risk assessments utilizing advanced biological data, either the dosimetric differences between the biological contexts of measurements (whole animal versus advanced biological assays) must be accounted for in explicit application of pharmacokinetic and pharmacodynamic models, or these differences treated as noise in empirical correlations that will reduce the strength and specificity of these correlations. Note that these differences are caused precisely by the strength of the advanced biological methods: their high volume and rapid throughput, characteristics that usually are obtained by making the measurements in a context different from that of a whole organism.
4. Effects, Predisposition, Cumulative Risk
The discussion turns next to a promising future direction for use of advanced biological data. In the traditional approach to regulation, a compound is considered to produce a particular, critical effect that is in and of itself adverse. That effect might be thyroid cancer or asthmatic attacks. The risk assessment is aimed at exploring all of the possible adverse effects a person might experience, and determining whether any of them are produced directly from exposures to the compound.
More recent research has shown that the effects of many risks agents may be indirect [20]. They may, for example, cause up or down regulation of genes, or genetic instability, that then predispose an individual to effects from other risk agents. This raises a complicated decision problem that has yet to be fully addressed in regulatory decisions. What is it that an environmental agency is protecting the public against? One thought is that it is protecting the public against specific effects: the critical effects that appear in traditional regulatory risk assessments such as those summarized in the USEPA’s Integrated Risk Information System.
There is, however, another potential approach to decisions based on how a compound causes indirect effects that don’t technically count as the critical effect for that particular compound. Imagine a case in which the oxidative stress of ozone up or down regulates certain genes that predispose an individual to effects from other air pollutants such as particulate matter (PM). The ozone doesn’t directly cause a disease from the PM, and will only show its influence if an individual is subsequently exposed to PM. The proximal cause of that disease is the PM. The ozone is a distal cause because it has contributed in moving the individual into a different state of sensitivity to PM. But distal cause doesn’t move the individual all the way to disease; so long as regulatory risk management is based primarily on control of proximal causes, this increased power of advanced biological information will be less evident.
In a sense, this situation is already reflected in regulatory risk practice. It is found in the idea of a sensitive subpopulation. One of the sensitive subpopulations for PM exposures is asthmatics. If ozone makes people more sensitive to compounds that cause asthmatic attacks, this is reflected in PM regulations by identifying a new subpopulation of people who are highly asthmatic. This shift of sensitivity won’t, however, necessarily be reflected in regulations on ozone; it will instead drive down the regulatory limit on PM where that limit is set on the basis of the sensitive subpopulation. Thus advanced biological information from one compound may be useful in defining whether that compound can move individuals to intermediate stages of disease, sensitizing them to the actions of other compounds that complete the transitions to full disease. A sensitive subpopulation for the second compound would then be defined as that subpopulation for which advanced biological information suggests movement to the intermediate stages has already taken place by exposure to the first compound.
When moving regulatory risk constructs backwards from adverse effect to the predisposing conditions for that effect, the increased role of advanced biological data will naturally cause a debate on the nature and aims of the regulatory process. It will cause regulators to ask more carefully whether their mission is to prevent a compound (e.g. ozone) from directly producing an unacceptably large risk of the critical effect for that compound, or whether its mission might include preventing an individual from being exposed to one particular compound (e.g. ozone) at levels that predispose that individual to the effects of yet other compounds (e.g. PM). The details of such a concept of attributable risk as applied in regulatory standards have yet to be addressed in policy, and so the rationality of this application is not yet established.
The issue is made more complicated by the nature of regulatory decisions, which generally are designed to apply nationally and equitably. If advanced biological information shows that there is significant interaction between compounds through sensitizing, then the risk caused by exposure to a compound such as ozone will depend on the magnitude of exposure to other compounds such as PM that allow this sensitivity to be expressed; i.e. the risk will be context dependent. This in turn raises the specter of a different allowed limit on exposure to ozone (as an example) in different parts of a country, depending on the exposures to other compounds such as PM. The regulatory process for human health risk is not yet structured to deal with this instance of interactions between compounds or the resulting context dependence of regulatory limits on exposure.
Applications of advanced biological information may be most evident first in cumulative risk assessment. At present, regulatory risk assessment for mixtures tends to be dominated by two approaches: dose additivity and effect additivity. Note first that both are based on the idea of additivity, therefore ignoring the possibilities of synergistic (the total effect of two compounds being greater than the sum of the two individual risks) or antagonistic (the total effect being less than the sum of the two) effects. Effect additivity has been applied to the clinical effect, introducing all of the limitations of developing the base of whole organism data required for such studies. This is a severe limitation when one considers the immense number of permutations of exposure to different compounds that can exist in complex environments where individuals might be exposed to hundreds of compounds simultaneously.
