World Journal of Vaccines
Vol.2 No.3(2012), Article ID:21525,18 pages DOI:10.4236/wjv.2012.23017
Characterization of Influenza H5N1 Nucleocapsid Protein for Potential Vaccine Design
![]()
Centre for Vaccine Evaluation, Biologics and Genetic Therapies Directorate, Health Canada-Santé Canada, Ottawa, Canada.
Email: *Mary.Hefford@hc-sc.gc.ca
Received March 20th, 2012; revised April 22nd, 2012; accepted May 25th, 2012
Keywords: Quadrant Influenza; Therapeutic Vaccine; Nucleocapsid Protein; Physicochemical Characterization
ABSTRACT
Avian influenza, subtype H5N1, causes occasional but serious infections in humans and efforts to produce vaccines against this strain continue. Current influenza vaccines are prophylactic and utilize the two major antigens, hemagglutinin and neuraminidase. Nucleocapsid protein (NP) is an attractive alternative antigen because it is highly conserved across all influenza strains, has been shown to increase the rate of viral clearance, and potential therapeutic vaccines would elicit cytotoxic T lymphocyte responses in an infected person. The NP antigen from H5N1 was characterized using a variety of physico-chemical methods to gain insights into both the biological and physical properties of the antigen which are important from a regulatory viewpoint when considering therapeutic vaccines. Results obtained to date show that NP is relatively unstable and indicate that the conformation of the H5N1 NP antigen is highly dependent upon purification procedure, buffer conditions, pH and the presence or absence of RNA. These factors will need to be clearly defined and taken into consideration when manufacturing and regulating NP vaccine preparations.
1. Introduction
Influenza is a highly contagious virus of the respiratory tract that, according to the World Health Organization (WHO), results in three to five million cases of severe illness and close to 250 - 500 thousand deaths annually [1]. Current vaccines against influenza are strain specific and prophylactic, utilizing the two major antigens of the influenza virus, Hemagglutinin (HA) and Neuraminidase (NA), in order to prevent infection. Variability in HA and NA proteins requires yearly identification of the three most common internationally circulating strains and production, distribution and testing of an annual vaccine. Several avenues of recent research have suggested that vaccines based on the viral nucleocapsid protein (NP) may provide additional, effective tools in the prevention and treatment of influenza. The nucleocapsid protein is a 56 kDa protein that binds the negative sense RNA segmented genome of the influenza virus. It’s primary function is to organize and act as an adapter between genome replication and transcription. A number of groups [2-5] have demonstrated that recombinant NP has the potential to promote cross-reactivity to various subtypes of influenza, and may be a valuable constituent to existing vaccines. NP has also been shown to stimulate a cytotoxic T lymphocyte response in both human and mice, resulting in quicker viral clearance and faster recovery from influenza infection [5]. Studies also demonstrate that NP can induce substantial titres of anti-NP antibodies which, while not neutralizing, were capable of increasing viral clearance and conferring hetero-subtypic immunity [6-8]. In addition, because NP is relatively conserved across all known influenza strains and has low sequence drift rate [9,10], vaccines based on NP might be effective against most influenza strains, thereby eliminating the necessity of annual production.
According to Volkin [11], there are two major considerations in the development of any vaccine. The first is the discovery of the biologic which elicits the desired therapeutic response. Several groups seem to believe that NP satisfies this criterion [6-8]. The second and equally important aspect in vaccine development is the translation of that biologic into a formulation which, while appropriate for both safe injection and bio-distribution, also confers stability, giving the product an acceptable shelf life [11]. It is this formulated vaccine product that must be carefully tested and controlled to maintain lot-to-lot consistency for product safety and efficacy. Because the properties of a biologic can be influenced by both the chemical nature of the product itself and the process used to manufacture and formulate it, proper characterization is essential. This paper describes the physico-chemical characterization of NP antigen derived from an avian influenza subtype (H5N1) in order to gain insights into the properties of that protein that might influence the quality, safety and efficacy of a potential NP-based therapeutic vaccine.
2. Materials and Methods
2.1. Chemicals and Reagents
LB broth and agar, phenylmethanesulfonylfluoride (PMSF), sodium phosphate monobasic monohydrate, sodium phosphate dibasic heptahydrate, imidazole, BCA reagents, 10 M NaOH, RNaseA, lysozyme, NaCl, KCl, adenosine, RNA (S. cerevisiae) and diethylpyrocarbonate (DEPC) were purchased from Sigma-Aldrich (Oakville, ON, Canada). Isopropyl β-D-1-thiogalactopyranoside (IPTG) was purchased from Roche (Laval, QC, Canada). Carbenicillin and Sypro® Ruby Red protein gel stain were purchased from Invitrogen (Burlington, ON, Canada). Sodium dodecyl sulfate (SDS) and ammonium persulfate (APS) were purchased from Bio-Rad (Mississauga ON, Canada). 10 M HCl was purchased from EMD Biosciences (Mississauga, ON, Canada). 18 M H2SO4 was purchased from BDH (Missisauga, ON, Canada). Vanadyl sulfate was purchased from Fischer (Ottawa, ON, Canada). Anti-His antibody for Western blot analysis was purchased from GE Healthcare (Baie d’Urfée, QC, Canada) as was the horseradish peroxidase-linked goat anti-Rabbit secondary antibody. Anti-NP antibody was a gift from Dr. Sean Li. All solutions were prepared using distilled and deionised water. Buffers were filtered through 0.2 µm membrane filters prior to use in FPLC/ HPLC.
2.2. Protein Expression and Purification
Sequence for the nucleocapsid gene from A/duck/yokohama/aq10/2003(H5N1) sub-cloned into the expression vector pQE2 (QIAGEN, Toronto, ON, Canada) was a gift from the laboratory of Dr. Sean Li (Health Canada). The pQE2 vector provided the 6xHis tag located at the N-terminal of the protein necessary for affinity chromatography. This construct was used to transform BL21 competent cells and each NP preparation was from a 1 L culture. The cells were harvested and lysed by lysozyme and sonication, following standard procedures. NP was recovered from the supernatant and isolated by FPLC using a HisTrap column, with a stepwise imidazole gradient (Buffer A: 50 mM Na3PO4, 200 mM KCl at pH 7.4 and Buffer B: 50 mM Na3PO4, 200 mM KCl, 200 mM Imidazole at pH 7.4). An aliquot of each 10 mL fraction collected was run on SDS-PAGE to determine which fraction contained NP. The purity of NP was estimated using densitometry. NP containing solutions were analyzed using SDS-PAGE and stained overnight using Sypro® stain. The NP band density was assessed as a percentage of the total lane density using a Gel-Doc XR™ with Quantity One 4.5.2 software (Bio-Rad). Fractions identified as containing NP were diluted 2X with Buffer B then combined for dialysis (MWCO 25KDa) against Buffer A in order to remove imidazole. Protein concentration was determined by BCA assay for each NP preparation prior to use in further experimentation. Concentration was also determined for one NP preparation using quantitative amino acid analysis performed by Dr. Rey Interior at the Sick Children’s Hospital (Toronto, ON).
2.3. Western Blot Analysis
NP identity was further verified by Western blot analysis. Briefly, two SDS-PAGE gels were electrophoresed. The first gel was stained with Sypro® stain. The second gel, in which the same sample was loaded twice, was cut in two. Both halves were transferred onto a PVDF membrane. One membrane was incubated with an anti-His antibody, while the other was simultaneously incubated with an anti-NP antibody. Standard procedures for washes, secondary antibody incubation and detection were used.
2.4. Circular Dichroism Spectropolarimetry (CD)
Protein samples, in 10 mM Na3PO4 at pH 7.4 (physiological conditions), were transferred in a 1.0 cm quartz cuvette and analyzed by CD with constant stirring. Circular dichroism spectral readings were taken using a JASCO J-815 Spectrometer with the following parameters: band width: 1 nm, response: 1 sec, sensitivity: standard, measurement range: 290 - 195 nm, pitch: 0.1 nm and scanning speed: 50 nm/min. During thermal denaturation the same parameters were used, as well as wavelength monitoring at 222 nm with a temperature slope of 1˚/min. The recorded data at 222 nm were deposited into Sigma Plot® graphing software for melting temperature determination. Full length CD spectra were recorded at 5˚C intervals as temperature increased from 20˚C to 85˚C. Spectra represent the cumulative average of 5 spectral accumulations. Raw spectral data (in mdeg) were then converted to molar ellipticity and/or mean residue ellipticity. In order to compute the amount of secondary structural elements within NP, specialized online algorithms on Dichroweb were used:
http://dichroweb.cryst.bbk.ac.uk/html/home.shtml or (http://www.ogic.ca/projects/k2d2/).
2.5. Fluorescence Spectroscopy
Fluorescence readings of NP samples were conducted using a Varian Cary Eclipse Fluorescence Spectrophotometer. Using a 1.0 cm quartz cuvette, the excitation wavelength was determined from an initial wavelength scan from 300 nm to 400 nm with an arbitrary excitation wavelength (280 nm) to obtain the emission maxima. This excitation wavelength was used for all fluorescence measurements pertaining to that particular sample. This method was used to determine the exact excitation wavelengths for every NP sample. Fluorescence readings were also acquired during thermal denaturation as described in the previous section. Data accumulated were deposited into Excel™ and analyzed using the method of Pace et al. [12] to determine fraction unfolded as temperature increases. Thermal denaturation data were collected under a variety of pH and salt concentration conditions.
2.6. pH Titrations
Immediately after recording the CD spectrum as per previous section, the optical density at both 280 nm and 214 nm was recorded using a spectrophotometer and then fluorescence emission spectra were recorded using a Varian Cary Eclipse Fluorescence Spectrophotometer. After recording all measurements at the initial pH value, the pH was manually adjusted to the next desired value (in increments of 0.5 pH units) by adding small aliquots of acid. Once the desired pH had been reached, the sample was allowed to sit for 5 minutes with constant stirring. The process of recording CD, OD280/214 and fluorescence was repeated. The process was repeated until the pH had been adjusted to pH 3.0. Once a pH of 3.0 had been reached, the entire protocol was repeated using base to bring the pH back to 7.5 in increments of 0.5 pH units. The resultant spectra obtained were corrected using blanks prepared to account for volume and effects of acid/base.
