Journal of Behavioral and Brain Science
Vol.07 No.02(2017), Article ID:74014,12 pages
10.4236/jbbs.2017.72006
High Mobility Group Box 1 and Traumatic Brain Injury
Seidu A. Richard1,2*, Wu Min1, Zhaoliang Su2, Huaxi Xu2
1Department of Neurosurgery, Affiliated Hospital of Jiangsu University (Jiangbin Hospital), Zhenjiang, China
2Department of Immunology, Jiangsu University, Zhenjiang, China

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 16, 2017; Accepted: February 6, 2017; Published: February 9, 2017
ABSTRACT
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. We identify High Mobility Group Box 1 (HMGB1) as a novel causative factor in the development of cerebral oedema, mediating coagulation, preventing secondary brain damage as well as serving as a novel therapeutic target to limit secondary neurological injury after TBI. As a prototypical danger associated molecular patterns (DAMP), HMGB1 is released from necrotic neurons via a NR2B-mediated mechanism during TBI. The secretion of HMGB1 requires severe injury and tissue hypoperfusion, and is associated with posttraumatic coagulation abnormalities, activation of complement and severe systemic inflammatory response. HMGB1 is clinically associated with elevated intracranial pressure (ICP) in patients and functionally promoted cerebral edema after TBI. The detrimental effects of HMGB1 is mediated at least in part between activation of microglial toll-like receptor 4 (TLR4) and the subsequent expression of the astrocytic water channel aquaporin-4 (AQP4). Anti-HMGB1mAb remarkably inhibited fluid percussion-induced brain edema by inhibiting HMGB1 translocation, protection of blood-brain barrier (BBB) integrity, suppression of inflammatory molecule expression and improvement of motor function. Our review demonstrates the pathological role of HMGB1 as well as the possible therapeutic valve of HMGB1 in patients with TBI.
Keywords:
HMGB1, TBI, Biomarker, Brain Edema, Coagulation, Anti-HMGB1 Therapy

1. Introduction
Traumatic Brain Injury (TBI) is the major cause of death and disability worldwide, with survivors living the rest of their lives with cognitive, motor, behavioural or speech-language disabilities [1] [2] . TBI, whether mild, moderate or severe, represents a pandemic disease that affects all nations with acute onset and progressing to chronic, debilitating health issues affecting far beyond the individual patients [1] [3] .
High Mobility Group Box 1 (HMGB1) is extremely preserved structure in various species and primitively known as a nuclear DNA-binding protein that takes part in the formation of nucleosomes and the regulation of gene transcription [4] [5] [6] . HMGB1 activates the innate immune system in response to tissue injury due to trauma, ischemia/reperfusion or infection since it is a prototypical DAMP molecule [7] . It is also well known that intracellular functions of HMGB1 include stabilization of nucleosome formation and facilitation of gene transcription as well as co-transcriptional factor with the ability to serve as a structural DNA-binding protein [8] . Kim et al. have further shown that HMGB1 services as a cytokine-like mediator in many pathogenic conditions such as delayed endotoxin lethality, acute lung injury, and transient focal cerebral ischemia [9] [10] [11] [12] .
Research has indicated that apoptotic cells do not release HMGB1, but instead retain it within their cells which is in total contrast to damage or necrotic cells which passively secretes HMGB1 [13] . The secret HMGB1 interacts with receptor for advanced glycation end (RAGE) products and members of the toll-like family of receptors (TLRs) including TLR2 and TLR4 hence mobilizes HMGB1 intracellularly into the extracellular compartments [14] , which serves as a danger signal that activates the immune system to the ubiquity of injured cells. The mobilization of HMGB1 from intracellular milieu into the extracellular milieu involves its release by immune cells. HMGB1 is hyper-acetylated in macrophages and monocytes which allows it to accumulate in the cytosol and hence prevent it from being translocated into the nucleus. In the cytosol HMGB1 is packaged into secretory lysosomes and secret out of the cell [15] .
A study by Klune et al. indicated that, in the Extracellular space, HMGB1 trigger the immune system by promoting dendritic cell maturation, accelerating release of inflammatory cytokines, activating neutrophils and monocytes, as well as natural killer (NK) cells [14] . In addition, research has demonstrated that extracellular HMGB1 accelerates tissue repair and regeneration [16] . Our review demonstrates the pathological role of HMGB1 in edema formation, coagulations as well as the possible therapeutic valve of HMGB1 in patients with TBI. We also evaluated the role of HMGB1 in secondary brain injury.
