Open Journal of Urology
Vol.2 No.2(2012), Article ID:19124,10 pages DOI:10.4236/oju.2012.22009
Replication of Prostate Cancer Risk Variants in a Danish Case-Control Association Study
1Department of Urology, Aarhus University Hospital Skejby, Aarhus, Denmark
2Department of Human Genetics, Aarhus University, Aarhus, Denmark
3Department of Molecular Medicine, Aarhus University Hospital Skejby, Aarhus, Denmark
Email: *diem.bentzon@dadlnet.dk
Received January 7, 2012; revised January 30, 2012; accepted February 24, 2012
Keywords: SNP; Family History; Cumulative Effect; Biochemical Relapse; Prostate Cancer Risk
ABSTRACT
Background: Prostate cancer is one of the main causes for cancer morbidity and mortality in Western countries. Recently, several single nucleotide polymorphisms (SNPs) associated with prostate cancer have been identified in genome-wide association studies and multiple variant models have been developed to predict prostate cancer risk. The association between genetic markers and clinico-pathological tumor variables has, however, been inconsistent. Methods and Materials: A total of 32 previously identified prostate cancer-associated risk SNPs were genotyped in 648 prostate cancer cases and 526 age-matched controls. Family history was obtained by questionnaire. Age at diagnosis, clinical tumor variables including preand postoperative PSA, Gleason score, and T stage were obtained from prospectively collected clinical data (Aarhus Prostate Cancer Study). The SNPs were genotyped using Sequenom and Taqman assays and associations between SNPs, prostate cancer risk, and clinico-pathological variables were assessed. Results: Seventeen SNPs were successfully replicated in our case-control study and the association estimates were consistent with previous reports. Four markers were excluded from further analysis due to linkage disequilibrium. The cumulative effect of having the disease-associated genotype at five SNPs (rs4430796, rs6983267, rs1859962, rs1447295 and rs16901979) increased the prostate cancer risk with odds ratio 6.02 (95% CI: 2.21 - 16.38; P = 1.0 × 10–4) in patients with any combination of ≥4 markers compared with patients without any of the five SNPs (P for trend = 1.0 × 10–4). Six markers were significantly associated with clinico-pathological variables: SNP rs2735839 (GG) at locus 19q13, which is in the KLK3 gene encoding PSA, was associated with high preoperative PSA (P = 0.04), low Gleason score (P = 0.01) and low T stage (P = 0.02). Variants rs5759167 (GG/GT) (22q13) and rs7679673 (CC/CA) (4q24) were correlated with low risk for biochemical relapse (P = 0.015 and P = 0.009, respectively), whereas rs6983267 (GG) (8q24) was significantly associated with biochemical recurrence (P = 0.045). In addition, variants rs6983267 (GG) and rs5759167 (GG/GT) were significantly associated with negative family history (P = 0.04 and P = 0.02, respectively). Conclusion: We replicated 17 previously identified prostate cancer-associated risk SNPs in a Danish case-control study and found a cumulative and significant association between five SNPs and prostate cancer. Overall, we noted significant associations between several prostate cancer-associated risk genotypes and less aggressive tumor variables, high level of PSA, and low risk for biochemical reccurrence.
1. Introduction
Prostate cancer is the most common cause of non-skin cancer morbidity and mortality in Western countries [1,2]. Family history is one of the strongest risk factors for prostate cancer in addition to age and ethnicity [3,4]. Based on large epidemiological cohort studies, familial prostate cancer accounts for 10% - 20% of all prostate cancer cases and hereditary prostate cancer for 5% - 10%. Except for early age at onset, no clinical patterns have been consistently associated with hereditary prostate cancer [5-13].
Since the introduction of PSA screening, more prostate cancer cases are diagnosed at a treatable stage. However, since not all cases are likely to progress it is important to distinguish the indolent from the aggressive form of prostate cancer.
Recently, more than 40 single nucleotide polymerphisms (SNP) associated with prostate cancer have been identified in genome-wide association studies [14-16]. Individually, these variants contribute only modestly to the risk; combined, they have cumulative risk [17-19]. However, findings on associations between genetic markers and clinico-pathological traits have been inconsistent [19-21].
In the present study, we evaluated 32 previously reported prostate cancer-associated risk variants in a casecontrol study comprising 526 control subjects and 648 cases diagnosed with prostate cancer.
2. Methods and Materials
2.1. Patients
Between 1997 and 2009, 648 patients with prostate cancer were recruited from the Central Denmark Region through the Department of Urology, Aarhus University Hospital, for the Aarhus Prostate Cancer Study. Prostate cancer patients between the ages of 36 and 77 were enrolled within few weeks of diagnosis. Of the 648 prostate cancer patients, 535 underwent retropubic or computerassisted laparoscopic radical prostatectomy for clinically localized prostate cancer (stage T1c-T2c).
In the current study, all 648 patients had DNA extracted from whole blood and provided information about prostate cancer in first-degree relatives. Demographic information and clinical characteristics including age at diagnosis, preoperative PSA, postoperative Gleason score and TNM (Tumor Node Metastasis) stage were obtained from the Aarhus Prostate Cancer Study database (Table 1). Based on preoperative PSA, preoperative T stage, and preoperative Gleason score the patients were grouped as low, intermediate, and high-risk according to D’Amico et al. [22]. Biochemical relapse was defined by a postoperative increase in PSA after radical prostatectomy based on a single measure of PSA ≥ 0.2 ng/ml.
Control individuals (n = 526) were patients either treated with percutaneous transluminal coronary angioplasty or enrolled in the DANish trial on Acute Myocardial Infarction-2 (DANAMI-2) [23]. All control subjects were agematched and information about their disease status was requested from the Danish Cancer Registry and the Cause of Death Registry; men diagnosed with prostate cancer were excluded from the control group. Written informed consent was obtained from both case patients and control patients and the Central Denmark Committee on Biochemical and Research Ethics and the Danish Data Protection Agency approved the study.
2.2. Selection of SNPs
Genotyping of eligible genetic variants included 32 SNPs with previously reported prostate cancer association (Table 2). The search strategy for the 32 prostate cancer-associated SNPs was as follows: genome-wide association studies on prostate cancer in the Pub Med database were selected. Only major association studies including cases diagnosed with prostate cancer and prostate cancer-free

Table 1. Clinico-pathological characteristics of the prostate cancer cases.
controls were included. SNPs reported as being signifycantly associated with prostate cancer risk and validated on at least one independent case-control study were included (genome-wide statistical significance level of 10–6) [24-39].
To optimize the genotyping regarding to cost, all candidate SNPs were combined into Sequenom assays. The two largest multiplex assays (i.e. with the highest numbers of SNPs incorporated) were analysed using the Sequenom platform. The remaining SNPs were genotyped using Taqman assays.
