Green and Sustainable Chemistry
Vol.04 No.02(2014), Article ID:45680,9 pages
10.4236/gsc.2014.42011
New One-pot Methods for Preparation of cyclohexanecarbonitrile―Green Chemistry Metrics evaluation for One-pot and similar Multi-step Syntheses
Jan Simbera, Richard Sevcik, Pavel Pazdera
Centre for Syntheses at Sustainable Conditions and Their Management, Faculty of Science, Masaryk University, Brno, Czech Republic
Email: pazdera@chemi.muni.cz
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 14 March 2014; revised 14 April 2014; accepted 22 April 2014
ABSTRACT
Three new multi-step one-pot processes for high-yielding cyclohexanecarbonitrile synthesis start- ing from cyclohexanone were developed for industrial application. In contrast to the current synthetic process, all of the processes described were designed to proceed completely in methanol as a uniform solvent and the key oxidation step can be realized either as stoichiometric or catalytic. Atom efficiency of processes is relatively advanced with high regioselectivity, reaction by-pro- ducts are either from environmental pool―carbon dioxide and nitrogen―or they can be reused― sodium chloride. Solvent―methanol―and other auxiliaries―cyclohexane, copper catalyst―can be reused after recycling as well. EcoScale for all three designed processes was evaluated and compared with current synthesis described in the past. Green chemistry metrics, including newly introduced evaluative tool―Sustainability Index of the Synthesis (SIS), were applied to evaluate design of described one-pot syntheses.
Keywords:
Cyclohexanecarbonitrile, one-Pot synthesis, EcoScale, EATOS, SIS

1. Introduction
Cyclohexanecarbonitrile (5) is a very important intermediate for chemical and pharmaceutical industry namely for preparation of pharmaceuticals [1] -[3] and also for some phytoeffector production [4] [5] . Common preparative methods such as substitution reactions on cyclohexyl halides, alcohol or on cyclohexyl alkyl- or arylsulfonates by alkali cyanides are not suitable processes for cyclohexanecarbonitrile industrial production. These substitution reactions are frequently accompanied by the side reactions such as elimination or isomerization resulting in a large number of side products [6] - [8] . Only when using tetramethylsilyl cyanide and cyclohexyl bromide in the presence of tetrabutylammonium fluoride, reaction afforded satisfactory results [9] [10] . Also reaction of cyclohexanole with tetrabutylammoniumcyanide (TBACN) proceeds very similarly [11] . To prepare (5) from cyclohexylhalogenide only indirect route based on the application of organometallic compounds in conjunction with p-toluensufonylcyanide [6] [12] , methyl(pyrid-2-yl)carbamic chloride [13] or pyrid-2-yl-cyanate [14] is possible. Another way to prepare (5) is reaction of cyclohexanole or cyclohexanethiole with TBACN under the action of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone [11] or 2,4,6-trichloro-1,3,5-triazine and TBACN in acetonitrile [15] . Rather unsuitable method for production of cykloalkancarbonitriles especially in terms of availability of starting materials and waste disposal is based on dehydratation reaction of cycloalkane carboxylic acids functional derivatives [16] [17] . Additional procedures for preparation of (5) are presented in literature [18] - [23] . Relatively a large number of these methods are not acceptable for (5) manufacturing but several industrial patents have been made though [24] - [27] . In the mentioned paper it is claimed that other oxidizing agents (tert- butylhypochlorite, 4-phenyl-4H-1,2,4-triazole-3,5-dione or Jones’s reagent) can be used instead of bromine but their application in industrial scale is irrelevant as well. Very suitable process for industrial production of (5) utilizes hydrogen cyanide addition on cyclohexene. There is also some catalytic hydrocyanation reactions [28] [29] generally on cyclopent-2-ene carbonitrile [30] and similar diene analogues [31] [32] described but the reaction products suffer from different amount of impurities which are ineligible in subsequent steps in pharmaceutical syntheses. However, catalytic hydrocyanation reactions on cyclohexene have similar disadvantages as well [33] - [35] . Some industrial patents dealing mostly with Ni0-phosphorus complexes catalysed HCN addition to different alkenes have been offered [31] [32] [36] - [40] . In these procedures, large amounts of Ni0-catalyst, complicated phosphorus ligand, Lewis acid (e.g. anhydrous zinc chloride) are necessary [41] - [43] , moreover all the auxiliaries are slowly decomposed and must be refuelled or recycled in a rather complicated way [44] . A promising three-step synthetic preparation process of (5) starting from accessible cyclohexanone was described in detail in Organic Syntheses [45] . In the first step, cyclohexanone (1) reacts with methyl hydrazinecarboxylate (2) in refluxing methanol giving methyl 2-(1-hydroxycyclohexyl) hydrazinecarboxylate. Then HCN was added into the mixture at 0˚C to yield methyl 2-(1-cyanocyclohexyl) hydrazinecarboxylate (3) which was isolated in very good yield 97% (Scheme 1). The first synthetic step affording (3) is really one-pot synthesis involving two partial steps.