2. HMGB1 as a Biomarker
King et al. demonstrated the presence of HMGB1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage [17] , while Zhou et al. observed elevation of HMGB1 in serum of patients with acute intracerebral hemorrhage (ICH) and associated this elevation with severity of ICH [18] . Both studies concluded that HMGB1 could be a promising biomarker in neurological injury patients. Yang and colleagues were the first researchers to show interest in HMGB1 as a biomarker of inflammation. They demonstrated adequately that HMGB1 was released in response to lipopolysaccharide (LPS) in mice. They again indicated that HMGB1 was released at a later time point (peak at 16 hours) as compared to the instant release of tumor necrosis factor alpha (TNF-α) and Interleukin 1 beta (IL-1β) after exposure to LPS [19] .
Peltz and colleagues in their study observed thirty times elevation of plasma HMGB1 levels after trauma compared to healthy controls during 1 h period of injury, which stabilized at 2 - 6 h post-injury [20] . Cohen and colleagues found a compelling distinction in plasma levels of HMGB1 among survivors and non- survivors from severe trauma. They also found a significant association with plasma levels of HMGB1 and numerous inflammatory mediators like interleukin-6(IL-6) and TNF-α, besides markers of endothelial cell activation like angiopoietin-2(Ang-2) and von Willebr and Factor (vWF) antigen. Their results indicated that HMGB1 was elevated with elevated plasma levels of vWF (P = 0.05 by both rank and trend, Spearman correlation r = 0.18, P = 0.02) and Ang-2 (P = 0.09 by rank but 0.01 by trend, Spearman correlation r = 0.23, P = 0.02) [21] .
Wang and associates found a cogent interrelationship between plasma HMGB1 absorption and austerity of acute traumatic brain injury [22] . Alicia and colleagues demonstrated that concentrations of HMGB1 are elevated in Cerebrospinal fluid (CSF) after trauma, and furthermore, these were separately related to poor outcomes [23] . They proposed that increased CSF HMGB1 is separately related to 6-month Glasgow Coma Scale (GOS) score 1 - 3 which reflects a poor outcome.
Zhang et al. observed that during trauma, circulating mitochondrial DNA (mtDNA) performs functions similar to DAMP which initiates systemic inflammatory response syndromes [24] . Walko and associates also demonstrated that there was a cogent interrelation between CSF mtDNA and HMGB1 concentrations at 24 h after trauma. They indicated that elevated CSF mtDNA concentrations were highly prognostic of poor outcomes of increased levels of CSF mtDNA and HMGB1 adumbrate a poor prognosis for children with TBI. They therefore concluded that CSF mtDNA could be a crucial prognostic biomarker [25] . They also proposed that although not ready for prime-time, the use of CSF mtDNA concentrations can assist in patient selection for clinical research, and opens up further lines of exploration as a potential therapeutic target. The prognostic values of HMGB1 were comparable to those of GCS scores, validating its ability as a new prognostic biomarker in TBI [25] .
3. Mechanisms of Secretion of HMGB1
Studies have shown that damage induced by trauma or ischemia can evoke HMGB1 secretion from necrotic tissue which may provide danger signal that alerts the immune system to the existence of injured cells [23] [26] . The traumatic events leading to the secretion of HMGB1 into the CSF also initiate the secretion of other DAMP into the CSF with successive activation of the innate immune response [25] . Laird et al. demonstrated that HMGB1 displayed a supreme neuronal localization within the mouse cerebral cortex and reduce immunoreactivity for HMGB1 was temporally associated with the loss of NeuN within the peri-contusional cortex, suggesting HMGB1 was released by injured neurons into the extracellular space after TBI [27] .
Zhang et al. demonstrated that HMGB1 levels decreased at lesion site and markedly increased in the plasma after percussion injury which significantly assent the conception that neuronal HMGB1 is released into extracellular space and diffuses into surrounding areas, including the blood stream [28] .
It has been observed that, over-stimulation of N-methyl-D-aspartate receptors (NMDA-R) by raised extracellular glutamate generate excitotoxicity, an important initiator of neuronal necrosis and edema after TBI [27] [29] . Studies have further demonstrated NMDA-R exist as a tetrameric protein receptor complex made up of two glycine-binding NR1 subunits and two glutamate-binding NR2 subunits that ascertain the biophysical properties [27] [30] .
It has been proven that NR2A and NR2B are the dominant subunits within the adult forebrain; so Laird et al. propose that NR2 subunit might explain the paradoxical roles of NMDA-R after TBI. They further demonstrated that activation of NR2B bolsters neuronal injury and accelerates neuronal HMGB1 release (Figure 1) to augment brain swelling, although the mechanism is still not well understood [27] . Bohmer and colleagues have indicated that there is accumulation of HMGB1 and Neuron-specific Enolase (NSE) within the CSF of neurotrauma patients. They further stated that after 72 hours of post-TBI there is acute elevation of NSE within CSF which was significantly associated with deterioration of brain death in severe TBI patients [31] . King and associates demonstrated that CSF contents of HMGB1 are associated with poor neurological outcomes of subarachnoid hemorrhage patients [32] .