2.3. SNP Genotyping
Genotyping of 18 SNPs was performed using the Sequenom MassARRAY Genotyping system (Sequenom, San Diego, CA) (Table 2). Primers for PCR and extension probes were designed using the MassARRAY Assay Design 3.1 software (Sequenom). Multiplex PCR was performed in 5 µl reactions containing 10 ng of genomic DNA, 1.25 × PCR buffer (Qiagen, Valencia, CA), 0.5 mM dNTP (Roche, Geneva, Switzerland), 100 nM of
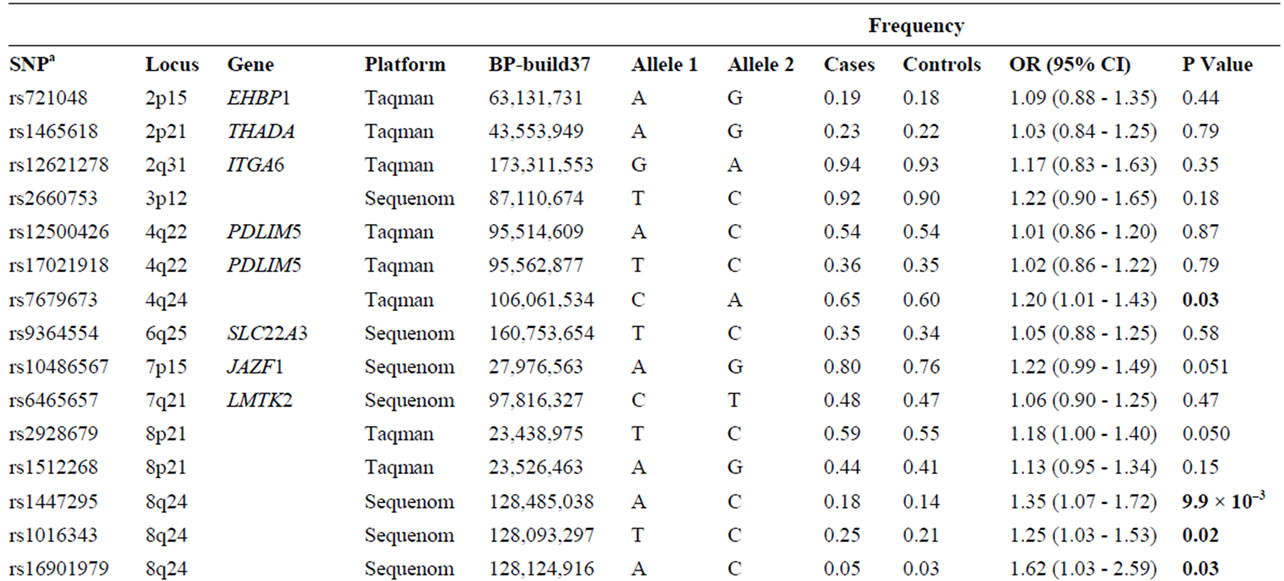
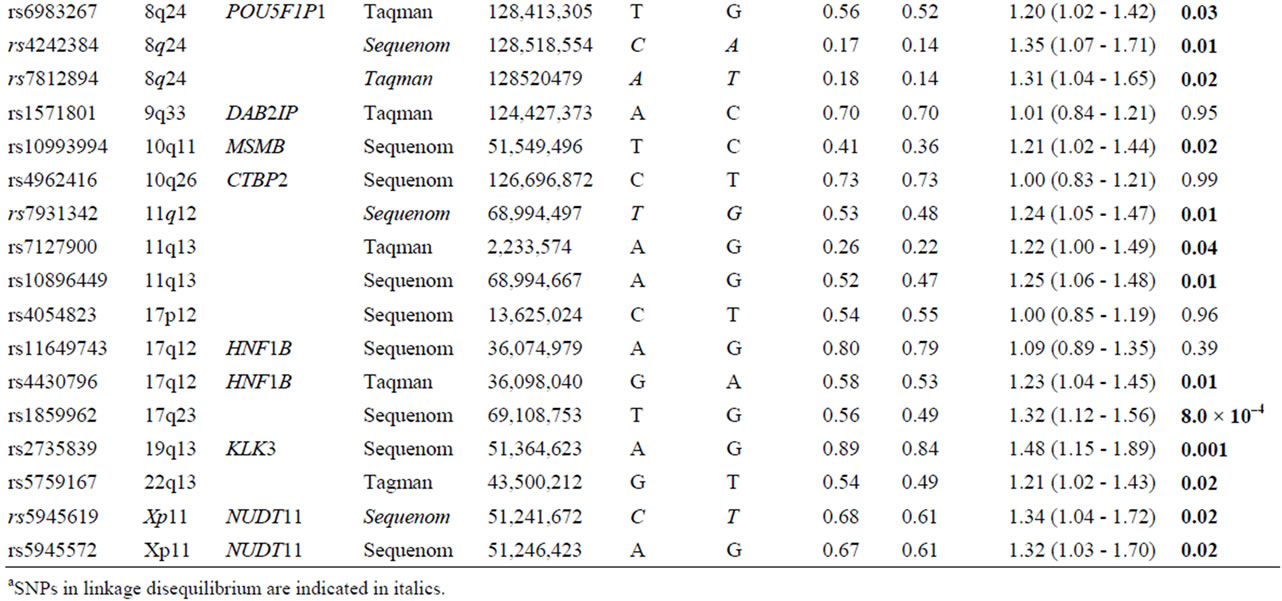
Table 2. List of 32 SNPs preselected from published literature.
each primer (Metabion, Martinsried, Germany) and 0.5U Taq polymerase (Qiagen), using the standard cycling conditions described by Sequenom. The PCR products were treated with arctic shrimp alkaline phosphatase (SAP) and the probe extension reaction (iPLEX) was carried out in accordance with the standard protocol (Sequenom). The iPLEX reactions were desalted using resin and spotted on a SpectroCHIP (Sequenom) using a nanodispenser. The samples were analyzed using a Bruker matrix assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometer (Sequenom) and the genotypes were determined using the MassARRAY Type 4.0 software (Sequenom). The primer sequences used can be obtained on request.
Fourteen SNPs were genotyped using commercial TaqMan genotyping assays from Applied Biosystems (Table 2). All genotyping reactions (5 µL total volume; 10 ng gDNA per reaction) were performed in 386-well plates on the Applied Biosystems 7900HT Fast RealTime PCR System using TaqMan Universal PCR Master Mix according to the manufacturer’s protocol (Applied Biosystems).
2.4. Statistical Analysis
Quality Control (QC) of the genotyping results was performed per individual and per marker. QC was conducted in PLiNK [40]. The individual DNA threshold call rate was set at > 0.8. For both cases and control, we used Fisher’s exact test to assess Hardy-Weinberg equilibrium for each SNP. Tests for pairwise linkage disequilibrium among variants in the control group were calculated by the Stata command pwld (David Clayton for Stata).