In the next synthetic step, the isolated (3) is oxidized with the bromine in two-phase system dichloromethane and saturated sodium hydrogen carbonate water solution. Methyl (1-cyanocyclohexyl) diazenecarboxylate (4) is then obtained in yield 93% (Scheme 2). After alkaline cleavage of (4) with sodium methanolate in methanol the final product (5) is formed. After extraction with pentane followed by fraction distillation the pure cyclohexanecarbonitrile is isolated in yields about 80% (Scheme 3).
Nevertheless, from the manufacturing point of view the procedure has some disadvantages, e.g. isolation of the intermediates, usage of different types of solvents, wide range of reaction temperatures, use of environmentally hazardous solvent (dichloromethane), economically inconvenient application of bromine as oxidizing agent and finally the process produces a lot of waste as well. For these reasons, there is still an interest in developing clean, fast, inexpensive and environmentally harmless preparation methods producing the desirable product in high yields. We would like to accomplish all the synthetic steps described above as one-pot reaction in a single solvent.
We have tried to compare both described three-step synthesis and our one-pot reaction in terms of green chemistry metrics using EcoScale [46] and EATOS [47] [48] tools especially for the reason that the metrics were not primarily designed for one-pot, domino and similar syntheses.
2. Results and discussions
2.1. One-Pot Synthesis
In our solution we unificated the solvent first, then suitable oxidazing agent was applied with regard to waste production and economy of the process. We also take into account easy and simple proceeding of all the reaction steps and simple separation of the product.
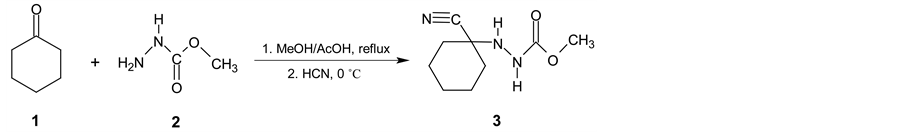
Scheme 1. First two reaction steps proceed subsequently as one-pot synthesis.
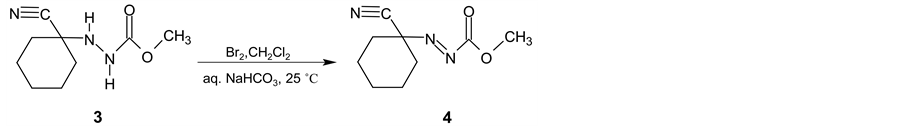
Scheme 2. Intermediate 3 oxidation using bromine.
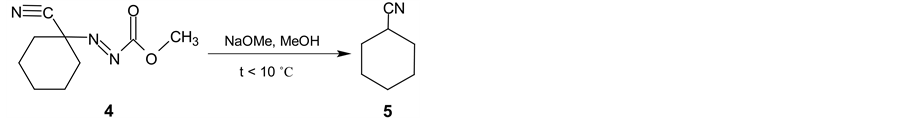
Scheme 3. Final alkaline cleavage of 4 using sodium methanolate in methanol.
Taking into account all the demands methanol seems to be a suitable solvent for all the reaction steps and the use of the chlorinated solvent is avoided. Sodium hypochlorite was selected as oxidizing agent for the middle stage of the synthesis (oxidation of (3) to (4)) because it has a number of advanteges. First of all it is a sufficient agent for intermediate (3) oxidation reacting under mild conditions in methanol (Scheme 2) and moreover its action produces acceptable sodium chloride as waste products. Another advantage of sodium hypochlorite is its adequate basicity which allows subsequent cleavage of compounds (4) yielding product (5) (Scheme 3) under gradual addition of sodium hypochlorite solution to the reaction mixture. Both reaction steps proceed gradually and generate nitrogen (environmentally neutral), carbon dioxide (contained in environmetal pool), methanol (contained in reaction pool as solvent with possibility of recyclation) and sodium chloride (possible as secondary raw material) as waste. In addition the reaction time is essentially shortened to ca 3 hours. Besides sodium hypochlorite, the hydrogen peroxide or oxygen have been tested as oxidizing agents but these agents require presence of auxilliary Cu2+-catalyst and pH of the mixture has to be kept between 8 - 9. Although the alternative oxidizing agents give comparable results as sodium hypochlorite, their usage in manufacturing has a few limitations. When using alternative oxidizing system, more complex technology is necessary and the Cu2+-catalyst has to be separated from the mixture and recycled (as usual for metal-containing waste waters).