2.7. Effects of Ionic Strength (NaCl Titration)
The effects of salt on NP secondary structure was assessed using CD in a similar manner to that previously described in the pH titration methodology. NP was sequentially titrated from 0.0 M to 1.0 M NaCl in 0.1 M intervals.
2.8. Examination of the Effects of RNA on NP Structure-RNase A Digest after “Anomalous” Spectra Recorded on CD
When an “anomalous” CD spectrum was obtained, the solution containing NP was divided into two equal volumes. One of these sub-fractions was incubated overnight at room temperature with gentle agitation, while the other sub-fraction was similarly incubated in the presence of RNase A (at approximately 1/50 th of the NP concentration as determined by BCA). Approximately 18 hours after RNase A addition, comparison CD spectra were obtained for the two sub-samples. The pH of each sub-sample was then adjusted to 3.0 using HCl and a new spectrum recorded. The pH was slowly changed back to 7.0 using NaOH and a new spectrum at this pH was obtained.
2.9. RNase-Free Work with and without Vanadyl Complex RNase Inhibitor
In order to keep RNase contamination to a minimum, the following preventative measures were followed: DEPC treated water was used throughout; all working surfaces, containers, equipment or instruments that came in contact with the protein preparation were wiped with RNase Erase® (MP Biomedicals) and then rinsed with DEPCtreated water; plasticware was treated with 0.1 N NaOH/ 1 mM EDTA overnight, then rinsed thoroughly with DEPCtreated water; all glassware was autoclaved; sterile, RNase free pipette tips were used.
A small molecule RNase inhibitor was made as described previously [13]. In order to verify that the vanadyl complex was formed and active, an RNase Alert® Lab test kit (Ambion) was used. This vanadyl complex was then added to the lysis buffer and the protein was purified as described above, with extra additions of vanadyl complex during sonication. Alternately, the purifycation protocol described above was repeated exactly except that, instead of adding vanadyl complex to the lysis buffer only, all buffers used in FPLC and dialysis were made with DEPC-treated water in an RNase-free manner, and all contained vanadyl complex.
2.10. NP Titration with Eukaryotic RNA
NP was purified and digested with RNase A as described above. The RNA nucleotide fragments and RNase A were then removed from the sample by dialysis against four 2 L changes of 10 mM Na3PO4 in DEPC treated RNase-free water using a 30 KDa molecular weight cut-off membrane. A stock solution of RNA (RNA from baker’s yeast, S. cerevisiae) was prepared at a working concentration of 1 mg/mL. NP was placed in a rectangular CD cuvette and RNA was added in successive, small aliquots until the concentration of RNA was 10× more than the measured concentration of NP (based on the assumption that 24 nt of RNA bind to each NP monomer) in an attempt to drive binding. After each addition of 2.5 uL of RNA, the sample was allowed to stir for 5 minutes then a CD spectrum was acquired. NP was titrated with RNA at both pH 7.0 and pH 5.5.
3. Results
3.1. Isolation and Verification of Nucleocapsid Protein
NP recombinantly produced in IPTG induced BL21 cells was isolated using metal affinity chromatography (IMAC) and FPLC fractions from this isolation were analysed using SDS-PAGE (Figure 1(a)). A band with an electrophoretic mobility consistent with protein of a molecular size of 60 kDa (which accurately compared with the molecular weight of 56 kDa for the expected (His)6-tagged NP) was routinely observed in all preparations. Identity of the band was confirmed by Western blot analysis using anti-NP antibody (Figure 1(b)). Some product degradation was occasionally observed in the more concentrated samples. Occasionally, such analysis of an NP preparation revealed a second, faster migrating band on SDSPAGE which was rich in histidines as shown by the presence of bands in Western blots probed with the anti-His antibody which was absent when the same blots were probed with anti-NP antibodies (data not shown). Densitometric analysis showed this isolation procedure produced NP at greater than 95% purity.
3.2. Verification of Proper Folding of NP (Circular Dichroism)
Every NP preparation (after dialysis into 10 mM Na3PO4) was analyzed by CD spectropolarimetry to ensure proper protein folding. In total, 15 preparations produced in the absence of RNase inhibitors were analyzed. Twelve of these 15 gave similar CD spectra at neutral pH, as indicated in blue in Figure 2 and herein referred to as the “predominant” spectrum. However, with 3 of the 15 preparations, a CD spectrum that looked quite different
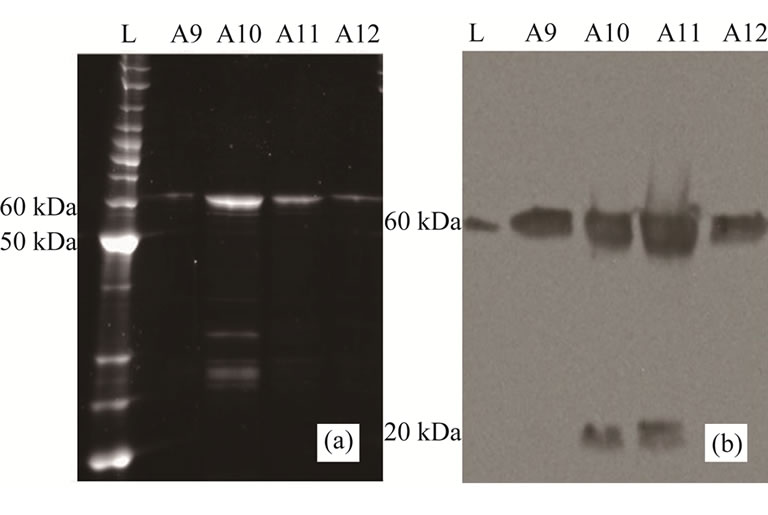
Figure 1. (a) Sypro®-stained SDS-PAGE analysis of purified NP (elution fractions from Ni-NTA column, Fractions A9-A12) of a representative protein preparation. A band at expected molecular size (60kDa: 56KDa NP + His Tag) and of purity > 90% is evident; (b) Western blot analysis of a replicate SDS-PAGE electrophoresed simultaneously with the gel shown in (a). A band at the expected molecular size (60 kDa) is evident in all fractions when the anti-NP antibody is used as a probe. Fractions 10 and 11 show some faster migrating material (potentially a degradation product).
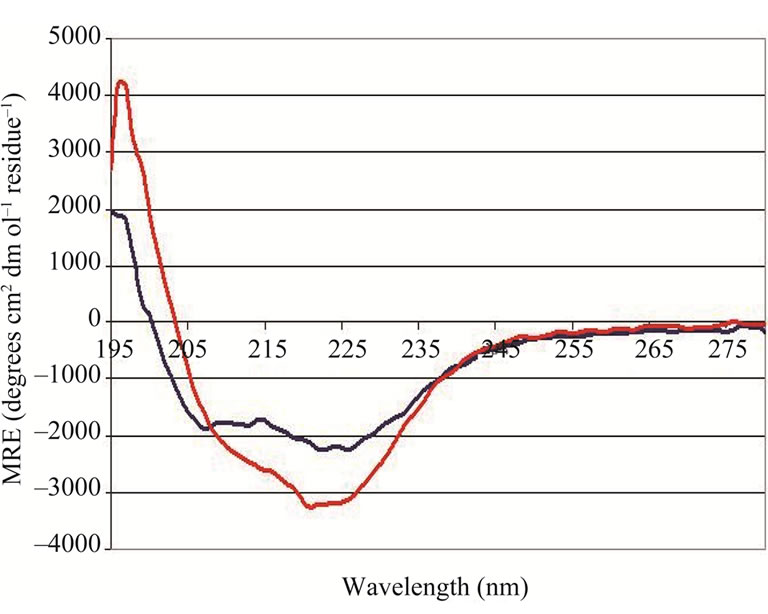
Figure 2. Representative CD spectra obtained for “predominant” (▬) and “anomalous” (▬) NP preparations at room temperature in 10mM Na3PO4, pH 7.4.
than the “predominant” spectrum was obtained. This spectrum, termed the “anomalous” spectrum, is depicted by the red trace in Figure 2. Interestingly, there were no obvious deviations or errors in the production process (in terms of growth conditions, isolation methods etc.) that could account for the “anomalous” result. The spectrum for the “predominant” preparations appears to have two distinct minima at roughly 222 nm and 208 nm, reminiscent of spectra from proteins with largely helical structures [14], while the “anomalous” spectrum appear to have only one minimum at 222 nm.
3.3. Response of “Predominant” Preparations of NP to Altered Solvent Conditions
Decreasing the solvent pH from near-physiological (7.4) to acidic conditions (3.0) using HCl, caused NP in “predominant” preparations to adopt an altered conformation(s) (Figure 3(a)). The increase in negative ellipticity around 222 nm observed in the CD spectrum at acid pH values suggests that the altered conformation may contain more helical or helix-like secondary structural elements than the conformation assumed at neutral pH. This conformational change was reversible: a spectrum similar to the original was obtained when the pH was returned to neutral by the gradual addition of base (Figure 3(b)). Similar, yet less dramatic increases in negative ellipticity were obtained when freshly prepared NP was titrated with 1 M H2SO4 (Figure 3(c) (with back titration in Figure 3(d))). Gradual addition of NaCl to predominant preparations at neutral pH (Figure 3(e)) also resulted in an increase in negative ellipticity at 222 nm (and presumably helical secondary structural content). However, the magnitude of the effect was considerably less than that observed during the acidification with HCl.