Assuming a multiplicative model, per-allele odds ratio (OR) and 95% confidence interval (CI) were calculated and allele frequency differences between 648 cases and 526 controls were tested using the Chi-square test df = 1- 2. Bonferroni correction for multiple testing was performed. Variants with significantly (P < 0.05) different allele frequencies between cases and controls were included in the genotype testing under the assumption of a dominant or recessive model by logistic regression controlled for age (continuous variable). ORs and 95% CIs for prostate cancer risk were calculated for individuals with risk genotypes compared with those with non-risk genotypes for each genetic model. The best-fitting model of each of the SNPs was conferred highest likelihood. Missing data were managed by omitting individuals with missing data in genotype counts.
The five variants presenting the best fitting genetic model with the highest likelihood were included in the cumulative effect model by assessing the presence of the prostate cancer-associated genotypes in each study subject. The five variants were rs16901979, rs6983267, rs4430796, rs1859962 and rs1447295 also previously tested by Zheng et al. [41]: one (1) was assigned for the presence of prostate cancer-associated genotypes of each of the five variants in each individual and zero (0) for non-presence. The cumulative effects of the five SNPs were tested by adding up the points for each SNP in each individual. Using logistic regression, the combinations of prostate cancer-associated genotypes (1, 2, 3, 4 and 5 SNPs) in each individual was estimated.
Associations between SNPs associated with prostate cancer risk and the clinico-pathological parameters were conducted on prostate cancer cases undergoing prostatectomy (n = 535).
The chi-square test was used in 2 × C contingency analysis for association between the prostate cancer-associated variants and preoperative PSA (<10 ng/ml; ≥10 ng/ml), postoperative T stage, postoperative Gleason score, D’Amico risk score, and family history (familial prostate cancer vs. sporadic prostate cancer). Postoperative T stage and Gleason score were stratified into two levels (≤T2c vs. >T2c and ≤6 vs. >6, respectively). The disease-free survival of the study subjects was shown in KaplanMeier plots. Time at risk was estimated from the date of surgery to the date of biochemical recurrence or prostate cancer-specific death, and case subjects without the events were censored at their last blood test. Log rank test (Mantel-Cox test) was used to compare the recurrence distribution of the genotypes. All Fischer’s exact tests and Chi-square tests in association analysis, logistic regression, Chi-square tests and Cochrane-Armitage test for trend were performed using intercooled Stata 10.0 (StataCorp LP, Texas, USA). The P-value significance level was set at <0.05.
3. Results
Thirty-two genetic variants were selected from published literature and genotyped for evaluation in our study population comprising 648 men diagnosed with prostate cancer and 526 heart patients as the control group. All 32 markers were polymorphic and in Hardy-Weinberg equilibrium. Thirty samples from the control group were genotyped in duplicates and the genotype concordance rate was above 99.5%.
Demographic information and clinical characteristics are listed in Table 1. Radical prostatectomy for clinically localized prostate cancer (cT2) was performed in 83% (535/648) of cases. Information about family history of prostate cancer in first-degree relatives was obtained from 535 (82.7%), of which 442 (82.5%) reported that none of their relatives were affected, whereas 94 (17.5%) stated at least one affected relative in the family (Table 1) The overall mean age of the prostate cancer group was 64.0 (95% CI: 63.5 - 64.6) years and 63.2 (95% CI: 62.5 - 64.1) years for control subjects.
3.1. Association Analysis of Alleles
Table 2 shows the unadjusted allelic ORs for each of 32 genetic variants comparing prostate cancer cases and control subjects. Four of the 32 markers were excluded due to linkage disequilebrium: SNPs rs78122894 and rs4242384 at 8q24 were filtered due to strong linkage with rs1447295 (r2 = 0.94; and r2 = 0.96, respectively); at chromosome 11, rs7931342 was excluded due to strong linkage with rs10896449 (r2 = 0.95). Finally, variants rs5945619 at Xq11 was in strong linkage with rs5945572 (r2 = 0.94) and therefore not included for further analysis. Variant rs5945572 at Xq11 was excluded from genotypic analysis due to only one allele. Thirteen SNPs exhibited significant ORs ranging from 1.20 to 1.62 (P = 8.0 × 10–4 - 0.04) including one marker at 4q24, 10q11, 11q13, 17q12, 17q23, 19q13, 22q13 and four at 8q24. After correction for multiple testing, only marker rs1859962 at 17q23 survived.
Variant rs16901979 was statistically significantly associated with prostate cancer risk presenting the highest OR at 8q24 and overall (1.62; 95% CI: 1.03 - 2.59; P = 0.03), whereas the OR of rs1447295 was the most statistically significant (1.35; 95% CI: 1.07 - 1.72; P = 9.9 × 10–3). The remaining markers at 8q24, rs6983267 and rs1016343 were significantly associated with prostate cancer risk (OR: 1.20; 95% CI: 1.02 - 1.42; P = 0.03 and OR: 1.25; 95% CI: 1.03 - 1.53; P = 0.02). Four SNPs each located at 4q24, 10q11, 19q13 and 22q13 showed a significant association with prostate cancer (rs7679673, rs10993994, rs2735839 and rs5759167), of which the rs2735839 that was previously reported to be associated with PSA exhibited the statistically most significant OR (1.45; 95% CI: 1.15 - 1.89; P = 0.001). At 11q23 (rs10896449 and rs7127900), 17q (rs4430796 and rs1859962) and Xq11 (rs5945572), five markers were significantly associated with prostate cancer risk with ORs ranging from 1.22 to 1.34 and P-values between 8.0 × 10–4 and 0.04.
Overall, 17 markers including four in linkage disequilibrium, were successfully replicated in the present study.
3.2. Best-Fitting Models
Thirteen markers were tested for the best-fitting genetic model (recessive or dominant); the model with the highest likelihood was considered the best fit for the particular marker. Ten out of 13 markers remained significant. The dominant model was the best-fitting model for the seven markers at 4q24, 10q11, 11q13, 17q23 and 22q23 (rs7679673, rs1447295, rs16901979, rs10993994, rs- 10896449, rs1859962 and rs5759167) (Table 3). The bestfitting model for rs6983267 (8q24), rs4430796 (17q12) and rs2735839 (19q13) was the recessive.
All 10 variants with significantly best-fitting genetic models were subjected to further analysis for correlation with clinico-pathological variables and for cumulative effects.
3.3. Cumulative Effect Model
Zheng et al. found a cumulative association between five of the SNPs genotyped in the present study and prostate cancer [41]. We tested this association on the basis of the best-fitting genetic models of the present study controlled for age. The five markers were rs4430796 (17q12), rs6983267 (8q24), rs1859962 (17q23), rs1447295 and rs16901979 (both at 8q24). Marker rs1859962 showed best fit under the dominant model in the current study in contrast to the recessive model in the study by Zheng et al. (dominant mode P = 9.6 × 10–4 versus recessive mode P = 0.025). The models were otherwise similar. The estimated ORs (95% CI) for men carrying any combination of 1, 2, 3 or ≥4 of the disease-associated genotypes at these five SNPs were 2.11 (1.26 - 3.52), 2.58 (1.55 - 4.31), 3.97 (2.22 - 7.12) and 6.02 (2.21 - 16.38), respectively, for prostate cancer compared to those without any risk variants (Table 4). The test for trend was statistically significant (p = 1.0 × 10–4).