Product (5) is then extracted from the reaction mixture using cyclohexane whereas the only extraction is sufficient (total product isolation >99%). Extraction mixture is then divided by the distillation and methanol and cyclohexane are easily recycled. Finally cyclohexanecarbonitrile (5) is obtained by the distillation under reduced pressure in high purity (>99.5%). Overall yield of our one-pot procedure is higher (>95%) when compared with Organic Syntheses multistep process giving (5) in total yield about 72%.
2.2. Green metrics Evaluation
EcoScale evaluations [46] as a benchmark for sustainability efficiency are very well described for simple one step syntheses, being aware of its ambiguity and limitations. Penalty points are granted according to EcoScale rules [46] taking into account several parameters such as yield, price and safety of the chemicals used, technical setup, temperatures used and ways of workup and purification. EcoScale does not follow reaction equations nor quantities of the chemicals used. When trying to apply the EcoScale evaluations to compare multi-step syntheses with similar syntheses realized as one-pot synthesis, or better as domino or other consecutive syntheses [49] performed in one synthetic step, distorted results may be obtained. We have got a few possibilities for evaluation. First of all we have to evaluate penalties for each item and determine EcoScale values for each reaction step (Table 1). Overall calculation can be done summing up EcoScale values for each step.
If calculating four-step synthesis as in this case, we can consider value of 400 points (P) as a maximum for the ideal process being decreased by penalties for the real one. But the declaration value of such a calculation can be controversial. Moreover EcoScale value always ranges between 0-100 P for a single process. Applying the calculation for cyclohexanecarbonitrile preparation according to Organic Syntheses [45] (OS) compared with our one-pot synthesis, higher EcoScale values are obtained for one-pot process (354 P) in comparison with OS procedure (282 P) as expected but we are about to claim that the difference is not very significant and does not reflect innovation/invention and simplification described in our contribution. Moreover when looking at penalties ratio of both of the processes, we can see that the final EcoScale values comparison does not respect the penalties ratio and therefore we can claim that the results calculated in this way can be distorted, especially when realizing that OS process suffers by several fold higher penalties than one-pot process almost in each step.
Comparing multi-step per partes synthesis transformed into one-pot procedure to prepare the same product in a similar way (as in this case), we can save not only penalties for intermediates work up and isolation and but penalties for handling hazardous chemicals when isolating or purifying intermediates as well. In the case of domino synthesis performed under the constant conditions (no more solvents, catalysts or other auxiliaries added, constant temperature and pressure), evaluation results should be even more advanced when compared with multi-step process. For that reason one-pot processes derived from original multi-step procedures are expected to be more favorable in terms of EcoScale metrics as well.
For better formulation of the reaction balance respecting penalties ratio a new indicator, Sustainability Index of the Synthesis (SIS), should be introduced. SIS can be then calculated according the equation (Equation (1)).
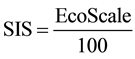 (1)
(1)
SIS can be then used as a favorable tool to evaluate overall balance of the one-pot multi-step process. First of all we have to take into account and recognize which reaction steps proceed subsequently in one stage and which ones are individual reaction steps necessitating intermediate isolation. If reactions proceed subsequently without intermediate isolation, we have to sum up penalties (P) for all of the subsequent stepsand then EcoScale value for multi-step one-pot synthesis is evaluated according to Equation (2).
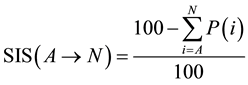 (2)
(2)
When intermediate is isolated EcoScale values and SIS are simply calculated as described by Equation (1). When all partial EcoScale and SIS values are calculated, SIS of the overall synthesis can be calculated by multiplying SIS values of each step (Equation (3)), similarly as it is done for overall yield of the multistep preparations [50] .
 (3)
(3)
Calculating overall yield, yields of each partial reaction (formulated as a real and theoretical yield ratio) have to be multiplied nor summed up to quantify the real balance of the compounds in the system. In the case of overall SIS evaluation we have to proceed in the same manner. Comparing these two methods of the overall balance calculation (Table 1) for Organic Syntheses process and our one-pot process yielding cyclohexanecarbonitrile, it can be clearly seen that calculation based on SIS respects penalties ratio (2.57) very well and that’s why we can claim that SIS calculations show benefits of innovation/invention more significantly than method based on simple sum of EcoScale values (Table 2). Moreover SIS value ranges between 0-1 and introducing this indicator, evaluation of the multi-step processes becomes easier.
In addition advantages of one-pot synthesis indicated by EcoScale and SIS values are supported by the results obtained by another metric tool known as EATOS (Environmental Assessment Tool for Organic Syntheses). Software which can be obtained online [48] evaluates processes in a slightly different manner according to chemical equations (Scheme 4) and parameters and amounts (see Experimental section for relevant numbers) of the chemical substances inserted (price, hazard symbols and phrases and various environmental and toxicological indicators). All computations are then performed by the EATOS software itself according to its rules. Be-

Table 1. Summary of EcoScale and SIS values for Organic Syntheses and one-pot procedures.