Kim and colleagues proposed that although the regulatory mechanism underlying HMGB1 secretion remain to be explicated, it is possible that in addition to bringing about early secretion from dying neurons, microglia, a major inflammatory cell type in the brain, might also actively release HMGB1 and initiates a loop of HMGB1 activation that augments the inflammatory process and exasperate brain injury. They therefore concluded that HMGB1 function pleiotropically
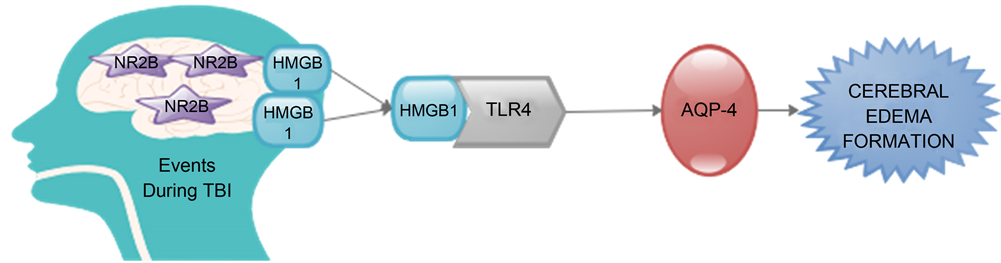
Figure 1. Activation of NR2B promotes neuronal injury and increases neuronal HMGB1 release. HMGB1 binds to TLR4 receipts thereby regulating AQP-4 and promotion of cerebral edema [27] .
in a sense that its effects are interconnected and affect many target cells in vivo, and that this multi-functionality and complexity may result in serious brain damage [12] . Treutiger et al. demonstrated that activation of human microvascular endothelial cells by HMGB1 resulted in the inauguration of many adhesion molecules, pro-inflammatory cytokines via RAGE in the activation of Mitogen- activated protein kinases (MAPKs) and nuclear factor kappa-light-chain- enhancer of activated B cells (NF-kB) [33] .
Okuma and colleagues demonstrated that intravenously administered recombinant HMGB1 dose-dependently aggravated the increase in Blood-Brain Barrier (BBB) permeability and motor impairment in rats receiving moderate percussion injury. They proposed that circulating endogenous HMGB1 performs an essential task in the advancement of BBB disruption and brain inflammation, principally through the stimulation of vascular endothelial cells and blood cells [34] . Shlosbeng et al. indicated that the reaction to HMGB1 could be responsible for the upstream of the secondary responses to TBI, provoking a cascade of activities championing the BBB disruption, brain edema, and brain injury [35] .
Diffuse Axonal Injury (DAI) is one of the most ubiquitous and essential pathologic physiognomies of TBI and it is mostly initiated by shearing forces championing the widespread tearing of axons [36] . Pang and associates indicated that HMGB1 is grossly released into the extracellular milieu after DAI. They suggest that this may result in the reduced HMGB1 level during TBI. They again indicated that at the late stage of DAI, astrocyte and microglia may be stimulated and discharge into the pro-inflammatory cytokines [37] .
4. HMGB1 and Brain Edema Formation
Laird and colleagues proposed that TLR4 mediates innate immune activation and edema development after TBI by using a translational research approach that incorporates patient CSF, human tissue culture, and pre-clinical TBI models to explicate the complex role of HMGB1-TLR4 signaling in the development of secondary neurovascular injury after TBI [27] . They also proposed the correlation between immune activation and the augmentation of post-traumatic brain edema and suggested that the release of neuronal HMGB1 may induce microglial activation [27] .
They further stated that a time dependent increase in microglial TLR4 elucidation occurred within the peri-contusional cortex, exhibiting both a temporal and spatial overlap with edema formation [27] . They demonstrated that Inhibition of TLR4 mitigated the delivery of AQP4 an astrocytic water channel involved in the evolution of cellular edema although the mechanism responsible for microglia interaction with astrocytes remained undetermined. Therefore they concluded that the unique involvement in HMGB1-TLR4 signaling is the regulation of AQP4 and promotion of cerebral edema (Figure 1), a primary cause of patient mortality after brain injury by establishing the mechanism responsible for microglial-astrocytic interactions increases acute neurovascular injury using pre-clinical models and human tissue cultures [27] .