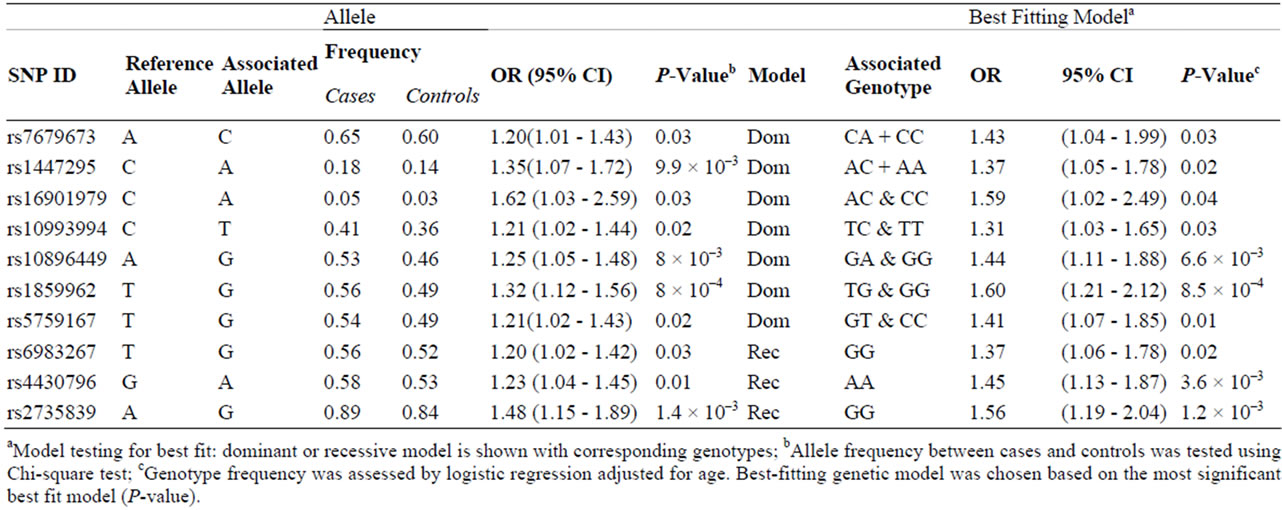
Table 3. Ten SNPs associated with prostate cancer were tested significant for the best fitting genetic model.
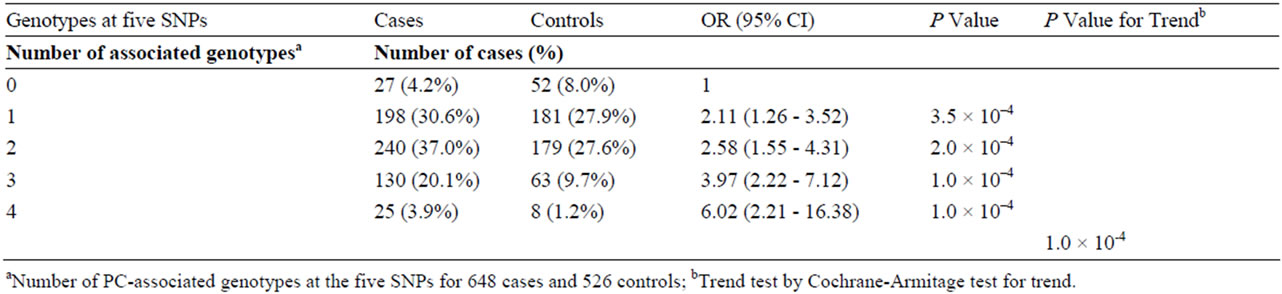
Table 4. The Cumulative effect of five SNPs associated with prostate cancer risk.
3.4. Clinico-Pathological Variables
The association between the 10 SNPs and clinico-pathological variables under the best fitting genetic model was assessed for prostatectomized cases. Variables tested were age at diagnosis, family history, PSA, postoperative Gleason score and T stage, and biochemical recurrence. Five markers with significant associations are summarized in Table 5. SNPs rs6983267 (GG) and rs5759167 (GG/GT) were significantly associated with negative family history (P = 0.04 and P = 0.02, respectively). SNP rs2735839(GG) and rs10896449(GG/GA) were significantly associated with high level of PSA (>10 ng/ml) (P = 0.04). Marker rs2735839 (GG) demonstrated a significant association with lower Gleason score (<7) (P = 0.0096). Lower T stage (≤T2c) was significantly associated with rs2735839 (GG) and rs4430796 (AA) (P = 0.02 and P = 0.03, respectively). Markers rs5759167 (GG/GT) and rs7679673 (CC/CA) demonstrated significant association with lower risk for biochemical recurrence (P = 0.015 and P = 0.009, respectively), whereas rs6983267 (GG) was significantly associated with biochemical relapse (P = 0.045) (Figure 1). None of the SNPs were associated with D’Amico score.