*SIS value calculated according to Equation (2) and Equation (3); **SIS value calculated according to Equation (1).
Table 2. Comparison of invention of one-pot procedures 1 - 3 for cyclohexanecarbonitrile preparation regarding OS proce- dure (all ratios are relative to OS values).
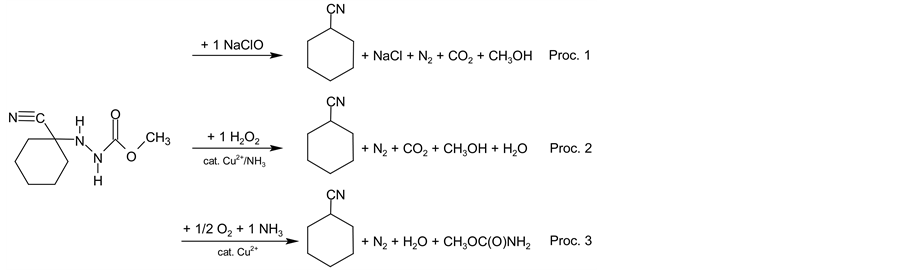
Scheme 4. Formal reaction equations for procedures 1 - 3 used for EATOS evaluations.
Sides basic enviromental indicators [51] such as mass index (S−1, in kg・kg−1) or enviromental factor (E, in kg・kg−1) we used EATOS to show economical benefits of our synthetic routes as well. Environmental indices (EI, given in PEI・kg−1) correspond with potential environmental impact (PEI) of the compouds involved in the procedure. While the first synthetic step is almost the same in both processes (Figure 1) we can see some differences given by the need of intermediate (3) separation especially. Although one-pot procedure requires more methanol in steps 1 and 2, it is more efficient because subsequent steps take advantage from it and moreover methanol is easily recycled (indicated by light green color in Figure 1) in the end. In case of subsequent reaction steps mass indexes and environmental factors (Figure 2) clearly show advantages of new synthetic routes similarly as described by EcoScale and SIS tools. For better clarity Organic syntheses step 3 and 4 were considered as one-pot process as well although these steps are carried out separately but it is not taken into account by EATOS software but it is evident when using EcoScale tool (Table 1). In EATOS diagrams (Figure 2) values of mass indexes and environmental factors of one-pot procedures 1 - 3 are several times (approx. 4 - 8 times) lower than corresponding values for Organic Syntheses process and these are in good agreement with results obtained by EcoScale and SIS. In addition satisfactory results of one-pot procedures from the view of atomic economy and environment are supported by lower costs as it is demostrated by EATOS in the Figure 3 (economy index CI, in EUR・kg−1). Prices of the chemicals are based on common price lists of the commercial suppliers (Sigma-Aldrich, Linde Gas) and although the prices can differ in dependence on the bulk and purity we can claim that the costs of one-pot procedures are approximately 3 times lower than costs for Organic Syntheses procedure. It is important to add that the costs presented in Figure 3 are based only on prices of the chemicals used and that is why the real costs will be higher because of investment in technology, energy and human resources. From this

Figure 1. EATOS diagram for steps 1 and 2 assessments (MU―One-pot pro- cedure, OS―Organic Syntheses procedure).

Figure 2. EATOS diagrams for one-pot procedures 1 (NaClO), 2 (H2O2) and 3 (O2) and Organic Syntheses (OS) steps 3 and 4 assessment.
point of view newly described one-pot syntheses are much more favourable than Oganic Syntheses procedure as well as it can be deduced from the EcoScale values especially.
3. Conclusion
In conclusion, we have developed an easy one-pot preparation procedure to obtain cyclohexanecarbonitrile from cyclohexanone using cheap and environmentally friendly oxidants (sodium hypochlorite, hydrogen peroxide or air). According to green chemistry metric tools―EcoScale, EATOS and newly introduced SIS―each of the three final oxidation procedures has its advantages and disadvantages but due to our experiences we consider oxidation procedure 1 using NaClO as the most suitable one for industrial use. However, all of the one-pot syntheses proceed under mild conditions in the single solvent with good (atom) economy and with high yields and moreover without hazardous waste, in other words, they are perfectly following Green Chemistry approaches and rules.

Figure 3. EATOS economy indexes for one-pot procedures 1 - 3 and Organic Syntheses (OS) steps 3 and 4.
4. Experimental
Cyclohexanone and cyclohexane (Acros Organics), methyl hydrazinecarboxylate, copper(II) chloride dihydrate,sodium hypochlorite solution (available chlorine 10% - 13%)and cyclohexanecarbonitrile as the standard (Sigma-Aldrich) were used as supplied.GC-MS was acquired on the HP 6890 plus/MS 5973N apparatus (Agilent Technologies). FTIR spectra were carried out on the Genesis (Unicam Mattson) spectrometeras a film of cyclohexanecarbonitrile.