 (a)
(a) (b)
(b) (c)
(c)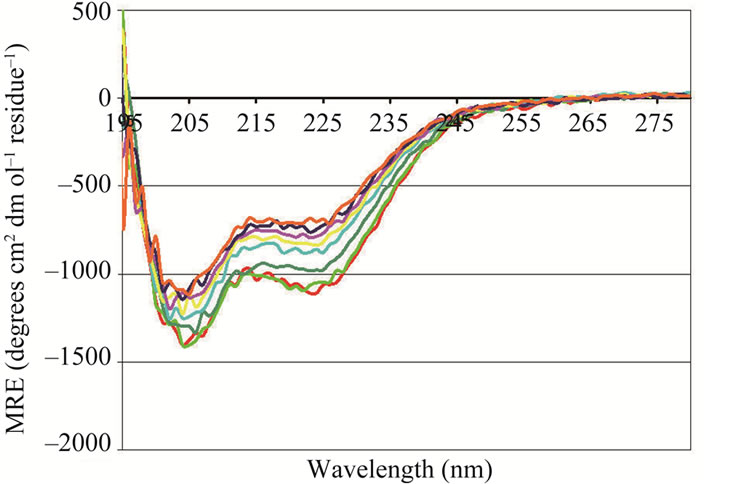 (d)
(d) (e)
(e)
Figure 3. Representative CD spectra obtained for “predominant” NP preparations when titrated with 1M HCl (a) and back-titrated with 1M NaOH (b) or with 1M H2SO4 (c) and back-titrated with 1M NaOH (d); (e) NP from a “predominant” preparation titrated with 5M NaCl at pH 7.4. ((a) to (d)) pH: (▬) 7.4 (▬) 7.0 (▬) 6.5 (▬) 6.0 (▬) 5.5 (▬) 5.0 (▬) 4.6 (▬) 4.0 (▬) 3.5 (▬) 3.0. (E) Final NaCl concentrations were as follows: (▬) 0.0 M (▬) 0.1 M (▬) 0.2 M (▬) 0.3 M (▬) 0.4 M (▬) 0.5 M (▬) 0.6 M (▬) 0.7 M (▬) 0.9 M (▬) 1.0 M. Samples were allowed to equilibrate for five minutes prior to taking CD measurements.
Fluorescent emission spectra were recorded throughout the titration to acid pH in order to assess tertiary structure (Figure 4(a)). Acidification of “predominant” preparations of NP resulted in a slight red shift of the intrinsic tryptophan at the emission maximum (from 336 nm at pH 7.4 to 338 nm at pH 3.0). Upon back titration, a compensatory blue shift was observed (Figure 4(b)). Titration with sulphuric rather than hydrochloric acid yielded similar results (Figures 4(c) to (e)).
3.4. Response of “Anomalous” Preparations of NP to Altered Solvent Conditions
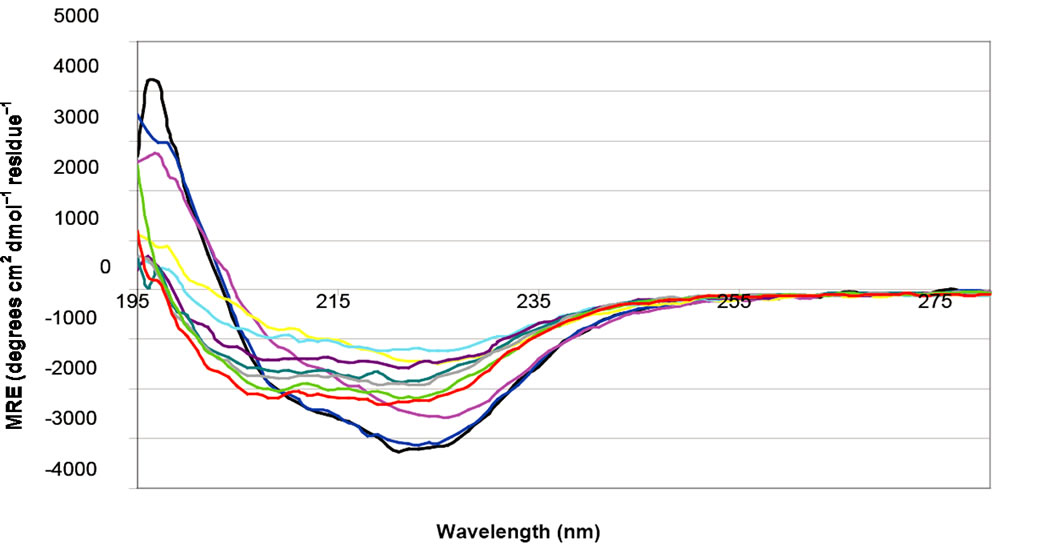
As noted above, routine preparation of NP occasionally yielded protein that gave rise to “anomalous” spectra at neutral pH. When “anomalous” NP preparations were titrated with HCl, an “anomalous”-shaped spectrum persisted until pH of 5.5, although the intensity of the signal (degree of negative ellipticity) decreased with increasing solution acidity. However, at pH values below 5.5, the shape of the CD spectra reverted to a profile more closely resembling that of the “predominant” preparations. At these lower pH values, ellipticity at 222 nm steadily increased with each further addition of acid (Figure 5(a)). The original “anomalous” spectrum is not seen upon reverse titration with NaOH. Rather, spectra appear similar to those obtained for a “predominant” preparation (Figure 5(b)).
Fluorescent emission spectra were recorded throughout the acid titration and the back-titration to neutral pH in order to assess the effects on tertiary structure of the
 (a)
(a)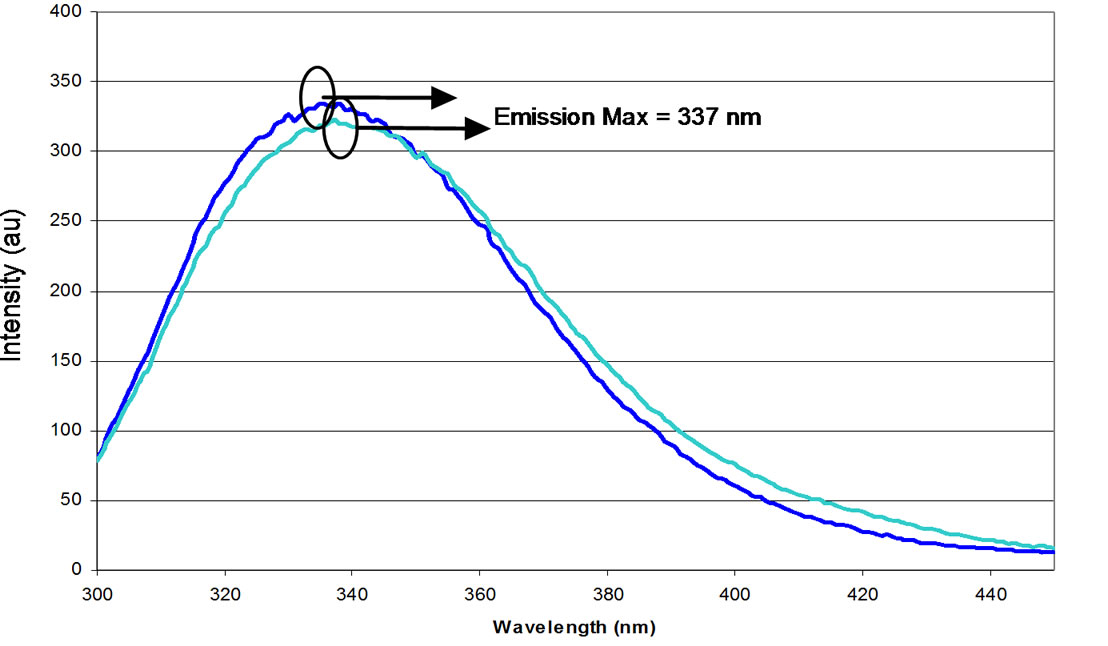 (b)
(b)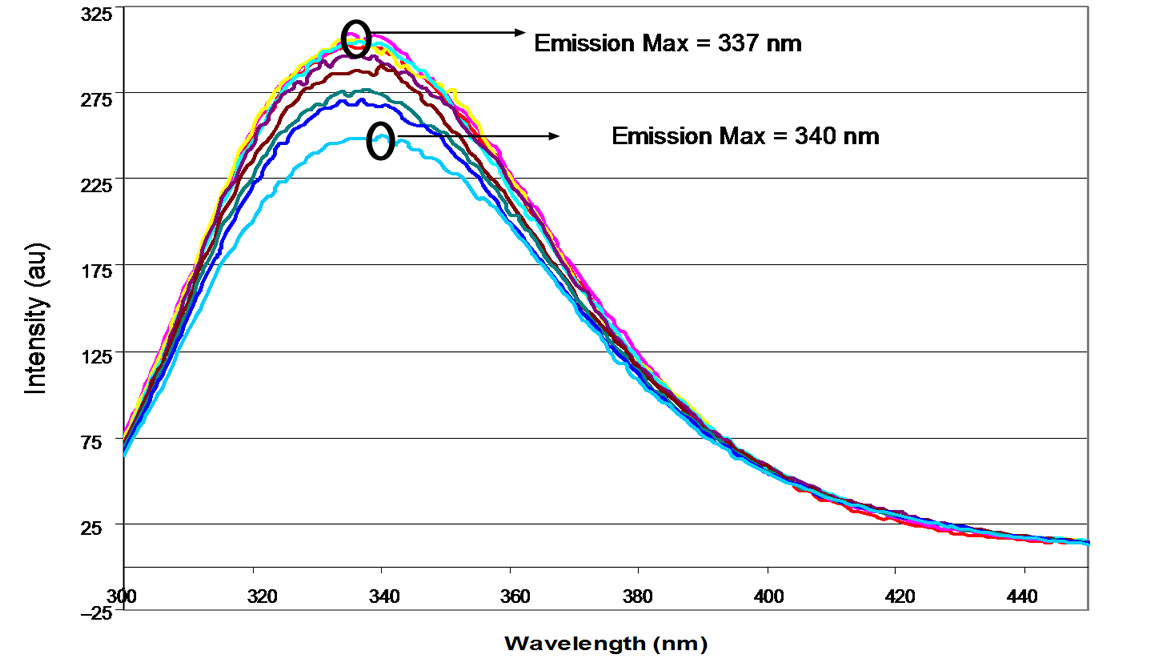 (c)
(c)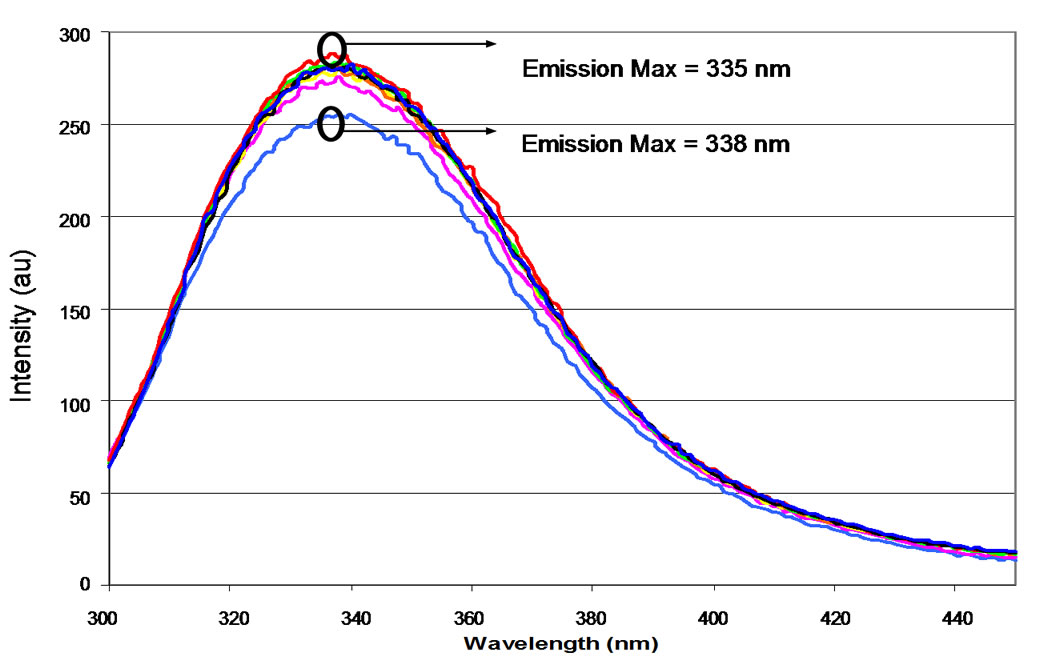 (d)
(d)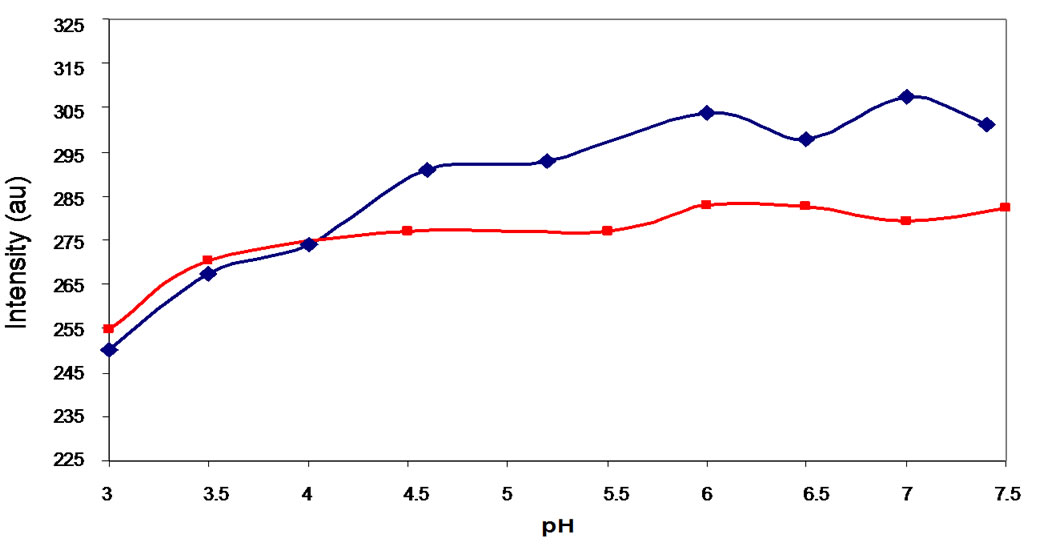 (e)
(e)
Figure 4. Characteristic fluorescence profiles of NP during titration with HCl (a) and back titration with NaOH (b) and of NP during titration with H2SO4 (c) and back titration with NaOH (D). (a) Titration performed on a predominant NP preparation from pH 7.0 to pH 3.0 followed by fluorescence spectroscopy; (b) Titration of a predominant NP preparation from pH 3.0 to pH 7.0 followed by fluorescence spectroscopy. pH: (▬) 7.4 (▬) 3.0; (c) Titration of a predominant NP preparation from pH 7.0 to pH 3.0 with H2SO4 followed by fluorescence spectroscopy; (d) Back-titration of a predominant NP preparation from pH 3.0.
 (a)
(a) (b)
(b)
Figure 5. Representative CD spectra obtained for anomalous NP preparations when titrated with 1M HCl (a) and 1M NaOH (b). pH: (▬) 7.5 (▬) 7.0 (▬) 6.5 (▬) 6.0 (▬) 5.5 (▬) 5.0 (▬) 4.5 (▬) 4.0 (▬) 3.5 (▬) 3.0.
“anomalous” NP preparations (Figures 6(a) and (b)). As was the case for the “predominant” preparation, a small but consistent red shift is observed upon decreasing the pH, with the wavelength shifting from 336 nm to 339 nm. However, upon addition of base the maximum did not shift, but remained at 339 nm throughout the titration. The changes in emission intensity at 340 nm relative to pH are shown in Figure 6(c).
3.5. RNA Interaction with NP: RNase A Digest