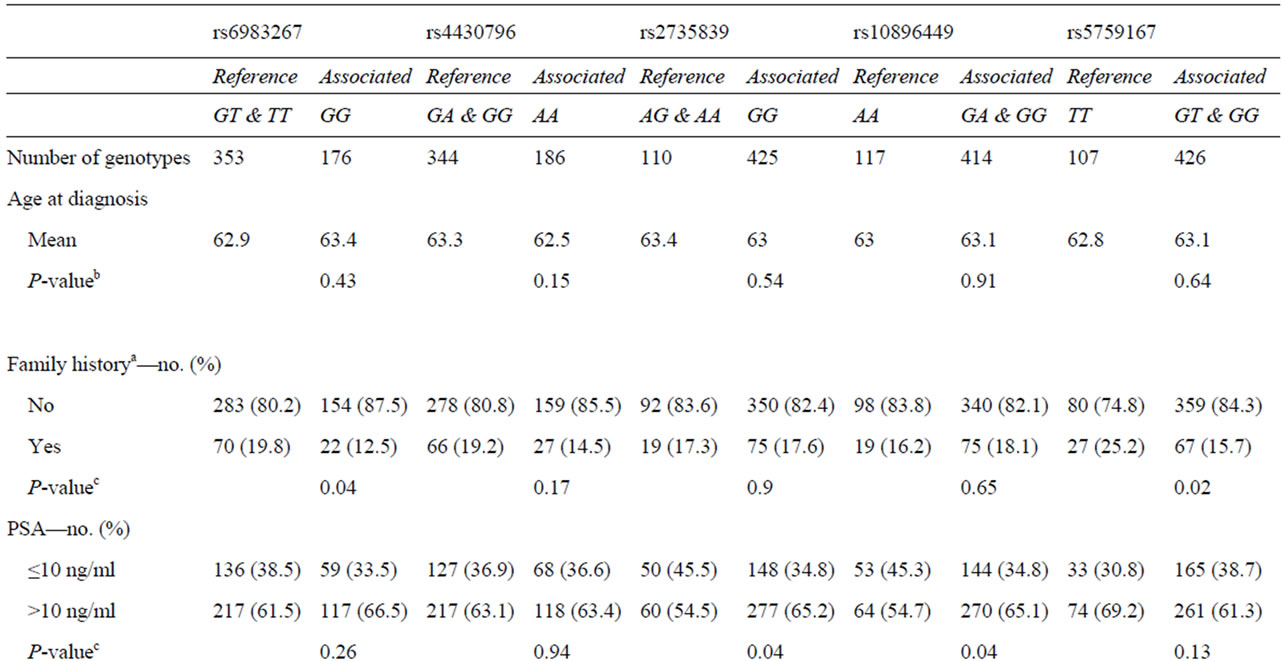
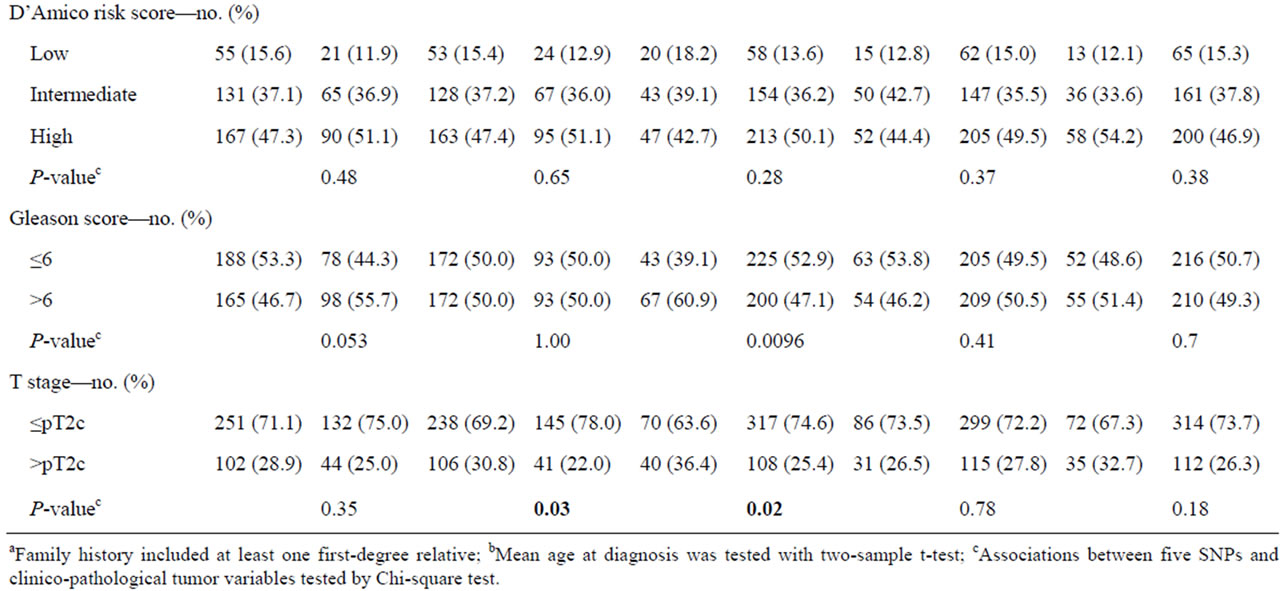
Table 5. Assocaitions between five SNPs and clinico-pathological parameters.
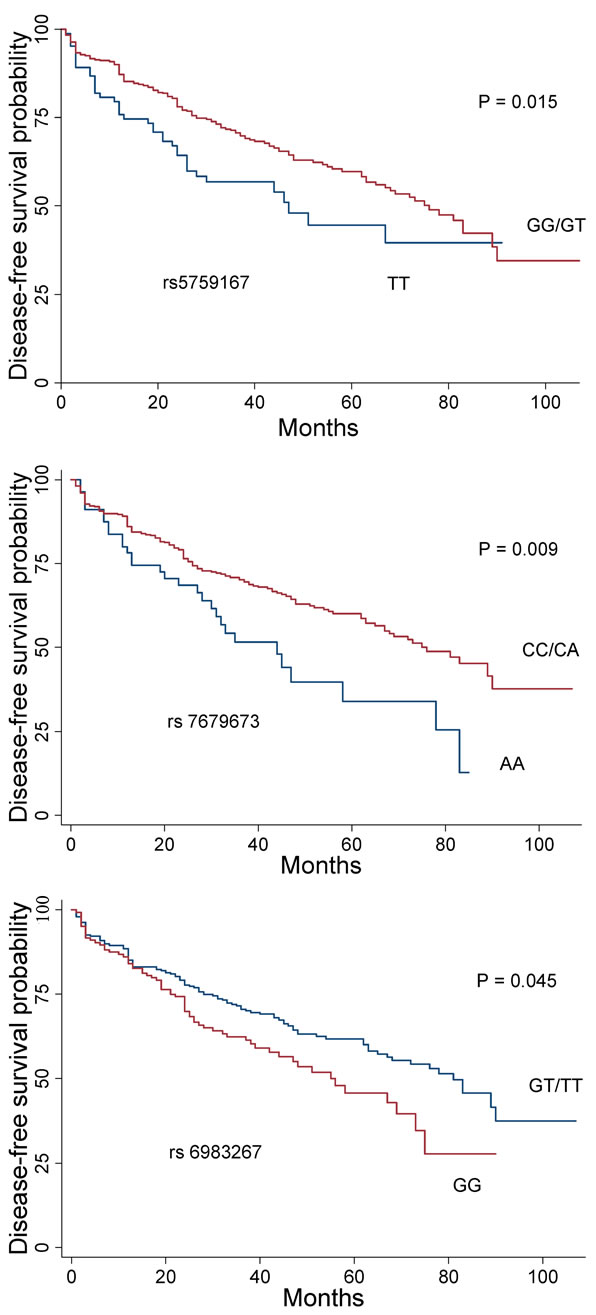
Figure 1. Kaplan-Meier plot of disease-free survival related to the genotype at SNPs rs6983267, rs7679673, and rs- 5759167. Red curves represent prostate cancer-associated genotypes under the dominant mode (rs7679673 and rs- 5759167, respectively), or recessive model (rs6983267). Blue curves are reference genotypes.
4. Discussion
In recent years, numerous association studies have provided information about the genetic foundation of prostate cancer and more than 40 genetic variants associated with the disease have been identified [16]. However, only a few have demonstrated a significant correlation between SNPs and clinico-pathological variables [25,41- 43]. In the present study, we genotyped 32 genetic variants in a Danish study population comprising 648 prostate cancer cases and 526 control subjects. We demonstrated that 17 genetic variants were associated with prostate cancer risk in our cohort, and that the association estimates (ORs) were similar to those previously reported [14-16]. In addition to confirming previous studies, we also observed several interesting and significant associations between clinico-pathological parameters and certain variants under either recessive or dominant models.