Computing of EATOS values was performed using EATOS Software pack [48] and detailed description of its application can be found in the attached software manual.
Procedures for one-pot preparation of cyclohexanecarbonitrile (5) [CAS 766-05-2].
4.1. General procedure
Cyclohexanone (65.2 g, 0.664 mol), methyl hydrazinecarboxylate (63.8 g, 0.709 mol) and glacial acetic acid (3.0 g, 0.050 mol) in methanol (150 ml) were refluxed in a 1500 ml three-necked flask equipped with a reflux condenser, thermometer, magnetic stirring bar and gas-exhaust tube as long as the concentration of cyclohexanone declined below 0.4% (GC) (ca 60 min). Then the reaction mixture was cooled to room temperature and liquid HCN (18.0 g, 0.666 mol) was added drop-wise over period of 1 hour. After HCN addition, methanol (210 ml) was added and then the mixture was stirred over 1 hour. Following this procedure a methanol solution of methyl 2-(1-cyanocyclohexyl)hydrazinecarboxylate (3) was obtained.
4.2. Oxidation procedure 1
In situ prepared solution of methyl 2-(1-cyanocyclohexyl)hydrazinecarboxylate (3) was heated to 45˚C and sodium hypochlorite solution (460 ml) was added over 3.5 hour at temperature 45˚C - 50˚C. After mixture had been stirred for another 30 min, water (150 ml) was added to dissolve precipitated sodium chloride. Extraction was followed by 30 minutes of stirring. Then cyclohexane (300 ml) was introduced into the reaction mixture being consequently stirred for another 30 minutes. After the separation of the organic phase cyclohexane was removed by distillation and may be recycled. Crude product is then distilled under reduced pressure. It was obtained 66.7 g (92%) of cyclohexanecarbonitrile (5).
4.3. Oxidation Procedure 2
Copper(II) chloride dihydrate (3.0 g, 0.018 mol) was added into methanol solution of in situ obtained methyl 2-(1-cyanocyclohexyl)hydrazinecarboxylate (3) (see general procedure). Solution prepared from hydrogen peroxide (30%, 75.0 g) and 28% aqueous ammonia (2 ml) in water (15 ml) was dripped into the reaction mixture over period of 3 hours. Temperature of the reaction mixture was kept at 40˚C - 45˚C and pH value between 8-9 by the controlled addition of 20% aqueous ammonia solution. After the completion of the reaction, cyclohexane (300 ml) was poured into reaction flask and the product was then separated and purified as described above (see oxidation procedure 1). It was obtained 65.9 g (91%) of cyclohexanecarbonitrile (5). (Water phase containing Cu2+ salt was treated with week acidic ion-exchanger to uptake metal ions).
4.4. Oxidation procedure 3
Freshly in situ prepared methanol solution of methyl 2-(1-cyanocyclohexyl)hydrazinecarboxylate (3) was mixed with the water solution (75 ml) of copper(II) chloride dihydrate (3.0 g, 0.018 mol) and aqueous ammonia (28%, 2 ml). A stream of oxygen (10 ml/min) was then bubbled through the prepared mixture for 10 hours at temperature 45˚C - 50˚C. The pH value of reaction mixture during the whole time was kept between 8 - 9 by the controlled addition of a mixture containing aqueous ammonia (20%) and methanol (1:5 vol.). When the reaction is finished (GC-control), cyclohexane (300 ml) was added and the product then separated and purified in the same way as described previously. It was obtained 64 g (89%) of cyclohexanecarbonitrile (5). (Water phase containing Cu2+ salt was processed as mentioned above).
Cyclohexanecarbonitrile (5) was identified by a comparison of its data such as bp, FTIR spectrum and by GC-MS with those of the standard. Boiling point (bp 75˚C - 76˚C /2.13 kPa; bp 69˚C - 70˚C /2.00 kPa [52]). FTIR spectrum (film of substance, wavenumber/cm−1: 2920, 2840 (νC-H), 2200 (νCN), 1450 (δC-H), 870 (γC-H), and GC-MS (M+calc/M+found; 109.0886/109.0880 Da).