Comparison of the “predominant” to the “anomalous” spectrum indicate approximately the same degree of ellipticity at 208 nm (Figure 2); however, the “anomalous” form has much deeper signal at approximately 220 nm. An increase in helical content in a protein would be expected to increase negative ellipticity at 220 nm with the concomitant increase at 208 nm and increase in sheet or random coil content would be expected to increase negative ellipticity at lower wavelengths (202 - 210 nm) and decrease the depth of signal at 220 nm [15]. Thus, the “anomalous” shape can not entirely be attributed simply to change in the relative proportions of typical protein secondary structural elements and alternate explanations
 (a)
(a) (b)
(b)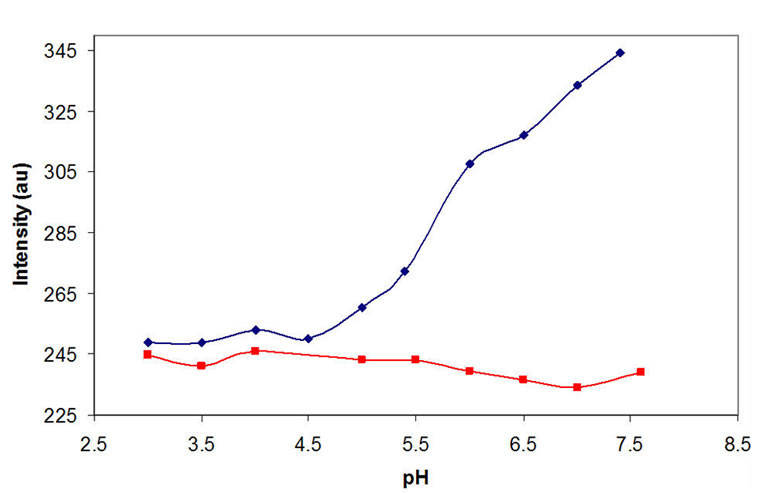 (c)
(c)
Figure 6. Characteristic fluorescence profiles of NP from an “anomalous” preparation during titration with sulphuric acid (a) and back titration with NaOH (b). (a) Titration of an anomalous NP preparation from pH 7.0 to pH 3.0 with H2SO4 followed by fluorescence spectroscopy; (b) Backtitration of an “anomalous” NP preparation from pH 3.0 to pH 7.0 following acidification with H2SO4. pH: (▬) 7.4 (▬) 7.0 (▬) 6.5 (▬) 6.0 (▬) 5.5 (▬) 5.0 (▬) 4.5 (▬) 4.0 (▬) 3.5 (▬) 3.0; (c) Change in emission intensity at 340 nm during acid (▬) and base titration (▬).
for the anomalous spectrum need to be considered. NP binds RNA, and it has been shown that some conformations of nucleic acids can give CD spectra with negative ellipticity in the region observed in spectrum of anomalous NP preparations [16]. Therefore, it was hypothesized that the “anomalous” preparations result from cellular RNA binding to NP during recombinant protein production or isolation process.
This hypothesis was tested when another preparation gave an “anomalous” CD spectrum at neutral pH: RNase A was added to determine if the binding of the protein to RNA could be responsible for the altered conformation/spectrum observed. Comparison of the CD spectra obtained for an “anomalous” preparation with and without RNase A treatment (Figures 7(a) and (b)) suggest that the atypical CD spectrum results from binding of NP protein to RNA.
 (a)
(a) (b)
(b)
Figure 7. Effects of RNase on secondary structure. An anomalous preparation at physiological pH as indicated by initial CD room temperature scan, was divided into two samples: one untreated sample (a) and one treated with RNase A to remove any residual RNA (b). After 18 hours, the spectra were recorded (▬). The pH was then decreased and a scan was taken at pH 3.0 using concentrated HCl (▬). The pH was then increased back to physiological pH before reading the final CD spectrum (▬).
3.6. RNase Free Preparations
To further test the hypothesis that RNA binding to NP is responsible for the anomalous spectra observed at neutral pH for some preparations of NP, a small molecule RNase inhibitor was used during the purification of recombinant NP in an effort to prevent degradation (and thus binding) of endogenous RNA. RNA-vanadyl complex was added either to all buffers used during the preparation or to only the lysis buffer used in the initial step of cell disruption. In both cases, the expected band for NP was obtained (Figures 8(a) and (b)). Addition of vanadyl complex at any stage during preparation appeared to result in higher yields of NP than without the vanadyl complex. However, this effect was less noticeable when the complex was added only to the lysis buffer, resulting in less NP (Figure 8(a)) than when the inhibitor was added into all the buffers during isolation (Figure 8(b)). Moreover, when the vanadyl complex was added at any point during the preparation, the “anomalous” spectrum (with a deep ellipticity valley at 222 nm) was obtained on CD analysis
 (c)
(c)
Figure 8. Sypro®-stained SDS-PAGE analysis of purified NP (elution fractions from Ni-NTA column, Fractions A10-B12) prepared with a vanadyl complex (RNase inhibittor) in the lysis buffer (a) and in all buffers (b) during isolation; (c) Representative CD spectrum of undigested NP after isolation in the presence of vanadyl complex (▬) and post RNase A digest (▬).