Considering the modest effect of each SNP on the heterogeneous and common prostate cancer disease, several thousands of subjects are typically required to detect such genetic variants. We were able to replicate 17 genetic markers in nine different chromosomal regions in our study population that was only 1/3 the size of larger association studies [41,43,44]. Although the ORs were similar with those estimated in larger studies, the width of the confidence intervals reflected the comparably modest size of our study population.
We evaluated the cumulative effects of five markers also previously tested by Zheng et al. [41]. Zheng et al. demonstrated that, individually, these five markers might only contribute a modest proportion of risk, but collectively, they have considerable composite risk. Our analysis of the cumulative effects of the five SNPs was conducted without including family history as a sixth risk factor, in contrast to Zheng et al. [41], because family history was not obtained from control subjects in the present study. We were, nevertheless, able to reproduce the cumulative association of the five genetic variants with prostate cancer risk [41]. In the work of Salina et al., it was claimed that the clinical relevance of cumulative effects of five genetic variants could not be established to predict more aggressive prostate cancer, as the SNPs were not correlated with the clinical and pathological tumor variables including age at diagnosis, preoperative PSA, tumor grade and stage [44]. In our study, we observed that two of the SNPs included in the cumulative model were correlated with clinico-pathological variables. The variant rs4430796(AA) was significantly associated with lower T stage (≤T2c). Furthermore, we found that rs6983267 (GG) was associated with negative family history implying a potentially protective effect of the risk genotype (GG) against familial/hereditary prostate cancer, the marker was, however, significantly correlated with biochemical relapse (Figure 1). In contrast, Freedman et al. observed a significant association between rs6983267 (GG) and a positive family history, advanced stage, and younger age, whereas Zheng and Sun found no association between this marker and family history, tumor grade and stage, age and PSA [24,31,45].
More interestingly, we found rs2735839 (GG) in the KLK3 gene encoding PSA to be associated with increased levels of PSA (>10 ng/ml), lower Gleason score (<7) and lower T stage (≤T2c). In men undergoing radical prostatectomy, Xu et al. reported that rs2735839 (GG) was significantly associated with less aggressive prostate cancer and low T stage similar to our findings; in healthy individuals, Xu et al. also found an association with an elevated pre-diagnostic PSA [46]. It is not yet elucidated how genetic variation in rs2735839 influence PSA levels, but our findings suggest the following scenario: triggered by increased PSA (>10 ng/ml), carriers of the described risk genotype were more likely to be diagnosed with an indolent type of prostate cancer that was less aggressive in terms of lower Gleason score (≤6) and lower T stage (≤T2c). It may be speculated that the increased PSA levels in these patients make them prone to diagnostic examinations like needle biopsy [43,47]. Consequently, prostate cancer screening can detect indolent prostate cancer in carriers of rs2735839 (GG). Having said that, Pomerantz et al. found no association between rs2735839 and PSA, while Penney et al. observed that rs2569735 in linkage disequilibrium with rs2735839 was associated with lower PSA levels [43,47]. Such conflicting discoveries with generally opposite SNP effects on phenotypes has been explained by differentially structured linkage disequilibrium across diverse populations when the interrogated genetic variant is closely related; this occurrence is called the flip-flop phenomenon [48].
Variant rs5759167 (GG/GT) was significantly associated with a negative family history of prostate cancer, similar to rs6983267, but with a low risk for biochemical recurrence. Like rs2735839, rs10896449 (GG/GA) was significantly associated with higher level of PSA (>10 ng/ml). Pomerantz et al. reported a statistically significant association between rs7679673 (CC/CA) and prostate cancer specific death, but in our study, the marker was associated with a significantly lower risk for biochemical recurrence [43]. Like Pomerantz et al., the associations between the prostate cancer-associated SNPs and D’Amico risk score were non-significant suggesting that the risk (associated) genotypes or non-risk (reference) genotypes may have poor value in predicting aggressiveness of prostate cancer (Table 5).
We acknowledge that there are several potential limitations of our study. As previously mentioned, our sample size was relatively modest, and particular variants that presented borderline significance or insignificance might reflect the limitation of our sample size. However, we were able to replicate several previously reported risk SNPs. In the current study, we did not adjust for possible ancestral admixture in the study population, as information about the origins of the patients was not provided. The effect of ancestral admixture would probably not influence the association estimates significantly because we have replicated 17 prostate cancer-associated risk SNPs reported in European case subjects [16]. However, we were not able to reproduce the remaining previously identified prostate cancer-associated risk SNPs. There may be following reasons: 1) the study was not powered to estimate the modest effect measure; 2) the SNPs might confer risk in one population but not the other; and 3) the SNP effect might vary from population to population due to gene-gene or gene-environment interactions. Only one SNP survived Bonferroni correction; we chose, however, to carry on with the 10 SNPs that were statistically significant under either the recessive or the dominant model, because the allele frequencies and ORs were consistent with previous reports. Our main concern has been whether the patients with myocardial infarction were appropriate control subjects for the prostate cancer cases. A recently published meta-analysis of 14 genome-wide association studies of coronary artery diseases has, however, at least temporarily, closed the discussion whether acute myocardial infarction patients as controls were appropriate [49]. None of the reviewed variants associated with coronary heart disease were correlated with prostate cancer.
5. Conclusion
We replicated 17 previously identified prostate cancerassociated risk SNPs in a Danish case-control study and found a cumulative and significant association between five SNPs and prostate cancer. Furthermore, we noted significant associations between several prostate cancerassociated risk genotypes and less aggressive tumor variables, high level of PSA, and low risk for biochemical recurrence.
REFERENCES
- A. Jemal, M. M. Center, C. DeSantis and E. M. Ward, “Global Patterns of Cancer Incidence and Mortality Rates and Trends,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 19, 2010, pp. 1893-1907.
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, “Estimates of Worldwide Burden of Cancer in 2008: GLOBOCAN 2008,” International Journal of Cancer, Vol. 127, No. 12, 2010, pp. 2893-2917.
- W. F. Page, M. M. Braun, A. W. Partin, N. Caporaso and P. Walsh, “Heredity and Prostate Cancer: A Study of World War II Veteran Twins,” The Prostate, Vol. 33, No. 4, 1997, pp. 240-245. doi:10.1002/(SICI)1097-0045(19971201)33:4<240::AID-PROS3>3.0.CO;2-L
- M. P. Zeegers, A. Jellema and H. Ostrer, “Empiric Risk of Prostate Carcinoma for Relatives of Patients with Prostate Carcinoma: A Meta-Analysis,” Cancer, Vol. 97, No. 8, 2003, pp. 1894-1903. doi:10.1002/cncr.11262
- B. S. Carter, G. S. Bova, T. H. Beaty, et al., “Hereditary Prostate Cancer: Epidemiologic and Clinical Features,” The Journal of Urology, Vol. 150, No. 3, 1993, pp. 797-802.