References
- Liu, F., Worthy, K.M., Lakshman, B., Alessio, G., Bottaro, D.P., Fisher, R.J. and Burke, T.R. (2007) Utilization of Achiral Alkenyl Amines for the Preparation of High Affinity Grb2 SH2 Domain-Binding Macrocycles by Ring-Closing Metathesis. Organic & Biomolecular Chemistry, 5, 367-372. http://dx.doi.org/10.1039/b611887a
- Yutaka, Y. and Morita, Y. (1985) 1,3-Oxazines and Related Compounds. X. Ring Transformation of 1,3-Oxazin- 4-Ones into Pyridine Derivatives. Chemical and Pharmaceutical Bulletin, 33, 975-981. http://dx.doi.org/10.1248/cpb.33.975
- Wu, Ch., Decker, E.R., Blok, N., Bui, H., You, T.J., Wang, J., Bourgoyne, A.R., Knowles, V., Berens, K.L., Holland, G.W., Brock, T.A. and Dixon, R.A.F. (2004) Discovery, Modeling, and Human Pharmacokinetics of N-(2-Acetyl-4,6- Dimethylphenyl)-3-(3,4-Dimethylisoxazol-5-Ylsulfamoyl)-Thiophene-2-Carboxamide (TBC3711), a Second Genera- tion, ETA Selective, and Orally Bioavailable Endothelin Antagonist. Journal of Medicinal Chemistry, 47, 1969-1986. http://dx.doi.org/10.1021/jm030528p
- Barluenga, J., Muniz, L., Iglesias, M.J. and Gotor, V. (1984) Synthesis of Hydrazone Derivatives by Reaction of Azines with Nitriles, and Their Transformation into Pyrazoles and Pyrimidones. Journal of the Chemical Society, Per- kin Transactions 1, 4, 611-614. http://dx.doi.org/10.1039/p19840000611
- Barluenga, J., Lopez, F., Palacios, F., Cano, F.H. and Concepción Foces-Foces de la, M. (1988) Reactivity and Che- moselectivity of Primary Z-β-Enamino-λ5-Phosphazenes towards Electrophiles. Journal of the Chemical Society, Per- kin Transactions 1, 8, 2329-2334. http://dx.doi.org/10.1039/p19880002329
- Roberts, J.D. and Chambers, V.C. (1951) Small-Ring Compounds. VIII. Some Nucleophilic Displacement Reactions of Cyclopropyl, Cyclobutyl, Cyclopentyl and Cyclohexyl p-Toluenesulfonates and Halides. Journal of the American Chemical Society, 73, 5034-5040. http://dx.doi.org/10.1021/ja01155a005
- Friedman, L. and Shechter, H. (1960) Preparation of Nitriles from Halides and Sodium Cyanide. An Advantageous Nucleophilic Displacement in Dimethyl Sulfoxide. The Journal of Organic Chemistry, 25, 877-879. http://dx.doi.org/10.1021/jo01076a001
- Thoman, Ch.J., Habeeb, T.D., Huhn, M., Korpusik, M. and Slish, D.F. (1989) Use of Polysorbate 80 (Tween 80) as a Phase-Transfer Catalyst. The Journal of Organic Chemistry, 54, 4476-4478. http://dx.doi.org/10.1021/jo00279a047
- Buchwald, S.L. and La Maire, S.J. (1987) A Regiospecific Method for the Hydrocyanation of Olefins. Tetrahedron Letters, 28, 295-298. http://dx.doi.org/10.1016/S0040-4039(00)95711-3
- Soli, E.D., Manoso, A.S., Patterson, M.C., De Shong, P., Favor, D.A., Hirschmann, R. and Smith III, A.B. (1999) Azide and Cyanide Displacements via Hypervalent Silicate Intermediates. The Journal of Organic Chemistry, 64, 3171-3177. http://dx.doi.org/10.1021/jo982302d
- Iranpoor, N., Firouzabadi, H., Akhlaghinia, B., and Nowrouzi, N. (2004) Conversion of Alcohols, Thiols, and Trime- thysilyl Ethers to Alkyl Cyanides Using Triphenylphosphine/2,3-Dichloro-5,6-Dicyanobenzoquinone/n-Bu4NCN. The Journal of Organic Chemistry, 69, 2562-2564. http://dx.doi.org/10.1021/jo035238v
- Klement, I., Lennick, K., Tucker, Ch.E. and Knochel, P. (1993) Preparation of Polyfunctional Nitriles by the Cyanation of Functionalized Organozinc Halides with p-Toluenesulfonyl Cyanide. Tetrahedron Letters, 34, 4623-4626. http://dx.doi.org/10.1016/S0040-4039(00)60640-8
- Lee, J.I. (1995) A Convenient Method for the Preparation of Nitriles and Carbodiimides Using N-Methyl-2-Pyridine- carbamoyl Chloride. Bulletin of the Korean Chemical Society, 16, 1143-1145.