(Figure 8(c)). This result is consistent with the hypothesis that the “anomalous” spectrum was the result of RNA binding NP. A sample of the “anomalous” preparation obtained was then digested overnight with RNase A, and the CD after digestion recorded. This spectrum resembled those obtained from “predominant” preparations (Figure 8(c)), again consistent with the hypothesis that the “anomalous” preparations contained bound RNA.
3.7. Deliberate Addition of RNase A to NP Preparations
To further investigate the interaction between NP and RNA, RNaseA was added to the lysis solution to a final concentration of 4 μg/mL and allowed to incubate for two hours after cell lysis by sonication. In each of the four replica of this experiment, no intact NP was recovered (data not shown).
3.8. RNA Titration
RNA-free preparations giving a “predominant” CD spectrum were titrated with yeast RNA to determine whether NP would bind the nucleic acid and an “anomalous” CD spectrum would be obtained. NP was titrated with increasing concentrations of RNA at both pH 7.0 and 5.5 (neutral pH and the point at which the anomalous spectrum began to behave as a predominant preparation, respectively). No change in the shape of the CD spectrum was observed in either case, even at concentrations of RNA that were calculated to result in 10-fold excess of nucleic acid binding units. Thus, NP folded in the absence of RNA did not appear to be able to bind any RNA (data not shown).
3.9. Stability of Secondary and Tertiary Structure Analyzed through CD and Fluorescence
The effect of salt concentration on the secondary and tertiary structure on both the “predominant” and “anomalous” forms of NP was examined using thermal denaturation assays followed by CD and fluorescence, respectively. Far UV CD studies showed that the melting temperature (TM) of the “predominant” preparation at pH 7.0 decreased when salt (NaCl) was added to a concentration of 150 mM (Figure 9(a)).
Normalized tryptophan fluorescence emission intensity at this same pH decreased with increasing temperature. However, it is well known that the quantum yield of tryptophan fluorescence is temperature dependent even for tryptophan free in solution [17]. Therefore, the effect of salt on quantum yield with increasing temperature must be viewed relative to the loss seen with free tryptophan in solution. With tryptophan free in solution, a consistent but non-linear loss of quantum yield with increasing temperature is observed (Figure 9(b), black traingles). On the other hand, the plot of quantum yield as a function of increasing temperature of the intrinsic tryptophan fluorescence from NP of the “predominant form” produces a biphasic curve at neutral pH (Figure 9(b)). At
 (a)
(a) (b)
(b)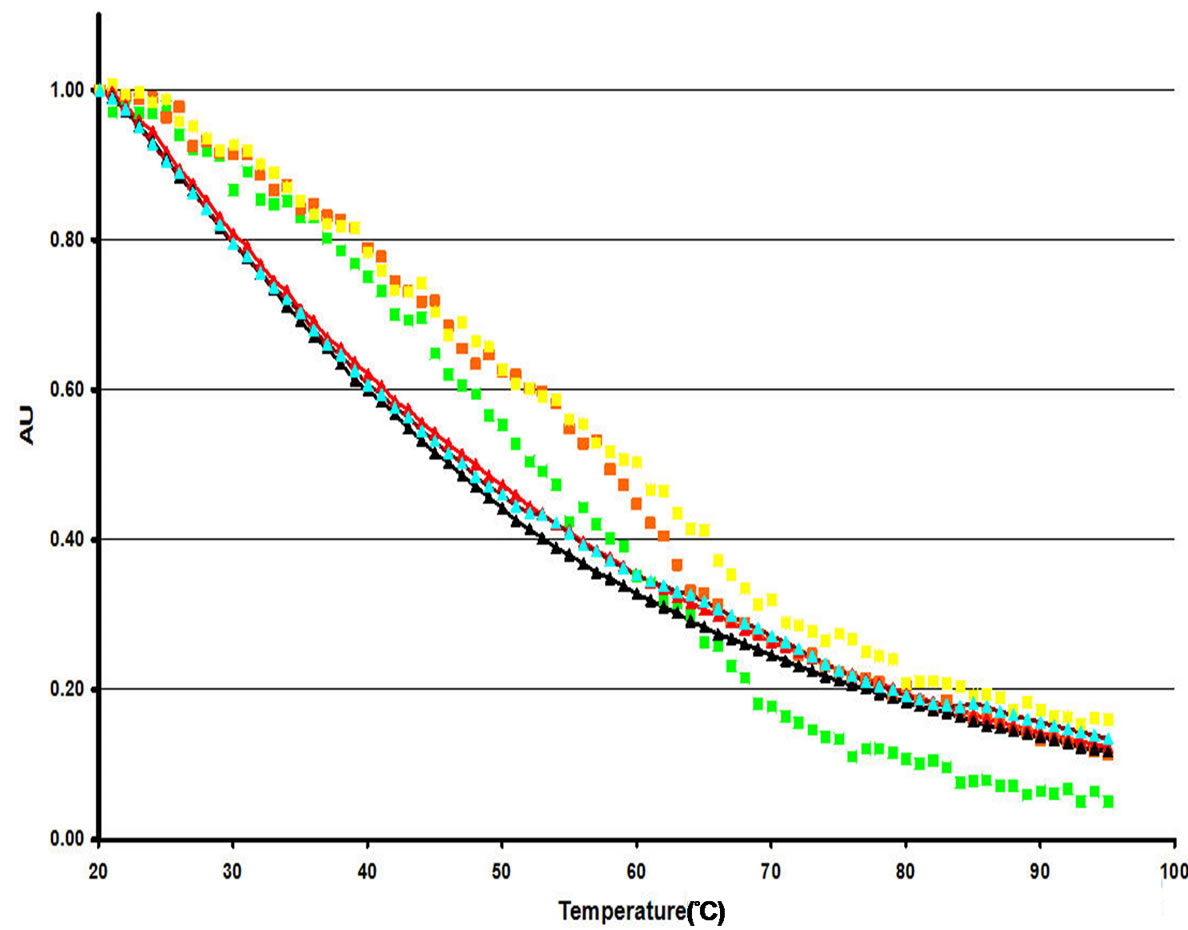 (c)
(c)
Figure 9. Secondary and tertiary structural analysis of “predominant” NP preparations analyzed through CD (a) and fluorescence spectroscopy (b and c). (a) Thermal denaturation was performed on “predominant” NP preparations under varying salt conditions ((n) 0 mM NaCl and (n) 150 mM NaCl)). Normalized tryptophan fluorescence emission intensity as a function of heat (25˚C to 95˚C) for “predominant” NP preparations at pH 7.0 (b) and pH 3.0 (c) at varying salt concentrations ((n) 0M NaCl, (n) 0.5 M NaCl, (n) 1.0 M NaCl). NP samples at both pH levels were compared to free tryptophan at varying salt concentrations ((►) 0 M NaCl, (►) 0.5 M NaCl, (►) 1.0 M NaCl).
lower temperatures, where the protein is expected to be folded, the loss in quantum yield is retarded relative to that seen in tryptophan free in solution. However, at higher temperatures the loss of quantum yields for NP in the “predominant form” is accelerated relative to that seen with free tryptophan in solution (Figure 9(b), green squares).
For tryptophan free in solution, increasing the salt concentration from 0 M to 0.5 M NaCl produces a slight delay in loss of quantum yield with temperature (Figure 9(b), triangles). A further increase in salt concentration from 0.5 M to 1.0 M NaCl had little additional effect (Figure 9(b), pink triangles to light blue triangles). The addition of salt had little effect on the quantum yield of the intrinsic tryptophan fluorescence for NP in the “predominant” form at low temperatures. However, at intermediate and higher temperatures, increasing salt appears to increasingly shield the intrinsic tryptophan fluorescence from a loss in quantum yield (Figure 9(b), red and yellow squares). At 0.5 M and 1.0 M NaCl, the point of inflection in the quantum yield curve for NP in the “predominant” form occurs at lower temperatures than in the absence of salt.
At low temperatures, the fluorescence results at pH 3.0 are very similar to those seen for the “predominant” form at pH 7.0: the loss of quantum yield with increasing temperature is less dramatic than that seen for tryptophan free in solution (Figures 9(b) and (c)). However, at higher temperatures there is much more loss of quantum yield at pH 3.0 than at pH 7.0 and the loss in observed fluorescence or exceeds (0 M NaCl, Figure 9(c), green squares) that of tryptophan free in solution (black triangles). As was the case for the “predominant” form at pH 7.0, salt appears to have little effect at lower temperatures. However, at higher temperatures, addition of 0.5 M NaCl delayed the temperature-induced onset of loss of tryptophan emission intensity relative to that seen in the absence of salt (Figure 9(c), pink vs. green squares). However, further of addition of salt (1.0 M NaCl final, Figure 9(c), yellow squares) seemed to reverse this effect to some extent.
The effect of salt on the temperature dependence of tryptophan fluorescence emission intensity in “anomalous” preparations was quite different than that observed for “predominant” preparations. At neutral pH, addition of 0.5 M NaCl hastened the onset of loss of emission intensity relative to preparations in which there was no salt (Figure 10(a)). An intermediate effect was observed at 1.0 M NaCl. At pH 3.0, the intensity versus temperature curve (Figure 10(b)) of “anomalous” preparations in the presence of 0.5 M NaCl was very similar to that containing no salt, whereas the addition of 1.0 M NaCl shifted the curve to the left (and the onset of loss of emission intensity occurring at lower temperatures). At neural pH (0.5 M NaCl, pH 7.0, for example), the plots of quantum yield versus temperature for the “anomalous” preparation actually appeared to be triphasic rather than biphasic. The same effect is not seen when the pH is decreased to 3.0.