- P. A. Kupelian, V. A. Kupelian, J. S. Witte, R. Macklis and E. A. Klein, “Family History of Prostate Cancer in Patients with Localized Prostate Cancer: An Independent Predictor of Treatment Outcome,” Journal of Clinical Oncology, Vol. 15, No. 4, 1997, pp. 1478-1480.
- G. S. Bova, A. W. Partin, S. D. Isaacs, et al., “Biological Aggressiveness of Hereditary Prostate Cancer: Long-Term Evaluation Following Radical Prostatectomy,” The Journal of urology, Vol. 160, No. 3, 1998, pp. 660-663. doi:10.1016/S0022-5347(01)62748-4
- A. Valeri, R. Azzouzi, E. Drelon, et al., “Early-Onset Hereditary Prostate Cancer Is Not Associated with Specific Clinical and Biological Features,” The Prostate, Vol. 45, No. 1, 2000, pp. 66-71. doi:10.1002/1097-0045(20000915)45:1<66::AID-PROS8>3.0.CO;2-W
- O. Bratt, J. E. Damber, M. Emanuelsson and H. Gronberg, “Hereditary Prostate Cancer: Clinical Characteristics and Survival,” The Journal of Urology, Vol. 167, No. 6, 2002, pp. 2423-2426. doi:10.1016/S0022-5347(05)64997-X
- S. V. Kotsis, S. L. Spencer, P. A. Peyser, J. E. Montie and K. A. Cooney, “Early Onset Prostate Cancer: Predictors of Clinical Grade,” The Journal of Urology, Vol. 167, No. 4, 2002, pp. 1659-1663. doi:10.1016/S0022-5347(05)65173-7
- E. Sacco, T. Prayer-Galetti, F. Pinto, et al., “Familial and Hereditary Prostate Cancer by Definition in an Italian Surgical Series: Clinical Features and Outcome,” European Urology, Vol. 47, No. 6, 2005, pp. 761-768. doi:10.1016/j.eururo.2005.01.016
- E. Spangler, C. M. Zeigler-Johnson, S. B. Malkowicz, A. J. Wein and T. R. Rebbeck, “Association of Prostate Cancer Family History with Histopathological and Clinical Characteristics of Prostate Tumors,” International Journal of Cancer, Vol. 113, No. 3, 2005, pp. 471-474.
- S. Pakkanen, P. M. Kujala, N. Ha, M. P. Matikainen, J. Schleutker and T. L. Tammela, “Clinical and Histopathological Characteristics of Familial Prostate Cancer in Finland,” BJU International, Vol. 109, No. 4, 2011, pp. 557-563.
- J. P. Ioannidis, P. Castaldi and E. Evangelou, “A Compendium of Genome-Wide Associations for Cancer: Critical Synopsis and Reappraisal,” Journal of the National Cancer Institute, Vol. 102, No. 12, 2010, pp. 846-858. doi:10.1093/jnci/djq173
- S. T. Kim, Y. Cheng, F. C. Hsu, et al., “Prostate Cancer Risk-Associated Variants Reported from Genome-Wide Association Studies: Meta-Analysis and Their Contribution to Genetic Variation,” The Prostate, Vol. 70, No. 16, 2010, pp. 1729-1738.
- Z. Kote-Jarai, A. A. Olama, G. G. Giles, et al., “Seven Prostate Cancer Susceptibility Loci Identified by a MultiStage Genome-Wide Association Study,” Nature Genetics, Vol. 43, No. 8, 2011, pp. 785-791. doi:10.1038/ng.882
- J. Beuten, J. A. Gelfond, J. L. Franke, et al., “Single and Multigenic Analysis of the Association between Variants in 12 Steroid Hormone Metabolism Genes and Risk of Prostate Cancer,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 18, No. 6, 2009, pp. 1869-1880.
- F. C. Hsu, J. Sun, Y. Zhu, et al., “Comparison of Two Methods for Estimating Absolute Risk of Prostate Cancer Based on Single Nucleotide Polymorphisms and Family History,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 19, No. 4, 2010, pp. 1083-1088.
- M. Aly, F. Wiklund, J. Xu, et al., “Polygenic Risk Score Improves Prostate Cancer Risk Prediction: Results from the Stockholm-1 Cohort Study,” European urology, Vol. 60, No. 1, 2011, pp. e1-e8. doi:10.1016/j.eururo.2011.01.017
- L. M. Fitzgerald, E. M. Kwon, J. S. Koopmeiners, C. A. Salinas, J. L. Stanford and E. A. Ostrander, “Analysis of Recently Identified Prostate Cancer Susceptibility Loci in a Population-Based Study: Associations with Family History and Clinical Features,” Clinical Cancer Research, Vol. 15, No. 9, 2009, pp. 3231-3237.
- N. J. Camp, J. M. Farnham, J. Wong, G. B. Christensen, A. Thomas and L. A. Cannon-Albright, “Replication of the 10q11 and Xp11 Prostate Cancer Risk Variants: Results from a Utah Pedigree-Based Study,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 18, No. 4, 2009, pp. 1290-1294.