- Koo, J.S. and Lee, J.I. (1996) 2-Pyridyl Cyanate: A Useful Reagent for the Preparation of Nitriles. Synthetic Commu- nications, 26, 3709-3713. http://dx.doi.org/10.1080/00397919608003787
- Wu, Y., Limburg, D.C, Wilkinson, D.E. and Hamilton, G.S. (2000) 1-Cyanoimidazole as a Mild and Efficient Electro- philic Cyanating Agent. Organic Letters, 2, 795-797. http://dx.doi.org/10.1021/ol0055263
- Akhlaghinia, B. and Roohi, E. (2005) A New and Convenient Method of Generating Alkyl Cyanides from Alcohols and Thiols Using 2,4,6-Trichloro[1,3,5]Triazine/n-Bu4NCN. Letters in Organic Chemistry, 2, 725-730. http://dx.doi.org/10.2174/157017805774717571
- Mlinarić-Majerski, K., Margeta, R. and Veljković, J. (2005) A Facile and Efficient One-Pot Synthesis of Nitriles from Carboxylic Acids. SyntLett, 13, 2089-2091. http://dx.doi.org/10.1055/s-2005-871967
- Hulkenberg, A. and Troost, J.J. (1982) An Efficient One-Pot Synthesis of Nitriles from Acid Chlorides. Tetrahedron Letters, 23, 1505-1508. http://dx.doi.org/10.1016/S0040-4039(00)87144-0
- Kidd, D.A.A., Robins, P.A. and Walker, J. (1953) Isomerisation of Ethyl α-2: 3: 4: 5-Tetrahydrobenzoylaceto-Acetate. Journal of the Chemical Society, 1953, 3244-3247. http://dx.doi.org/10.1039/jr9530003244
- Sivakumar, M., Senthilkumar, P. and Pandit, A.B. (2001) Ultrasound Enhanced PTC Conversion of Benzamide to Ben- zonitrile. Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemi- stry, 31, 2583-2587. http://dx.doi.org/10.1081/SCC-100105382
- Sasson, R. and Rozen, S. (2005) From Azides to Nitriles. A Novel Fast Transformation Made Possible by BrF3. Or- ganic Letters, 7, 2177-2179. http://dx.doi.org/10.1021/ol050523h
- Kamogawa, H., Kanzawa, A., Kadoya, M., Naito, T. and Nanasawa, M. (1983) Conversions of Carbonyl Compounds via Their Polymeric Sulfonylhydrazones into Alkenes, Alkanes, and Nitriles. Bulletin of the Chemical Society of Japan, 56, 762-765. http://dx.doi.org/10.1246/bcsj.56.762
- Barton, D.H.R., Bévière, S.D., Chavasiri, W., Doller, D. and Hu, B. (1993) New Alkane Functionalization Reactions Based on Gif-Type Chemistry in the Presence of Alkali Metal Salts. Tetrahedron Letters, 34, 1871-1874. http://dx.doi.org/10.1016/S0040-4039(00)91950-6
- The Dow Chemical Company (1978) US Patent No. 4079075.
- Bayer, A.G. (1980) US Patent No. 4189580.
- Bayer, A.G. (1991) US Patent No. 5049693.
- Bayer, A.G. (1988) EP Patent No. 0138141.
- McElvain, S.M. and Starn Jr., R.E. (1955) Ketene Acetals. XXXIV. Tetra- and Pentamethylene Ketene Acetals. Jour- nal of the American Chemical Society, 77, 4571-4577. http://dx.doi.org/10.1021/ja01622a040
- Guillemin, J.C., Breneman, C.M., Joseph, J.C. and Ferris, J.P. (1998) Regioselectivity of the Photochemical Addition of Ammonia, Phosphine, and Silane to Olefinic and Acetylenic Nitriles. Chemistry―A European Journal, 4, 1074- 1082. http://dx.doi.org/10.1002/(SICI)1521-3765(19980615)4:6<1074::AID-CHEM1074>3.0.CO;2-B
- Kleinpeter, E., Köhler, H., Tschierske, C. and Zaschke, H. (1988) Conformational Influences on the Liquid-Crystalline Behaviour of 1,4-Disubstituted Cyclohexanes. Journal für Praktische Chemie, 330, 484-486. http://dx.doi.org/10.1002/prac.19883300323
- Pont, D. (1970) US Patent No. 3496217.
- Pont, D. (1973) US Patent No. 3766237.