4. Discussion
The main objective of this study was to characterize the nucleocapsid protein (NP) antigen of the avian H5N1 influenza strain using physico-chemical methods in order to gain insights into the properties of the antigen that might influence the quality, safety and efficacy of a therapeutic vaccine. In order to fully characterize NP, the recombinant protein was first produced in E. coli. Twelve of the fifteen (80%) initial NP preparations (produced without RNase inhibition) resulted in CD spectra (the “predominant” spectrum) with two distinct minima at approximately 222 nm and 208 nm, reminiscent of spectra from proteins with high α-helical content [14]. Despite the fact that secondary structure deconvolution programs failed to give an estimate of secondary structure content that produced a satisfactory fit to the experimental data, these CD data do indicate that the purified protein was successfully refolded. In contrast to the two peaks of negative ellipticity observed in the spectra of NP samples, CD spectra of random coil/extended protein chains exhibit little or slightly positive ellipticity between 210 nm and 240 nm and either a single, sharp minimum at approximately 198 nm or a single, more shallow minimum centered between 195 nm and 205 nm [14]. Spectra resembling those expected for random coil conformation could be obtained from NP protein through thermal denaturetion (see below) indicating that the spectrum in Figure 2 was the result of a folded structure that was lost upon heating. The available X-ray crystal structure of NP [18] also indicates that the protein is rich in α-helix (48.6% α-helix, 4.8% β-strand and 46.6% loop). The remaining three preparations (20%) of NP (“anomalous” preparations) produced without deliberate RNase inhibition resulted in CD spectra with one distinct minimum at 222 nm. The negative ellipticity at 222 nm suggests that, similar to the “predominant” preparations, the protein is largely helical; however, the absence of an equally predominant minimum at 208 nm suggests that the conformation of these “anomalous” preparations differ from that of the “predominant” preparation.
The observed spectral differences between the “anomalous” and “predominant” preparations cannot be explained by changing the relative proportion of other common protein secondary structural elements (a-helix, β-sheet, turn or random coil), all of which give signifycant negative ellipticity at lower wavelengths [15]. The fact that NP”s primary biological function is to bind viral RNA and research indicating that nucleotides (RNA/DNA)
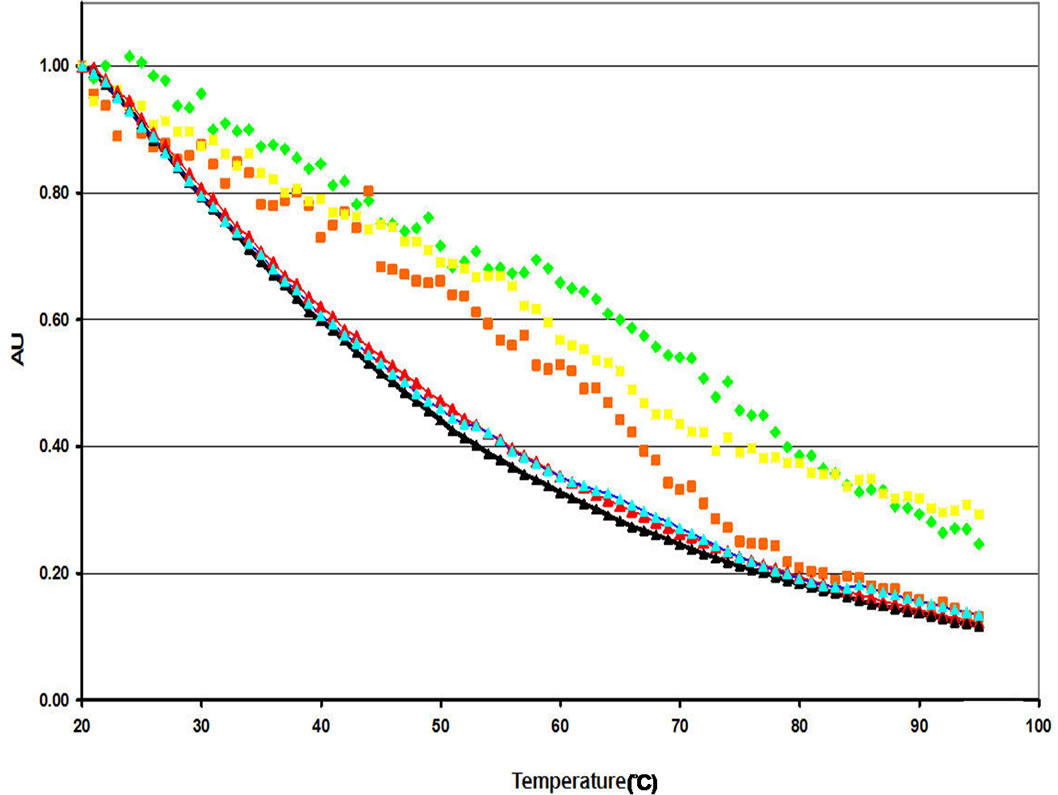 (a)
(a) (b)
(b)
Figure 10. Tertiary structural analysis of “anomalous” NP preparations analyzed through fluorescence spectroscopy at pH 7.0 (a) and pH 3.0 (b). Normalized tryptophan fluorescence emission intensity as a function of heat (25˚C to 95˚C) for “anomalous” NP preparations at pH 7.0 (a) and pH 3.0 (b) at varying salt concentrations ((n) 0 M NaCl, (n) 0.5 M NaCl, (n) 1.0 M NaCl). NP samples at both pH levels were compared to free tryptophan in solution at varying salt concentrations ((►) 0 M NaCl, (►) 0.5 M NaCl, (►) 1.0 M NaCl).
in certain conformations produce a characteristic negative ellipticity between 230 nm and 250 nm [16] suggested that the “anomalous” spectrum resulted from RNA binding to NP during purification. Therefore, the next NP preparation that gave an “anomalous” CD spectrum after purification was subject to RNaseA digestion. Overnight incubation of “anomalous” NP samples with RNaseA resulted in a CD spectrum that was reminiscent of the predominant preparation with a minima at both 222 nm and 208 nm. These data indicate that NP has the propensity to bind RNA during the purification process, and that RNA binding impacts the CD spectrum and possibly the secondary structure of NP.
Data from two other lines of experimental probing support this conclusion. Firstly, RNaseA was deliberately added into the lysis buffer prior to disruption of the recombinant E. coli cells expressing NP with the expectation that a preparation giving the “predominant” CD spectrum would result. Interestingly, in all three preparations where RNaseA was added to the lysis buffer, RNaseA addition precluded the isolation of any NP, suggesting that at least small amounts of RNA maybe required for successful isolation of NP. Secondly, four preparations were executed in an RNase-free environment (utilizing DEPC treated water, sterile pipette tips, an established RNase-free working area and the addition of a small molecule RNase inhibitor (vanadyl ribonucleoside complex) [13] to the lysis solution) with the expectation that the inhibition of RNase enzymes would result in an “anomalous” preparation and CD spectrum. This was indeed the case: each of the preparations where the inhibitor was included gave “anomalous” spectra, again indicating that it is the binding of RNA to NP that results in the alteration in NP and/or RNA conformation and the “anomalous” CD spectra observed.
Unfortunately, attempts to reproduce the “anomalous” form of NP by adding total yeast RNA to RNA-free NP were unsuccessful. While it has been suggested that influenza NP binds RNA in a non-sequence-specific fashion [19], demonstrations of RNA binding to influenza NP by this group [20] and others [9] have been accomplished only when using a virus-specific oligonucleotide sequence. Further experiments using influenza-specific RNA as a titrant may resolve this issue. The conclusion from the RNase A and RNase-free experiments that the “anomalous” spectrum results from the presence of RNA is bolstered by literature indicating that recombinant NP from other negative stranded RNA viruses (e.g., Ebola, Vesicular Stomatitis Virus, Rabies) also have the propensity to bind RNA of their expression system [20-23]. The data we present here indicate that association of NP and RNA may well result in observable changes in influenza NP structure and such association must be tightly monitored by both vaccine manufacturers and regulators of NPbased vaccines for human use.
Regulatory guidance advises the full characterization of the quality (physico-chemical) attributes of therapeutic proteins, including the protein”s response to stress conditions like exposure to extremes of temperature and pH [24] where proteins are likely to unfold. Acid-induced unfolding of a protein is driven by the protonation of ionizable side chains of amino acids comprising the polypeptide which leads to intramolecular charge repulsion and subsequent loss of secondary and tertiary structure [25]. The CD spectrum of the “predominant” form of NP in its RNA-free state at pH 7.0 (Figure 2 blue trace) contains two distinct negative peaks at 222 nm and 208 nm suggesting high α-helical content, content one would expect to lose on acid-induced unfolding of the protein. However, rather than seeing the intensity of these peaks decrease upon solution acidification, a dramatic increase in negative ellipticity was unexpectedly observed. Thus “predominant” NP preparations, when titrated with acid, can be driven into an acid-induced state which exhibits increased negative ellipticity at both minima, suggesting an increased helical structure compared to the neutral state (Figure 3(a)). Although these results are surprising, there are reports in the literature in which proteins do not unfold when exposed to acidic conditions but rather assume higher ordered acid-induced conformations [25,26]. The acid-induced conformational change in the “predominant” form of NP is globally reversible.
Acid titrations of the “anomalous” preparations were also performed. Addition of either HCl or H2SO4 resulted in the retention of the overall anomalous spectral profile (single minimum at 222 nm) until pH of 5.5 although the intensity of the signal decreased as the solution became more acidic. This loss of ellipticity as the pH decreased is consistent with a loss in regular secondary structure elements. However, once the pH was decreased to below 5.5, the trend was interrupted and NP displayed instead the profile seen with the “predominant” form at lower pH values: one marked by the increased negative ellipticity and defined minima at both 222 nm and 208 nm. This change in “anomalous” to “predominant” profile suggests that, during acidification, NP releases its bound RNA and takes on a conformation (with increased helical content) similar to that of the predominant preparation at pH 3.0. In addition, the “anomalous” spectrum initially observed at pH 7.0 is not seen upon reverse titration with NaOH; rather, the spectrum achieved on back titration appears to be similar to that observed for a “predominant” preparation. Indeed, the spectrum at pH 7.0 after acidification of an “anomalous” preparation overlays closely with the spectrum of a “predominant” NP sample prior to acidification, indicating that, upon acidification, the “anomalous” preparation releases its RNA but fails to reincorporate that RNA when the pH is returned to neutral values.