The rapid, high throughput nature of advanced biological information means assays can be performed on many of these permutations, examining the ability of these mixtures to produce any of the intermediate stages of disease such as biomarkers in Figure 1. The assumption must be made that how a cell was caused to display a given biomarker does not affect the prognostic ability of that advanced biological effect for higher stages of the disease, an assumption that has not been tested to date. And all of the issues raised earlier on the nature of rationality in using prognostic tools apply. But if those issues can be resolved reliably, giving reasonable confidence that they provide a sound and protective basis for regulatory risk assessments, the ability to conduct cumulative risk assessment will be greatly improved.
5. Error Rates
We turn finally to the issue of under and over development of margins of safety and of precaution. In decision problems, there are two kinds of errors in applying advanced biological information that weaken the rationality of a specific decision process relying on that information [21]:
Type I error, which in this context means stating that a compound will produce an adverse effect—based on a positive result from the advanced biological data—when it does not in fact produce that effect.
Type II error, which in this context means stating that a compound will not produce an adverse effect—based on a negative result from the advanced biological data— when it in fact produces that effect.
Both of these types of error have consequences. Type II errors can arise either because the statistical properties of the data collection were inadequate to identify an effect at some stage of the pathway to disease, or because the compound does produce changes that are part of the causal chain or network leading to the adverse effect but not by causing changes in the particular stage, biomarker, genetic change, etc. analysed in the advanced biological data. The result in either case is a false negative.
Type I errors appear to be less problematic at first glance. If anything, they lead to incorrectly identifying a compound as posing a risk. They will arise either because the statistical properties of the data collection were inadequate, producing a false positive, or because the measurement is of a precursor effect with low specificity.
The argument might be that these Type I errors are acceptable—more so than Type II—because they are more precautionary and lead to protection of public health even if they lead to incorrect beliefs. But Type I errors do have adverse consequences. They cause limited resources to be devoted to compounds that are of little or no risk, diverting those resources from other compounds posing higher risk. They cause regulatory costs to an economy, which in turn has health implications due to economic inefficiencies that can decrease welfare [22]. They are not, in short, truly precautionary.
As mentioned, the promise of advanced biological methods lies in their rapid and high throughput nature, enhancing their ability to explore the web of diverse causal events leading to a suite of diseases. This means many dozens of assays could be performed on a given compound. On the surface, this appears to be a laudable advance. A richer base of scientific information should improve the evidential base for risk assessments and reduce the need to apply uncertainty factors related to inadequate data. However, there remain unresolved issues of how to treat problems of “data dredging” in dealing with rich bodies of data, in which decision rules as to when a body of data suggests a risk exists can lead to false negatives and false positives in such decisions.
In traditional risk assessment, this issue is addressed through the criterion of statistical significance. The NOELs and LOELs described previously are not simply levels of exposure at which an elevation in effect is observed, but rather levels where that elevation has statistical significance. There are well established methods for calculating statistical significance in a single measurement [23]. The very rich set of data on advanced biological effects, however, means that one must understand not only the rates of false positives and false negatives in a particular measurement, but the rate in complex ensembles of measurements across diverse endpoints. The rationality of applying such arrays of data in risk assessment requires development of decision rules that recognise that despite a rate of false positives that is low for any single measurement (the classical role of measures of statistical significance), there can be a much higher probability that one or more of the assays will show a false positive in the array. Even if there is no true relationship between exposure to a compound and disease, the partial randomness of assay results will produce at least one or more false positives in a suitably large array.
The frequency and implications (for decisions) of both Type I and Type II errors in complex arrays must be characterised for the use of advanced biological information and methods if the rationality of their use is to be strong, and suitable decision rules developed analogous to the use of statistical significance in traditional risk assessment. At present, neither rates of errors nor decision rules are well characterized for arrays of advanced biological information.
6. Conclusions and Discussion
The discussion in this paper suggests that advanced biological data might play a significant role in improving the rationality of regulatory assessments and decisions, but that there remain important issues to be resolved. The most significant conclusions are that:
• It will be problematic if methods of dealing with uncertainty developed for application to whole organism data, such as application of traditional uncertainty factors to Points of Departure, are applied uncritically to PODs obtained from advanced biological data. Such an approach would be likely to lead to unnecessary degrees of conservatism in risk-based standards, and given strong legal scrutiny.