Stein et al. demonstrated that the translocation of HMGB1 in neurons appears to be a paradox common to both ischemic and traumatic insults preferentially, the secondary brain ischemia due to brain edema in TBI [38] . Okuma and associates demonstrated that the treatment of TBI rats with anti-HMGB1-neutraliz- ing mAb fiercely subdued acute brain edema, as notice on T2-weighted MRI, associated with the collateral reduction in BBB permeability, structural changes in capillary blood vessels, up-regulation of matrix metalloproteinase-2/9 (MMP- 2/9) activity, and elucidation of inflammatory molecules which was initial proposed by other researchers [34] . They therefore suggested that HMGB1 released from the TBI lesion caused the protein exudation by directly affecting the microvasculature in surrounding areas.
Studies have further shown that activation of MMP-2/9 and inducible nitric oxide synthase (iNOS) accelerates the digestion of tight junction-associated protein complex and extracellular matrix along with basal lamina and the inflammatory response, respectively, has been proposed to partake in TBI [39] [40] . Moreover, the inhibition of MMPs or iNOS by specific inhibitors was shown to contribute to the decrease of TBI to some extent [40] . Thus, the coordination of both MMP-2/9 and iNOS may elucidate the protective effects of antiHMGB1mAb. Hergenroeder et al. also indicated that increase levels of CSF and serum concentrations of IL-6, a microglial-derived cytokine, significantly associated with neuronal injury and increased ICP after TBI in human [41] .
5. HMGB1 and Coagulation
Thrombin is a pro-inflammatory serine protease that is well distinguished from its crucial task as the resolute protease in the coagulation pathway. Thrombin and HMGB1 are cardinal molecules of the robust host defence systems that assemble during innate immune process, coagulation, and inflammation. Barry et al. proposed that they may play essential roles in the BBB disruption since both are pro-inflammatory and both are known to disrupt vascular barriers in other tissues [42] .
Cohen and colleagues stated that there is association between plasma levels of HMGB1 and activation of protein C pathway that they have initially proven to be promoted by tissue injury and hypoperfusion. They indicated that the association is particularly interesting in light of the recent discovery that HMGB1 binds in vitro to the lectin domain of thrombomodulin (TM) [21] . Abeyama and colleagues demonstrated that TM could interact with HMGB1 and perform the role as a pool for induction of HMGB1 in the plasma [43] . Esmonreported that TM is an anti-inflammatory protein via its beleaguerment of thrombin, and its induction into protein C and Thrombin induced fibrinogen inhibitor (TAFI) [44] .
Cohen and colleagues further indicated that whether TM after fettering with HMGB1 would still continue its capacity to induce protein C is questionable, although protein C induction is ancillary to the Gla domain of TM while HMGB1 is restrained to its lectin domain [21] . Ito and associates demonstrated that, administration of HMGB1 precipitated fibrin deposition and protracted clotting times in healthy rats. They demonstrated that HMGB1-bound TM decreases the ability of thrombomodulin to induce protein C in vitro. [45]
Cohen and colleagues again stated that the release of HMGB1 in the plasma is not adequate to prohibit the induction of the protein C pathway and the augmentation of coagulopathy within 45 minutes after explicit trauma-hemorrhage. They concluded that in addition to the cytokine-like effect of HMGB1 via the TLR4 and RAGE receptors, extracellular HMGB1 could also mitigate the defective induction of the protein C observed after severe trauma [21] . We therefore suggest that further studies with TBI patients as well as mouse models on trauma-hemorrhage that characterizes the discovery in trauma patients is needed to demonstrate this new function of extracellular HMGB1 after severe trauma.
6. HMGB1 and Secondary Brain Injury
Gao and colleagues proposed that HMGB1 might be released from damaged cells and at the long run partake in the pathophysiologic process of secondary brain injury after TBI [4] . Yamada and Maruyama initially proposed that HMGB1 can either be passively released from injured and necrotic cells or actively released by certain immune cells [46] . Gao and colleagues again showed that HMGB1 was also released into the cytoplasm of reactive phagocytic microglia at later stages after TBI. They also found that neutrophils quickly infiltrated into the core of the lesion after TBI. They therefore concluded that HMGB1 might also participate in later stages of inflammation after TBI [4] . Experimental study has shown that complement and alarmins are endogenous trigger for trauma-associated inflammation, HMGB1 appears to be one of the essential mediators in generating this posttraumatic sterile inflammation via receptors such as TLR4 and RAGE [26] .