During the course of this work, the lab group of PangChui Shaw and collaborators published an extensive analysis of the link between NP oligomerization and its ability to form ribonucleoprotein (RNP) particles. Their original paper [18] described the first X-ray structure of H5N1 NP and showed that recombinant NP (at 2.5 mg/mL), in the absence of RNA, existed as a mixture of trimer and tetramer. These authors propose that homo-oligomerization is facilitated through the insertion of the tail loop domain of one NP protomer into the body domain of a neighboring NP protomer at the “insertion groove”. This hypothesis is supported by their more recent work [20] demonstrating that mutations which disrupt the tail loop structure or abrogate the ability to form a salt bridge between R416 in the tail loop of one protomer and E339 in the insertion groove of another protomer were both deficient in ribonucleoprotein (RNP) activity (transcription-replication activity) and had lost the ability to oligomerize. Size exclusion chromatography experiments also indicate that, in the presence of RNA, the amount of wild type NP in the trimeric/tetrameric forms is greatly diminished, and largely replaced by higher oligomeric forms of NP complexed with RNA [20].
The ability to form a salt bridge depends upon the positive charge on R416 and a negative charge on E339, a condition which presumably existed in both “predominant” and “anomalous” preparations at pH 7.4. Acid titration of either form should result in the protonation and concomitant loss of charge on E339. Analogous to the E339A mutant [20], deprotonated E339 can no longer engage in a salt bridge and NP at pH 3.0 should be monomeric thus explaining the observed change in CD spectrum for the “predominant” form of NP on acid titration. The results presented in this paper in conjunction with Chan and coworkers” published work indicate that the monomeric form of NP is more helical than the trimeric/tetrameric form of NP in the absence of RNA (i.e., the RNA-free “predominant” form of NP we isolated). The “anomalous” spectrum at pH 7.4 represents higher oligomeric forms of NP complexed with RNA also isolated by Chan and coworkers. Acidification of this RNA-complexed (“anomalous”) form of NP results in protonation of E339, which in turn disrupts the salt bridge maintaining the interaction of NP protomers in this higher oligiomeric state and results in the loss of RNA from the complex and formation of the same monomeric NP seen on acid titration of the “predominant”, RNA-free form. Because experimental conditions used in our present study did not preclude the activity of advantageous RNases, the RNA once released from the NP-RNA complex is presumably digested and no longer available for reincorporation when the pH is returned to neutral. Thus back-titration of the “anomalous” preparation results in a CD spectrum reminiscent of the “predominant” form at pH 7.4 rather than the original “anomalous” spectrum.
During the course of the acid titrations described above, changes in tertiary structure of each type of NP preparation (“predominant” and “anomalous”) was monitored using the molecule’s tryptophan residues as probes. Titration of the “predominant” preparations with either HCl or H2SO4 resulted in a red shift (shifting of the emission maximum to higher wavelengths), indicating that the aromatics that were buried inside the hydrophobic pocket of the protein are becoming more exposed to the hydrophilic (aqueous solvent) environment as the pH is decreased. Bringing the pH back to 7.0 with NaOH results in a blue shift which indicated that, as the pH was raised, NP adopted a global fold that results in the burial of the aromatics into the hydrophobic core of NP.
Fluorescence profiles of the “anomalous” preparations displayed different fluorescent profiles throughout the titrations. When the “anomalous” preparations were acidified, a red shift was indeed observed. However, the “anomalous” preparation also gave a peak around 420 nm that appeared to increase in intensity as the pH was decreased. In contrast to the observations for the “predominant” form, no shift in emission maxima was observed on back-titration of the “anomalous” form. The additional peak at 420 nm did not decrease in intensity, and can not be attributed to the presence of RNA in the sample. While the source of the fluorescence emission at 420 nm is not clear, the results of the fluorescence data from the acid titration of the “anomalous” preparations confirms the conclusions of the CD data for this moiety: the conformation of the “anomalous” preparation before and after acid titration is not the same and neutralizing the pH does not restore the original “anomalous” conformation and/or RNA binding. These data, viewed in conjunction with acidification experiments performed using “anomalous” preparations that had been RNase A-digested, lend further credence to the postulation that acidification of “anomalous” preparations leads to the irreversible loss of bound RNA and a concomitant conformational change.
Both CD data (an indicator of secondary structure) and normalized tryptophan fluorescence emission intensity (one indicator of tertiary structure) suggested that the addition of salt may decrease the stability of the local environment of the “predominant”, RNA-free form of NP at neutral pH. The CD data clearly indicate that addition of salt shifts the melting curve to the left, indicating protein destabilization. Fluorescence data at neutral pH indicate that tertiary structure is lost concomitantly with secondary structure both in the presence and absence of salt. At lower temperatures (where the protein is expected to be folded) in the absence of salt the loss in quantum yield is actually retarded relative to that seen in tryptophan free in solution indicating that the protein structure is shielding the tryptophan from solvent quenching. At higher temperatures, the loss of quantum yield actually occurs more rapidly than for tryptophan which is free in solution, indicating that in the unfolded protein, side chains from other residues are also contributing to quenching. The fact that the discontinuity in the rate of loss of quantum yield (break in the curve) occurs at approximately the same temperature as the TM observed for NP in the “predominant” form upon thermal denaturation followed by CD reinforces the conclusion that the discontinuity in the quantum yield curve is due to concerted protein unfolding upon increasing temperature. For tryptophan in NP in the “predominant” form, the addition of salt has very little effect on the quantum yield at low temperatures, where the protein is expected to be folded. However, at intermediate temperatures, where the protein is beginning to unfold, and at higher temperatures, where the protein is likely to be completely unfolded, increasing salt appears to increasingly shield the tryptophan fluorescence from quenching. At 0.5 M and 1.0 M NaCl, the break in the quantum yield curve for NP in the “predominant” form occurs at lower temperatures than in the absence of salt, indicating that salt is destabilizing the protein. While technical limitations precluded the use of higher concentrations of salt, the CD data at 0.15 M NaCl also indicate that salt destabilizes NP in the “predominant” form. The fact that the effects of salt on the “predominant” form of NP were different at pH 3.0 compared to those observed at pH 7.0 reinforces the conclusion that the folded structure of NP is very different in the presence of acid.
Fluorescence data indicate that the effects of salt in combination with temperature for the “anomalous” (RNAbound) preparation of NP at neutral pH are quite complex. Not only is the effect of salt different than for the “predominant” preparation at this same pH, the curves resulting from plotting normalized fluorescence intensity against temperature appear to be triphasic for the “anomalous” preparation rather than biphasic. These data are consistent with the conclusion (from the RNase A digestion experiments) that the “anomalous” form of NP results from binding of RNA to the protein. As the temperature increases, in the absence of salt, one might expect that dissociation of the RNA from the protein (with a concomitant conformational change) occurs before (at lower temperatures) the complete unfolding of the protein. The presence of salt would be expected to influence the binding of the negatively charged RNA to the positively charged arginine residues in binding groove between the head and body domain of NP. Theory [27] suggests that lower salt concentrations might be expected to be stabilizing, whereas higher salt concentrations would be expected to interrupt the charge-charge interactions and thus destabilize the binding. While the data indicate differences between the fluorescence intensity vs temperature plots for “anomalous” preparation at pH 7.0 in the presence of various amounts of salt, it is difficult to draw conclusions as one does not know a priori what the effect of RNA binding to NP will be on the quantum yield of tryptophan fluorescence. Further experiments with smaller graduations in concentration covering a wider range of salt concentrations would need to be performed to elucidate this effect. The data here, however, do help support the presence of RNA in the “anomalous” form at pH 7.0 but not pH 3.0.
5. Conclusions
The research presented here is, to the best of our knowledge, the first attempt to characterize the influenza H5N1 NP antigen using physico-chemical methodology that includes CD and fluorescence. Overall, the data presented here demonstrate that NP is a dynamic protein that can adopt several different conformations. Even when the expression and purification protocol remain ostensibly unchanged, the NP product can give at least two different CD spectra (“predominant” and “anomalous”), suggesting it can adopt at least two different conformations. Acidification of NP solutions (“predominant” or “anomalous”) results in yet a third, structured conformation. The data also show that the “anomalous” spectrum/conformation is most likely the result of RNA binding to NP. Since the “anomalous” spectrum cannot be reproduced by mathematically summing the CD spectrum of RNA and that of NP in the “predominant” form, these findings refute the notion [19] that the nucleocapsid protein does not change conformation when binding RNA. Taken in conjunction with the published literature, the data also suggest that the monomeric form of NP (achieved here by acidification rather than mutation to interrupt an essential salt bridge) is conformationally distinct and, indeed, more highly helical than the trimeric/tetrameric (“predominant”) form in the absence of RNA of NP. Previous researchers have suggested that, despite structural differences among individual NP molecules in the unit cell, the structure of one NP protomer in the trimeric crystal is representative of NP structure in both its monomeric and trimeric/tetrameric/higher oligomeric forms [18]. The results presented here suggest that this is not the case: self-association of NP may well be accompanied by a fairly major structural change. These data, therefore, form the basis of a different and perhaps more complete understanding of how the influenza virus achieves nucleocapsid particle formation and binds RNA for packaging or exposes it to replication during the viral life cycle.