- Goertz, W., Kamer, P.C.J., van Leeuwen, P.W.N.M. and Vogt, D. (2001) Asymmetric Nickel-Catalyzed Hydrocyana- tion of Vinylarenes by Applying Homochiral Xantphos Ligands. Chemistry―A European Journal, 7, 1614-1618. http://dx.doi.org/10.1002/1521-3765(20010417)7:8<1614::AID-CHEM16140>3.0.CO;2-E
- Reetz, M.T. and Li, X. (2006) Asymmetric Hydrogenation of β-Keto Esters Using Chiral Diphosphonites. Advanced Synthesis & Catalysis, 348, 1157-1160. http://dx.doi.org/10.1002/adsc.200606093
- Reetz, M.T. and Li, X. (2006) An Efficient Catalyst System for the Asymmetric Transfer Hydrogenation of Ketones: Remarkably Broad Substrate Scope. Journal of the American Chemical Society, 128, 1044-1045. http://dx.doi.org/10.1021/ja057357t
- Bäckvall, J.E. and Andell, O.S. (1984) Stereochemistry and Mechanism of the Nickel-Catalysed Hydrocyanation of Conjugated Dienes. Journal of the Chemical Society, Chemical Communications, 1984, 260-261. http://dx.doi.org/10.1039/c39840000260
- Backvall, J.E. and Andell, O.S. (1986) Stereochemistry and Mechanism of Nickel-Catalyzed Hydrocyanation of Ole- fins and Conjugated Dienes. Organometallics, 5, 2350-2355. http://dx.doi.org/10.1021/om00142a030
- Saha, B. and RajanBabu, T.V. (2006) Nickel(0)-Catalyzed Asymmetric Hydrocyanation of 1,3-Dienes. Organic Letters, 8, 4657-4659. http://dx.doi.org/10.1021/ol062002f
- Gaspar, B. and Carreira, E.M. (2007) Mild Cobalt-Catalyzed Hydrocyanation of Olefins with Tosyl Cyanide. Ange- wandte Chemie International Edition, 46, 4519-4522. http://dx.doi.org/10.1002/anie.200700575
- Goertz, W., Kammer, P.C.J., van Leeuwen, P.W.N.M. and Vogt, D. (1997) Application of Chelating Diphosphine Li- gands in the Nickel-Catalysed Hydrocyanation of Alk-l-Enes and ω-Unsaturatedfatty Acid Esters. Chemical Communi- cations, 1997, 1521-1522. http://dx.doi.org/10.1039/a702811c
- Göethlich, A.P.V., Tensfeldt, M., Rothfuss, H., Tauchert, M.E., Haap, D., Rominger, F. and Hofmann, P. (2008) Novel Chelating Phosphonite Ligands: Syntheses, Structures, and Nickel-Catalyzed Hydrocyanation of Olefins. Organome- tallics, 27, 2189-2200. http://dx.doi.org/10.1021/om701140c
- Wilting, J., Janssen, M., Müller, C., Lutz, M., Spek, A.L. and Vogt, D. (2007) Binaphthol-Based Diphosphite Ligands in Asymmetric Nickel-Catalyzed Hydrocyanation of Styrene and 1,3-Cyclohexadiene: Influence of Steric Properties. Advanced Synthesis & Catalysis, 349, 350-356. http://dx.doi.org/10.1002/adsc.200600315
- Burello, E., Marion, P., Galland, J.C., Chamard, A. and Rothenberg, G. (2005) Ligand Descriptor Analysis in Nickel- Catalysed Hydrocyanation: A Combined Experimental and Theoretical Study. Advanced Synthesis & Catalysis, 347, 803-810. http://dx.doi.org/10.1002/adsc.200404363
- Invista North America S.A.R.L. (2005) US Patent No. 6936171.
- Wender, P.A., Eissenstat, M.A., Sapuppo, N. and Ziegler, F.E. (1978) Nitriles from Ketones: Cyclohexanecarbonitrile. Organic Syntheses, 58, 101. http://dx.doi.org/10.15227/orgsyn.058.0101
- Van Aken, K., Strekowski, L. and Patiny, L. (2006) EcoScale, a Semi-Quantitative Tool to Select an Organic Prepara- tion Based on Economical and Ecological Parameters. Beilstein Journal of Organic Chemistry, 2. http://dx.doi.org/10.1186/1860-5397-2-3
- Eissen, M.M. (2001) Bewertung der Umweltverträglichkeit Organisch-Chemischer Synthesen. Ph.D. Thesis, Universi- ty of Oldenburg, Oldenburg. http://docserver.bis.uni-Oldenburg.de/publikationen/bisverlag/2002/eisbew01/eisbew01.html
- Eissen, M.M. and Metzger, J.O. (2002) EATOS, Environmental Assessment Tool for Organic Syntheses. http://www.chemie.uni-oldenburg.de/oc/metzger/eatos/
- Hudlicky, T. and Reed, J.W. (2007) The Way of Synthesis: Evolution of Design and Methods for Natural Products. Wiley-VCH, Weinheim, 101.
- Hudlicky, T. and Reed, J.W. (2007) The Way of Synthesis: Evolution of Design and Methods for Natural Products. Wiley-VCH, Weinheim, 92.
- Eissen, M.M. and Metzger, J.O. (2002) Environmental Performance Metrics for Daily Use in Synthetic Chemistry. Chemistry―A European Journal, 8, 3580-3585. http://dx.doi.org/10.1002/1521-3765(20020816)8:16<3580::AID-CHEM3580>3.0.CO;2-J
- Orere, D.M. and Reese, C.B. (1977) Conversion of Aldehydes and Ketones into Nitriles Containing an Additional Car- bon Atom. Journal of the Chemical Society, Chemical Communications, 1977, 280-281. http://dx.doi.org/10.1039/c39770000280