• Any use of advanced biological data will require a significant research effort to examine their prognostic ability across diverse settings (species, levels of biological organisation, compounds, exposure levels, exposure routes), with well defined sets. The science is in only the earliest stages of this process of developing and assessing the required database.
• For advanced biological data to be fully useful in informing Semi-Empirical Extrapolation from high to low exposures, or across species and exposure routes, it will be necessary to replace simple curve fitting models (such as the multistage model) with more “first-principle” models in which terms have meaning that can show where specific bodies of data can be used to constrain model forms and/or parameter va- lues. It will be crucial to incorporate pharmacokinetic and pharmacodynamic considerations into such models.
• Regulatory decisions are often based on the concept of Weight of Evidence (WOE [24]), where the analyst considers not one body of data but rather the cumulative impact of multiple—potentially conflicting— bodies of data. WOE determinations are already complex and subjective (however informed they might be by expert judgment), with external reviewers of a regulatory decision often unable to understand how a particular claim in a risk assessment is supported by the full body of evidence. Since advanced biological data can be generated in large numbers, there is even greater opportunity to selectively report those studies that are supportive of a claim to risk (or against such a claim). A more formal WOE procedure will be needed if the addition of advanced biological data is not to further cloud this picture.
• At least in the short term, meaning over the next decade, advanced biological data are not likely to supplant traditional whole organism data, or be used when the latter are not available, but will rather be supplementary to the traditional data. The most obvious examples are in using the advanced biological data to improve extrapolation equations and in identifying sensitive subpopulations.
• The shortest term applications of advanced biological data are likely to be in rapid screening approaches such as the first step in the CCL process. This is because the conservatism built into such data (since they are data on precursor effects that are not fully established to lead reliably to the critical adverse effects of regulatory interest) makes them at present a less contentious means of screening than of providing quantitative estimates of risk.
Finally, a decision framework is needed to define how and when advanced biological information can supplant or supplement traditional, whole organism data. A candidate decision framework for the use of advanced biological data is shown in Figure 3 below. This framework was developed initially for the NexGen programme of the US EPA [15], which is leading the US effort at incorporating such data (called Next Generation or NexGen) into the regulatory process. It contains within it all
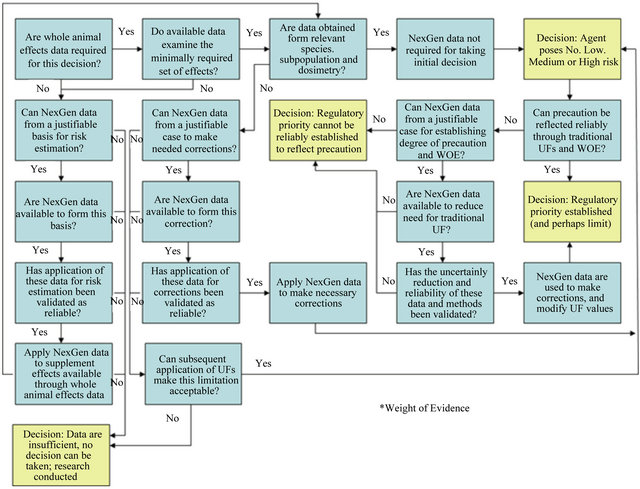
Figure 3. A candidate decision framework for determining when advanced biological data might play a role in risk assessment, and the nature of that role. Development of the decision rules and analytic methodologies for the various Yes/No decisions in the framework remains the challenge of future research if advanced biological data are to enhance the rationality of risk assessments. “Corrections” in this figure refer to dosimetric, exposure-response and other corrections applied to the results of whole organism data.
of the potential applications of advanced biological data within a full risk assessment.
7. Acknowledgements
The author acknowledges the US Environmental Protection Agency for providing significant support for the work reported here, via a subcontract through ICF International. Recognition is also given to the valuable discussions with Ila Cote and Annie Jarabek of the US Environmental Protection Agency.
REFERENCES
- National Research Council, “Science and Decisions: Advancing Risk Assessment,” National Academy Press, Washington DC, 2009.
- D. Crawford-Brown, “The Precautionary Principle in Environmental Regulations for Drinking Water,” Environmental Science and Policy, Vol. 14, No. 4, 2011, pp. 379- 387. doi:10.1016/j.envsci.2011.02.002
- European Commission, “Communication from the Commission on the Precautionary Principle,” COM(2000)1, Brussels, 2 February 2000.
- CFR, “Protection of Environment, CFR Parts 1-49, Code for Federal Regulations,” US Environmental Protection Agency, Washington DC, 2003.
- European Parliament, “Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH),” 2006.