Laird and colleagues implicated NR2B as a crucial initiating factor in bolstering cerebral innate immune responses, bridging acute excitotoxicity with secondary neurological injury [27] . Kim and colleagues indicated that HMGB1 might aggravate brain damage by both an autocrine and a paracrine mechanism [12] . Gao and colleagues further demonstrated that RAGE was expressed in microglia at later stages after TBI, which indicates that microglia might be triggered by HMGB1 through its interaction with RAGE. They are of the view that HMGB1 plays a vital role in later stages of inflammation after TBI [4] . Venereau et al. demonstrated that the release of HMGB1 in inflammation supports the concept of HMGB1 as an endogenous danger signal alerting the immune system to the ubiquity of inflammation and necrosis [47] . Walko and colleagues suggested that DAMPs play a feasible role in either inflammation or other aspects of the secondary injury response to pediatric TBI [25] .
7. Therapeutic Potentials HMGB1
Okuma and colleagues reported that the pharmacological effects of anti-HMGB1 therapy comprises preservation of the BBB structurally and functionally, inhibition of several features of major inflammatory molecules and interdiction of MMP-2/9 activity. The diverse interconnecting and dependence on each other could substantiate the remarkable efficacy of anti-HMGB1mAb therapy in TBI [34] . In line with the conspicuous effects of anti-HMGB1mAb on brain edema, this treatment decreased the upsurge in BBB permeability, MMP2/9 activity, and emergence of major inflammatory molecules, including TNF-α and iNOS [34] . Okuma and colleagues again indicated that the therapy also drastically ameliorated the acute motor impairments initiated by percussion injury. They concluded that anti-HMGB1mAb protected the BBB from disruption, control of inflammatory responses and maintenance of central nervous system motor sensory function [34] .
Chaichana et al. confirmed indeed that, anti-HMGB1mAb drastically inhibited the release of plasminogen activator inhibitor-1(PAI-1), a marker for vascular endothelial cell activation and neutrophil elastase suggesting that the interaction between vascular endothelial cells and blood cells was reduced [48] through the clearance of circulating HMGB1 by specific mAb. Shlosberg and associates are of the view that because uncontrolled brain edema usually champions brain herniation which is the main causes of death in patients after TBI [35] , early treatment with anti-HMGB1mAb might be a promising strategy for TBI and other complications such as post-traumatic epilepsy [49] and impairment in cognitive function [50] . Guo and colleagues have also indicated that TBI is a risk factor for Alzheimer disease [51] . Okuma and colleagues are of the view that because the auspicious effects of anti-HMGB1mAb remain for at least 1 to 2 weeks in terms of neuronal cell death and motor sensory function, it is feasible that anti-HMGB1mAb therapy might inhibit the secondary brain injury emanating from TBI, and thereby greatly enhance the prognosis of this condition and decrease the incidence of complications [34] .
8. Conclusion
HMGB1 is an early arbitrator of sterile inflammation which is expressed in the plasma within 45 minutes after severe trauma in humans, during severe tissue injury and tissue hypoperfusion. HMGB1 is well known to be linked with post- traumatic coagulation abnormalities, activation of complement and severe systemic inflammatory response. While anti-HMGB1mAb efficiently decreases acute brain edema after TBI through preservation of the BBB and suppression of inflammatory responses, NR2B has been incriminated as an important initiating factor enhancing cerebral innate immune responses, bridging acute excitotoxicity with secondary neurological injury. Also CSF mtDNA is unique DAMP in TBI which appears to be a useful biomarker that predicts neurological outcomes after TBI.
Conflict of Interest
The authors declare no conflict of interest.
Cite this paper
Richard, S.A., Min, W., Su, Z.L. and Xu, H.X. (2017) High Mobility Group Box 1 and Traumatic Brain Injury. Journal of Behavioral and Brain Science, 7, 50-61. https://doi.org/10.4236/jbbs.2017.72006
References
- 1. Namas, R., Ghuma, A., Hermus, L., et al. (2009) The Acute Inflammatory Response in Trauma/Hemorrhage and Traumatic Brain Injury: Current State and Emerging Prospects. Libyan Journal of Medicine, 4, 97-103.
https://doi.org/10.4176/090325 - 2. Perel, P., Edwards, P., Wentz, R. and Roberts, I. (2006) Systematic Review of Prognostic Models in Traumatic Brain Injury. BMC Medical Informatics and Decision Making, 6, 38.
https://doi.org/10.1186/1472-6947-6-38 - 3. Kauvar, D.S. and Wade, C.E. (2005) The Epidemiology and Modern Management of Traumatic Hemorrhage: US and International Perspectives. Critical Care, 9, S1.
https://doi.org/10.1186/cc3779 - 4. Gao, T.-L., Yuan, X.-T., Yang, D., et al. (2012) Expression of HMGB1 and RAGE in Rat and Human Brains after Traumatic Brain Injury. Journal of Trauma and Acute Care Surgery, 72, 643-649.