In addition to adding to our understanding of the boilogy of the influenza virus, the data in this paper also have ramifications for the production, regulation and quality assurance of potential therapeutic vaccines that use NP protein as the antigen. Vaccines, and indeed all biologics, are subjected to strict regulatory scrutiny. Manufacturers must demonstrate that the material in a lot released for sale today is identical to the lot that was demonstrated to be safe and effective in clinical trials performed to achieve licensure. For vaccines, even those that are therapeutic rather than prophylactic, the conformation of the antigen or protein component is extremely important. NP, when used in trial vaccines, has been shown to induce effective antibodies [7,8]. Conformational changes in proteins can expose different epitopes and result in changes in the immune response to the protein. NP is an internal protein and not exposed on the surface of the virus and the antibodies elicited by NP appear to be non-neutralizing (i.e., they do not result in capture and death of the whole virus). Therefore, one might argue that a conformational change, even one that results in the induction of a different antibody, may not alter the therapeutic effect of eliciting CTL response. Even if this argument proves to be true, efficacy is only one part of the regulatory consideration. Safety is an equally, if not more, important consideration. When an NP-based vaccine is eventually licensed, the manufacturers will have had to demonstrate, through clinical trials, that the non-neutralizing antibody response raised by the NP antigen does not result in untoward effects and that the vaccine is safe. Subsequent lots of vaccine that present a different conformation of NP (because of different isolation procedures, differences in oligomeric state or the amount of RNA bound or differences in the pH or salt concentration of the buffer) run the risk of eliciting a different antibody response-one that has not been demonstrated to be safe through clinical trials.
In a recent expert review, Volkin and Middaugh have suggested that the application of physico-chemical methods could provide a basis for “[treating] vaccines as well-defined physico-chemical substances in which particular structural changes can be detected and used to reduce or eliminate deleterious events” [11]. While these authors envision such characterization as a means of enhancing the rational design of stable formulation, the approach is equally applicable in a regulatory or quality assurance context. The use of physico-chemical characterization is the norm for other biologics using recombinant proteins, but these newer techniques are only beginning to see application in vaccine evaluation. The results presented here demonstrate the utility of physico-chemical methods in the complete characterization of potential vaccines.
REFERENCES
- World Health Organization, Influenza-Seasonal Fact Sheet 211. Available via WHO Media Centre, 2009. http://www.who.int/mediacentre/factsheets/fs211/en/index.html
- J. A. Wiley, R. J. Hogan, D. L. Woodland and A. G. Harmsen, “Antigen-Specific CD8(+) T Cells Persist in the Upper Respiratory Tract Following Influenza Virus Infection,” The Journal of Immunology, Vol. 167, No. 6, 2001, pp. 3293-3299.
- G. F. Rimmelzwaan and A. D. Osterhaus, “Cytotoxic T Lymphocyte Memory: Role in Cross-Protective Immunity against Influenza?” Vaccine, Vol. 13, No. 8, 1995, pp. 703-705.
- A. J. McMichael and B. A. Askonas, “Influenza VirusSpecific Cytotoxic T Cells in Man; Induction and Properties of the Cytotoxic Cell,” European Journal of Immunology, Vol. 8, No. 10, 1978, pp. 705-711. doi:10.1002/eji.1830081007
- C. Gschoesser, G. Almanzar, U. Hainz, J. Ortin, D. Schonitzer, H. Schild, M. Saurwein-Teissl and B. GrubeckLoebenstein, “CD4+ and CD8+ Mediated Cellular Immune Response to Recombinant Influenza Nucleoprotein,” Vaccine, Vol. 20, No. 31-32, 2002, pp. 3731-3738. doi:10.1016/S0264-410X(02)00355-9
- D. M. Carragher, D. A. Kaminski, A. Moquin, L. Hartson and T. D. Randall, “A Novel Role for Non-Neutralizing Antibodies against Nucleoprotein in Facilitating Resistance to Influenza Virus,” The Journal of Immunology, Vol. 181,No. 6, 2008, pp. 4168-4176.
- M. W. LaMere, H. T. Lam, A. Moquin, L. Haynes, F. E. Lund, T. D. Randall and D. A. Kaminski, “Contributions of Antinucleoprotein IgG to Heterosubtypic Immunity against Influenza Virus,” The Journal of Immunology, Vol. 186, No. 7, 2011, pp. 4331-4339. doi:10.4049/jimmunol.1003057
- M. W. Lamere, A. Moquin, F. E. Lee, R. S. Misra, P. J. Blair, L. Haynes, T. D. Randall, F. E. Lund and D. A. Kaminski, “Regulation of Antinucleoprotein IgG by Systemic Vaccination and Its Effect on Influenza Virus Clearance,” Journal of Virology, Vol. 85, No. 10, 2011, pp. 5027-5035. doi:10.1128/JVI.00150-11
- Q. Ye, R. M. Krug and Y. J. Tao, “The Mechanism by Which Influenza A Virus Nucleoprotein Forms Oligomers and Binds Rna,” Nature, Vol. 444, No. 7122, 2006, pp. 1078-1082. doi:10.1038/nature05379
- S. Roy, G. P. Kobinger, J. Lin, J. Figueredo, R. Calcedo, D. Kobasa and J. M. Wilson, “Partial Protection against H5N1 Influenza in Mice with a Single Dose of a Chimpanzee Adenovirus Vector Expressing Nucleoprotein,” Vaccine, Vol. 25,No. 39-40, 2007, pp. 6845-6851. doi:10.1016/j.vaccine.2007.07.035
- D. B. Volkin and C. R. Middaugh, “Vaccines as Physically and Chemically Well-Defined Pharmaceutical Dosage Forms,” Expert Review of Vaccines, Vol. 9, No. 7, 2010, pp. 689-691. doi:10.1586/erv.10.73
- N. C. Pace, B. A. Shirley and J. A. Thomson, “Measuring the Conformational Stability of a Protein”. http://dasher.wustl.edu/bio5325/reading/pace-review-87.pdf
- B. L. Pasloske, “Chapter 8: Ribonuclease Inhibitors,” In: C. H. Schein, Ed., Methods in Molecular Biology: Nuclease Methods and Protocols, Humana Press Inc., Totowa, p. 105.
- N. J. Greenfield, “Using Circular Dichroism Collected as a Function of Temperature to Determine the Thermodynamics of Protein Unfolding and Binding Interactions,” Nature Protocols, Vol. 1, No. 6, 2006, pp. 2527-2535.
- N. J. Greenfield, “Methods to Estimate the Conformation of Proteins and Polypeptides from Circular Dichroism Data,” Analytical Biochemistry, Vol. 235, No. 1, 1996, pp. 1-10. doi:10.1006/abio.1996.0084
- M. I. Barría, A. González, J. Vera-Otarola, U. León, V. Vollrath, D. Marsac, O. Monasterio, T. Pérez-Acle, A. Soza and M. López-Lastra, “Analysis of Natural Variants of the he Patitis C Virus Internal Ribosome Entry Site Reveals That Primary Sequence Plays a Key Role in Cap-Independent Translation,” Nucleic Acids Research, Vol. 37, No. 3, 2009, pp. 957-971. doi:10.1093/nar/gkn1022
- J. A. Gally and G. M. Edelman, “The Effect of Temperature on the Fluorescence of Some Aromatic Amino Acids and Proteins,” Biochimica et Biophysica Acta, Vol. 60, No. 3, 1962, pp. 499-509. doi:10.1016/0006-3002(62)90869-7
- A. K. Ng, H. Zhang, K. Tan, Z. Li, J. H. Liu, P. K. Chan, S. M. Li, W. Y. Chan, S. W. Au, A. Joachimiak, T. Walz, J. H. Wang and P. C. Shaw, “Structure of the Influenza Virus A H5N1 Nucleoprotein: Implications for RNA Binding, Oligomerization, and Vaccine Design,” The FASEB Journal, Vol. 22, No. 10, 2008, pp. 3638-3647. doi:10.1096/fj.08-112110
- A. K. Ng, J. H. Wang and P. C. Shaw, “Structure and Sequence Analysis of Influenza A Virus Nucleoprotein,” Science in China Series C: Life Sciences, Vol. 52, No. 5, 2009, pp. 439-449. doi:10.1007/s11427-009-0064-x
- W. H. Chan, A. K. Ng, N. C. Robb, M. K. Lam, P. K. Chan, S. W. Au, J. H. Wang, E. Fodor and P. C. Shaw, “Functional Analysis of the Influenza Virus H5N1 Nucleoprotein Tail Loop reveals Amino Acids That Are Crucial for Oligomerization and Ribonucleoprotein Activities,” Journal of Virology, Vol. 84, No. 14, 2010, pp. 7337-7345. doi:10.1128/JVI.02474-09
- F. Iseni, A. Barge, F. Baudin, D. Blondel and R. W. Ruigrok, “Characterization of Rabies Virus Nucleocapsids and Recombinant Nucleocapsid-Like Structures,” Journal of General Virology, Vol. 79, Part 12, 1998, pp. 2909- 2919.
- T. Noda, K. Hagiwara, H. Sagara and Y. Kawaoka, “Characterization of the Ebola Virus Nucleoprotein-RNA Complex,” Journal of General Virology, Vol. 91, Part 6, 2010, pp. 1478-1483.
- T. J. Green, S. Macpherson, S. Qiu, J. Lebowitz, G. W. Wertz and M. Luo, “Study of the Assembly of Vesicular Stomatitis Virus N Protein: Role of the P Protein,” Journal of Virology, Vol. 74,No. 20, 2000, pp. 9515-9524. doi:10.1128/JVI.74.20.9515-9524.2000
- Food and Drug Administration, Department of Health and Human Services, “International Conference on Harmonisation; Guidance on Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process; Availability. Notice,” Federal Register, Vol. 70, No. 125, 2005, pp. 37861-37862.
- A. L. Fink, L. J. Calciano, Y. Goto, T. Kurotsu and D. R. Palleros, “Classification of Acid Denaturation of Proteins: Intermediates and Unfolded States,” Biochemistry, Vol. 33, No. 41, 1994, pp. 12504-12511. doi:10.1021/bi00207a018
- E. Ahmad, S. Fatima, M. M. Khan and R. H. Khan, “More Stable Structure of Wheat Germ Lipase at Low pH than Its Native State,” Biochimie, Vol. 92, No. 7, 2010, pp. 885-893. doi:10.1016/j.biochi.2010.03.023
- T. Creighton, “Proteins: Structures and Molecular Properties,” W.H. Freeman and Company, New York, 1997.
NOTES
*Corresponding author.