- US Senate, “Reauthorization of the Toxic Substances Control Act: Hearings before the Subcommittee on Toxic Substances, Research, and Development of the Committee on Environment and Public Works,” United States Senate, One Hundred Third Congress, Second Session, May 1 2011.
- D. Crawford-Brown, “Risk-Based Environmental Decisions: Methods and Culture,” Kluwer Academic Publishers, Norwell, 1999. doi:10.1007/978-1-4615-5227-7
- M. Dourson, S. Felter and D. Robinson, “Evolution of Science-Based Uncertainty Factors in Noncancer Risk Assessment,” Regulatory Toxicology and Pharmacology, Vol. 24, No. 2, 1996, pp. 108-120. doi:10.1006/rtph.1996.0116
- D. Crawford-Brown, “Mathematical Methods of Environmental Risk Modeling,” Kluwer Academic Publishers, Norwell, 2001. doi:10.1007/978-1-4757-3271-9
- S. Moolgavkar, “The Multistage Theory of Carcinogenesis and the Age Distribution of Cancer in Man,” Journal of the National Cancer Institute, Vol. 61, No. 1, 1978, pp. 49-52.
- D. Crawford-Brown, “Scientific Models of Human Health Risk Analysis in Legal and Policy Decisions,” Law and Contemporary Problems, Vol. 64, No. 4, 2001, pp. 63-81. doi:10.2307/1192291
- M. Mebust, D. Crawford-Brown, W. Hofmann and H. Schoelnberger, “Testing of a Biologically-Based Exposure-Response Model from in vitro to in vivo Conditions,” Regulatory Toxicology and Pharmacology, Vol. 35, No. 1, 2002, pp. 72-79. doi:10.1006/rtph.2001.1516
- D. Crawford-Brown, “The Concept of Sound Science in Risk Management Decisions,” Risk Management: An International Journal, Vol. 7, No. 3, 2005, pp. 7-20.
- D. Crawford-Brown and K. Brown, “A Framework for Judging the Carcinogenicity of Risk Agents,” Risk: Health, Safety and Environment, Vol. 8, No. 2, 1997, p. 307.
- I. Cote, “Challenges for Use of New Science in Risk Assessment and Regulation,” Presentation to Standing Committee on Use of Emerging Science for Environmental Decision Making, Washington DC, 2010.
- M. Anderson and D. Krewski, “Toxicity Testing in the 21st Century: Bringing the Vision to Life,” Toxicological Sciences, Vol. 107, No. 2, 2009, pp. 324-330. doi:10.1093/toxsci/kfn255
- T. K. Nayak and S. Kundu, “Calculating and Describing Uncertainty in Risk Assessment: The Bayesian Approach,” Human and Ecological Risk Assessment, Vol. 7, No. 2, 2001, pp. 307-328. doi:10.1080/20018091094385
- J. Wilson, “Advanced Methods for Dose-Response Assessment: Bayesian Approaches, Final Report,” Resource for the Future, Washington DC, 2001. doi:10.1289/ehp.0901249
- K. Crump, C. Chen, W. Chiu, T. Louis, C. Portier, R. Subramaniam and P. White, “What Role for Biologically Based Dose-Response Models in Estimating Low-Dose Risk?” Environmental Health Perspectives, Vol. 118, No. 5, 2010, pp. 585-588.
- D. Krewski, V. Hogan, M. C. Turner, P. L. Zeman, I. McDowell, N. Edwards and J. Losos, “An Integrated Framework for Risk Management and Population Health,” Human and Ecological Risk Assessment, Vol. 13, No. 6, 2007, pp. 1288-1312. doi:10.1080/10807030701655798
- J. Lemons, K. Shrader-Frechette and C. Cranor, “The Precautionary Principle: Scientific Uncertainty and Type I and Type II Errors,” Foundations of Science, Vol. 2, No. 2, 1997, pp. 207-236. doi:10.1023/A:1009611419680
- R. Raucher, S. Rubin, D. Crawford-Brown and M. Lawson, “Benefit-Cost Analysis for Drinking Water Standards: Efficiency, Equity, and Affordability Considerations in Small Communities,” Journal of Benefit-Cost Analysis, Vol. 1, No. 1, 2011, pp. 1-22.
- D. E. Savitz, “Is Statistical Significance Testing Useful in Interpreting Data?” Reproductive Toxicology, Vol. 7, No. 2, 1993, pp. 95-100. doi:10.1016/0890-6238(93)90242-Y
- European Chemicals Agency, “Practical Guide 2: How to Report Weight of Evidence,” Helsinki, 2010.