https://doi.org/10.1097/TA.0b013e31823c54a6 - 5. Park, J.S., Arcaroli, J., Yum, H.-K., et al. (2003) Activation of Gene Expression in Human Neutrophils by High Mobility Group Box 1 Protein. American Journal of Physiology-Cell Physiology, 284, C870-C879.
https://doi.org/10.1152/ajpcell.00322.2002 - 6. Thomas, J. (2001) HMG1 and 2: Architectural DNA-Binding Proteins. Biochemical Society Transactions, 29, 395-401.
https://doi.org/10.1042/bst0290395 - 7. Bianchi, M.E. (2007) DAMPs, PAMPs and Alarmins: All We Need to Know about Danger. Journal of Leukocyte Biology, 81, 1-5.
https://doi.org/10.1189/jlb.0306164 - 8. Huttunen, H. and Rauvala, H. (2004) Amphoterin as an Extracellular Regulator of Cell Motility: From Discovery to Disease. Journal of Internal Medicine, 255, 351-366.
https://doi.org/10.1111/j.1365-2796.2003.01301.x - 9. Kim, J.-B., Choi, J.S., Yu, Y.-M., et al. (2006) HMGB1, a Novel Cytokine-Like Mediator Linking Acute Neuronal Death and Delayed Neuroinflammation in the Postischemic Brain. The Journal of Neuroscience, 26, 6413-6421.
https://doi.org/10.1523/JNEUROSCI.3815-05.2006 - 10. Wang, H., Vishnubhakat, J.M., Bloom, O., et al. (1999) Proinflammatory Cytokines (Tumor Necrosis Factor and Interleukin 1) Stimulate Release of High Mobility Group Protein-1 by Pituicytes. Surgery, 126, 389-392.
https://doi.org/10.1016/S0039-6060(99)70182-0 - 11. Abraham, E., Arcaroli, J., Carmody, A., Wang, H. and Tracey, K.J. (2000) Cutting Edge: HMG-1 as a Mediator of Acute Lung Inflammation. The Journal of Immunology, 165, 2950-2954.
https://doi.org/10.4049/jimmunol.165.6.2950 - 12. Kim, J.B., Lim, C.M., Yu, Y.M. and Lee, J.-K. (2008) Induction and Subcellular Localization of High-Mobility Group Box-1 (HMGB1) in the Postischemic Rat Brain. Journal of Neuroscience Research, 86, 1125-1131.
https://doi.org/10.1002/jnr.21555 - 13. Scaffidi, P., Misteli, T. and Bianchi, M.E. (2002) Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature, 418, 191-195.
https://doi.org/10.1038/nature00858 - 14. Klune, J.R., Dhupar, R., Cardinal, J., Billiar, T.R. and Tsung, A. (2008) HMGB1: Endogenous Danger Signaling. Molecular Medicine, 14, 476-484.
https://doi.org/10.2119/2008-00034.Klune - 15. Bonaldi, T., Talamo, F., Scaffidi, P., et al. (2003) Monocytic Cells Hyperacetylate Chromatin Protein HMGB1 to Redirect It towards Secretion. The EMBO Journal, 22, 5551-5560.
https://doi.org/10.1093/emboj/cdg516 - 16. Bianchi, M.E. and Manfredi, A.A. (2007) High-Mobility Group Box 1 (HMGB1) Protein at the Crossroads between Innate and Adaptive Immunity. Immunological Reviews, 220, 35-46.
https://doi.org/10.1111/j.1600-065X.2007.00574.x - 17. King, M.D., Laird, M.D., Sangeetha, S., et al. (2010) Elucidating Novel Mechanisms of Brain Injury Following Subarachnoid Hemorrhage: An Emerging Role for Neuroproteomics. Neurosurgical Focus, 28, E10.
https://doi.org/10.3171/2009.10.focus09223 - 18. Zhou, Y., Xiong, K.-L., Lin, S., et al. (2010) Elevation of High-Mobility Group Protein Box-1 in Serum Correlates with Severity of Acute Intracerebral Hemorrhage. Mediators of Inflammation, Online.
- 19. Yang, R., Harada, T., Mollen, K.P., et al. (2006) Anti-HMGB1 Neutralizing Antibody Ameliorates Gut Barrier Dysfunction and Improves Survival after Hemorrhagic Shock. Molecular Medicine, 12, 105-114.
https://doi.org/10.2119/2006-00010.Yang - 20. Peltz, E.D., Moore, E.E., Eckels, P.C., et al. (2009) HMGB1 Is Markedly Elevated within 6 Hours of Mechanical Trauma in Humans. Shock, 32, 17-22.
https://doi.org/10.1097/SHK.0b013e3181997173 - 21. Cohen, M.J., Brohi, K., Calfee, C.S., et al. (2009) Early Release of High Mobility Group Box Nuclear Protein 1 after Severe Trauma in Humans: Role of Injury Severity and Tissue Hypoperfusion. Critical Care, 13, R174.