- V. D’Amico, R. Whittington, S. B. Malkowicz, et al., “Biochemical Outcome after Radical Prostatectomy, External Beam Radiation Therapy, or Interstitial Radiation Therapy for Clinically Localized Prostate Cancer,” The Journal of the American Medical Association, Vol. 280, No. 11, 1998, pp. 969-974. doi:10.1001/jama.280.11.969
- H. R. Andersen, T. T. Nielsen, T. Vesterlund, et al., “Danish Multicenter Randomized Study on Fibrinolytic Therapy versus Acute Coronary Angioplasty in Acute Myocardial Infarction: Rationale and Design of the Danish Trial in Acute Myocardial Infarction-2 (DANAMI-2),” American Heart Journal, Vol. 146, No. 2, 2003, pp. 234-241. doi:10.1016/S0002-8703(03)00316-8
- M. L. Freedman, C. A. Haiman, N. Patterson, et al., “Admixture Mapping Identifies 8q24 as a Prostate Cancer Risk Locus in African-American Men,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 103, No. 38, 2006, pp. 14068-14073. doi:10.1073/pnas.0605832103
- L. T. Amundadottir, P. Sulem, J. Gudmundsson, et al., “A Common Variant Associated with Prostate Cancer in European and African Populations,” Nature Genetics, Vol. 38, No. 6, 2006, pp. 652-658. doi:10.1038/ng1808
- D. Duggan, S. L. Zheng, M. Knowlton, et al., “Two Genome-Wide Association Studies of Aggressive Prostate Cancer Implicate Putative Prostate Tumor Suppressor Gene DAB2IP,” Journal of the National Cancer Institute, Vol. 99, No. 24, 2007, pp. 1836-1844. doi:10.1093/jnci/djm250
- J. Gudmundsson, P. Sulem, V. Steinthorsdottir, et al., “Two Variants on Chromosome 17 Confer Prostate Cancer Risk, and the One in TCF2 Protects against Type 2 Diabetes,” Nature Genetics, Vol. 39, No. 8, 2007, pp. 977-983. doi:10.1038/ng2062
- J. Gudmundsson, P. Sulem, A. Manolescu, et al., “Genome-Wide Association Study Identifies a Second Prostate Cancer Susceptibility Variant at 8q24,” Nature Genetics, Vol. 39, No. 5, 2007, pp. 631-637. doi:10.1038/ng1999
- C. A. Haiman, N. Patterson, M. L. Freedman, et al., “Multiple Regions within 8q24 Independently Affect Risk for Prostate Cancer,” Nature Genetics, Vol. 39, No. 5, 2007, pp. 638-644. doi:10.1038/ng2015
- M. Yeager, N. Orr, R. B. Hayes, et al., “Genome-Wide Association Study of Prostate Cancer Identifies a Second Risk Locus at 8q24,” Nature Genetics, Vol. 39, No. 5, 2007, pp. 645-649. doi:10.1038/ng2022
- S. L. Zheng, J. Sun, Y. Cheng, et al., “Association between Two Unlinked Loci at 8q24 and Prostate Cancer Risk among European Americans,” Journal of the National Cancer Institute, Vol. 99, No. 20, 2007, pp. 1525-1533. doi:10.1093/jnci/djm169
- R. A. Eeles, Z. Kote-Jarai, G. G. Giles, et al., “Multiple Newly Identified Loci Associated with Prostate Cancer Susceptibility,” Nature Genetics, Vol. 40, No. 3, 2008, pp. 316-321. doi:10.1038/ng.90
- J. Gudmundsson, P. Sulem, T. Rafnar, et al., “Common Sequence Variants on 2p15 and Xp11.22 Confer Susceptibility to Prostate Cancer,” Nature genetics, Vol. 40, No. 10, 2008, pp. 281-283. doi:10.1038/ng.89
- G. Thomas, K. B. Jacobs, M. Yeager, et al., “Multiple Loci Identified in a Genome-Wide Association Study of Prostate Cancer,” Nature Genetics, Vol. 40, No. 3, 2008, pp. 310-315. doi:10.1038/ng.91
- A. Al Olama, Z. Kote-Jarai, G. G. Giles, et al., “Multiple Loci on 8q24 Associated with Prostate Cancer Susceptibility,” Nature Genetics, Vol. 41, No. 10, 2009, pp. 1058-1060. doi:10.1038/ng.452
- R. A. Eeles, Z. Kote-Jarai, A. A. Al Olama, et al., “Identification of Seven New Prostate Cancer Susceptibility Loci through a Genome-Wide Association Study,” Nature Genetics, Vol. 41, No. 10, 2009, pp. 1116-1121. doi:10.1038/ng.450
- J. Gudmundsson, P. Sulem, D. F. Gudbjartsson, et al., “Genome-Wide Association and Replication Studies Identify Four Variants Associated with Prostate Cancer Susceptibility,” Nature Genetics, Vol. 41, No. 10, 2009, pp. 1122-1126. doi:10.1038/ng.448
- M. Yeager, N. Chatterjee, J. Ciampa, et al., “Identification of a New Prostate Cancer Susceptibility Locus on Chromosome 8q24,” Nature Genetics, Vol. 41, No. 10, 2009, pp. 1055-1057. doi:10.1038/ng.444
- W. Y. Wang, B. J. Barratt, D. G. Clayton and J. A. Todd, “Genome-Wide Association Studies: Theoretical and Practical Concerns,” Nature Reviews Genetics, Vol. 6, No. 2, 2005, pp. 109-118. doi:10.1038/nrg1522
- S. Purcell, B. Neale, K. Todd-Brown, et al., “PLINK: A Tool Set for Whole-Genome Association and PopulationBased Linkage Analyses,” American Journal of Human Genetics, Vol. 81, No. 3, 2007, pp. 559-575. doi:10.1086/519795
- S. L. Zheng, J. Sun, F. Wiklund, et al., “Cumulative Association of Five Genetic Variants with Prostate Cancer,” The New England Journal of Medicine, Vol. 358, No. 9, 2008, pp. 910-919. doi:10.1056/NEJMoa075819
- A. K. Kader, J. Sun, S. D. Isaacs, et al., “Individual and Cumulative Effect of Prostate Cancer Risk-Associated Variants on Clinicopathologic Variables in 5895 Prostate Cancer Patients,” The Prostate, Vol. 69, No. 11, 2009, pp. 1195-1205. doi:10.1002/pros.20970
- M. M. Pomerantz, L. Werner, W. Xie, et al., “Association of Prostate Cancer Risk Loci with Disease Aggressiveness and Prostate Cancer-Specific Mortality,” Cancer Prevention Research, Vol. 4, No. 5, 2011, pp. 719-728. doi:10.1158/1940-6207.CAPR-10-0292
- C. A. Salinas, J. S. Koopmeiners, E. M. Kwon, et al., “Clinical Utility of Five Genetic Variants for Predicting Prostate Cancer Risk and Mortality,” The Prostate, Vol. 69, No. 4, 2009, pp. 363-372. doi:10.1002/pros.20887
- J. Sun, B. L. Chang, S. D. Isaacs, et al., “Cumulative Effect of Five Genetic Variants on Prostate Cancer Risk in Multiple Study Populations,” The Prostate, Vol. 68, No. 12, 2008, pp. 1257-1262. doi:10.1002/pros.20793
- J. Xu, S. D. Isaacs, J. Sun, et al., “Association of Prostate Cancer Risk Variants with Clinicopathologic Characteristics of the Disease,” Clinical Cancer Research, Vol. 14, No. 18, 2008, pp. 5819-5824.
- K. L. Penney, F. R. Schumacher, P. Kraft, et al., “Association of KLK3 (PSA) Genetic Variants with Prostate Cancer Risk and PSA Levels,” Carcinogenesis, Vol. 32, No. 6, 2011, pp. 853-859. doi:10.1093/carcin/bgr050
- P. I. Lin, J. M. Vance, M. A. Pericak-Vance and E. R. Martin, “No Gene Is an Island: The Flip-Flop Phenomenon,” American Journal of Human Genetics, Vol. 80, No. 3, 2007, pp. 531-538. doi:10.1086/512133
- H. Schunkert, I. R. Konig, S. Kathiresan, et al., “LargeScale Association Analysis Identifies 13 New Susceptibility Loci for Coronary Artery Disease,” Nature Genetics, Vol. 43, No. 4, 2011, pp. 333-338. doi:10.1038/ng.784
NOTES
*Corresponding author.