https://doi.org/10.1186/cc8152 - 22. Wang, K.-Y., Yu, G.-F., Zhang, Z.-Y., Huang, Q. and Dong, X.Q. (2012) Plasma High-Mobility Group Box 1 Levels and Prediction of Outcome in Patients with Traumatic Brain Injury. Clinica Chimica Acta, 413, 1737-1741.
https://doi.org/10.1016/j.cca.2012.07.002 - 23. Au, A.K., Aneja, R.K., Bell, M.J., et al. (2012) Cerebrospinal Fluid Levels of High-Mobility Group Box 1 and Cytochrome C Predict Outcome after Pediatric Traumatic Brain Injury. Journal of Neurotrauma, 29, 2013-2021.
https://doi.org/10.1089/neu.2011.2171 - 24. Zhang, Q., Raoof, M., Chen, Y., et al. (2010) Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature, 464, 104-107.
https://doi.org/10.1038/nature08780 - 25. Walko, T.D., Bola, R.A., Hong, J.D., et al. (2014) Cerebrospinal Fluid Mitochondrial DNA—A Novel DAMP in Pediatric Traumatic Brain Injury. Shock, 41, 499-503.
https://doi.org/10.1097/SHK.0000000000000160 - 26. Fink, M.P. (2007) Bench-to-Bedside Review: High-Mobility Group Box 1 and Critical Illness. Critical Care, 11, 229.
https://doi.org/10.1186/cc6088 - 27. Laird, M.D., Shields, J.S., Sukumari-Ramesh, S., et al. (2014) High Mobility Group Box Protein-1 Promotes Cerebral Edema after Traumatic Brain Injury via Activation of Toll-Like Receptor 4. Glia, 62, 26-38.
https://doi.org/10.1002/glia.22581 - 28. Zhang, J., Takahashi, H.K. and Liu, K., et al. (2011) Anti-High Mobility Group Box-1 Monoclonal Antibody Protects the Blood-Brain Barrier from Ischemia-Induced Disruption in Rats. Stroke, 42, 1420-1428.
https://doi.org/10.1161/STROKEAHA.110.598334 - 29. Ankarcrona, M., Dypbukt, J.M. and Bonfoco, E., et al. (1995) Glutamate-Induced Neuronal Death: A Succession of Necrosis or Apoptosis Depending on Mitochondrial Function. Neuron, 15, 961-973.
https://doi.org/10.1016/0896-6273(95)90186-8 - 30. Cull-Candy, S.G. and Leszkiewicz, D.N. (2004) Role of Distinct NMDA Receptor Subtypes at Central Synapses. Science Signaling, 2004, re16.
https://doi.org/10.1126/stke.2552004re16 - 31. Böhmer, A.E., Oses, J.P., Schmidt, A.P., et al. (2011) Neuron-Specific Enolase, S100B, and Glial Fibrillary Acidic Protein Levels as Outcome Predictors in Patients with Severe Traumatic Brain Injury. Neurosurgery, 68, 1624-1631.
https://doi.org/10.1227/NEU.0b013e318214a81f - 32. King, M.D., McCracken, D.J., Wade, M.F., et al. (2011) Curcumin Attenuates Hematoma Size and Neurological Injury Following Intracerebral Hemorrhage in Mice. Journal of Neurosurgery, 115, 116-123.
https://doi.org/10.3171/2011.2.JNS10784 - 33. Treutiger, C., Mullins, G., Johansson, A.S., et al. (2003) High Mobility Group 1 B-Box Mediates Activation of Human Endothelium. Journal of Internal Medicine, 254, 375-385.
https://doi.org/10.1046/j.1365-2796.2003.01204.x - 34. Okuma, Y., Liu, K., Wake, H., et al. (2012) Anti-High Mobility Group Box-1 Antibody Therapy for Traumatic Brain Injury. Annals of Neurology, 72, 373-384.
https://doi.org/10.1002/ana.23602 - 35. Shlosberg, D., Benifla, M., Kaufer, D. and Friedman, A. (2010) Blood-Brain Barrier Breakdown as a Therapeutic Target in Traumatic Brain Injury. Nature Reviews Neurology, 6, 393-403.
https://doi.org/10.1038/nrneurol.2010.74 - 36. Li, X.-Y. and Feng, D.-F. (2009) Diffuse Axonal Injury: Novel Insights into Detection and Treatment. Journal of Clinical Neuroscience, 16, 614-619.
https://doi.org/10.1016/j.jocn.2008.08.005 - 37. Pang, H., Huang, T., Song, J., et al. (2016) Inhibiting HMGB1 with Glycyrrhizic Acid Protects Brain Injury after DAI via Its Anti-Inflammatory Effect. Mediators of inflammation, 2016, Article ID: 4569521.
https://doi.org/10.1155/2016/4569521 - 38. Stein, S.C., Graham, D.I., Chen, X.-H. and Smith, D.H. (2004) Association between Intravascular Microthrombosis and Cerebral Ischemia in Traumatic Brain Injury. Neurosurgery, 54, 687-691.
https://doi.org/10.1227/01.NEU.0000108641.98845.88 - 39. Higashida, T., Kreipke, C.W., Rafols, J.A., et al. (2011) The Role of Hypoxia-Inducible Factor-1α, Aquaporin-4, and Matrix Metalloproteinase-9 in Blood-Brain Barrier Disruption and Brain Edema after Traumatic Brain Injury: Laboratory Investigation. Journal of Neurosurgery, 114, 92-101.
https://doi.org/10.3171/2010.6.JNS10207 - 40. Khan, M., Im, Y.-B., Shunmugavel, A., et al. (2009) Administration of S-Nitrosog-lutathione after Traumatic Brain Injury Protects the Neurovascular Unit and Reduces Secondary Injury in a Rat Model of Controlled Cortical Impact. Journal of Neuroinflammation, 6, 32.
https://doi.org/10.1186/1742-2094-6-32 - 41. Hergenroeder, G.W., Moore, A.N., McCoy, J.P., et al. (2010) Serum IL-6: A Candidate Biomarker for Intracranial Pressure Elevation Following Isolated Traumatic Brain Injury. Journal of Neuroinflammation, 7, 19.
https://doi.org/10.1186/1742-2094-7-19 - 42. Festoff, B.W., Sajja, R.K., van Dreden, P. and Cucullo, L. (2016) HMGB1 and Thrombin Mediate the Blood-Brain Barrier Dysfunction Acting as Biomarkers of Neuroinflammation and Progression to Neurodegeneration in Alzheimer’s Disease. Journal of Neuroinflammation, 13, 194.
https://doi.org/10.1186/s12974-016-0670-z - 43. Abeyama, K., Stern, D.M., Ito, Y., et al. (2005) The N-Terminal Domain of Thrombomodulin Sequesters High-Mobility Group-B1 Protein, a Novel Antiinflammatory Mechanism. The Journal of Clinical Investigation, 115, 1267-1274.
https://doi.org/10.1172/JCI22782 - 44. Esmon, C. (2005) Do-all Receptor Takes on Coagulation, Inflammation. Nature Medicine, 11, 475-477.
https://doi.org/10.1038/nm0505-475 - 45. Ito, T., Kawahara, K., Nakamura, T., et al. (2007) High-Mobility Group Box 1 Protein Promotes Development of Microvascular Thrombosis in Rats. Journal of Thrombosis and Haemostasis, 5, 109-116.
https://doi.org/10.1111/j.1538-7836.2006.02255.x - 46. Yamada, S. and Maruyama, I. (2007) HMGB1, a Novel Inflammatory Cytokine. Clinica Chimica Acta, 375, 36-42.
https://doi.org/10.1016/j.cca.2006.07.019 - 47. Venereau, E., Schiraldi, M., Uguccioni, M. and Bianchi, M.E. (2013) HMGB1 and Leukocyte Migration during Trauma and Sterile Inflammation. Molecular Immunology, 55, 76-82.
https://doi.org/10.1016/j.molimm.2012.10.037 - 48. Chaichana, K.L., Pradilla, G., Huang, J. and Tamargo, R. (2010) Role of Inflammation (Leukocyte-Endothelial Cell Interactions) in Vasospasm after Subarachnoid Hemorrhage. World Neurosurgery, 73, 22-41.
https://doi.org/10.1016/j.surneu.2009.05.027 - 49. Tomkins, O., Feintuch, A., Benifla, M., et al. (2011) Blood-Brain Barrier Breakdown Following Traumatic Brain Injury: A Possible Role in Posttraumatic Epilepsy. Cardiovascular Psychiatry and Neurology, 2011, Article ID: 765923.
https://doi.org/10.1155/2011/765923 - 50. Silver, J.M., McAllister, T.W. and Arciniegas, D.B. (2009) Depression and Cognitive Complaints Following Mild Traumatic Brain Injury. American Journal of Psychiatry, 166, 653-661.
https://doi.org/10.1176/appi.ajp.2009.08111676 - 51. Guo, Z., Cupples, L., Kurz, A., et al. (2000) Head Injury and the Risk of AD in the MIRAGE Study. Neurology, 54, 1316-1323.
https://doi.org/10.1212/WNL.54.6.1316