Case Reports in Clinical Medicine
Vol.05 No.12(2016), Article ID:72938,13 pages
10.4236/crcm.2016.512067
A Fatal Case of Chronic Eosinophilic Leukemia Not Otherwise Specified (CEL-NOS) in a 19-Year-Old Male with Acute Transformation into Blast Crisis
Basheer Al-Sanouri1, Yahya Maslamani2, Ibrahim Al-Sanouri3
1College of Science, Michigan State University, Lansing, MI, USA
2Department of Adult Critical Care Medicine, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
3Department of Pulmonary and Critical Care, Allergy and Immunology, Gunderson Health System, University of Wisconsin, La Crosse, WI, USA
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: November 30, 2016; Accepted: December 19, 2016; Published: December 22, 2016
ABSTRACT
Chronic eosinophilic leukemia (CEL) is a rare disorder that is characterized by hypereosinophilia with increased number of blood or marrow blasts (>5% and <20%). CEL is distinguished from hypereosinophilic syndrome (HES) by the presence of eosinophilic clonality. Chronic eosinophilic leukemia not otherwise specified (CEL-NOS) diagnosis is made when no fusion genes are detected by most modern molecular testing, particularly the most common fusion gene FIP1L-1/PDGFRA (Factor Interacting with PAP like-1/Platelet-De- rived Growth Factor Receptor Alpha). This disease is very rare, and its description in the literature is not well characterized. We report a fetal case of severe CEL-NOS in a 19-year-old male who presented with a plethora of clinical features consists of constitutional symptoms, pancytopenia, intravascular thrombosis, acute stroke and endomyocardial infiltrates. The course of his disease was aggressive and resistant to conventional treatment. After a short period of improvement, an acute transformation into blast crisis (BC) had occurred. The diagnosis was confirmed by morphology and immunophenotyping of bone marrow biopsy. The patient eventually died of heart failure and sepsis. To our knowledge this is the first case report of fatal CEL-NOS trans- forming into severe blast crisis.
Keywords:
Hypereosinophilic Syndrome (HES), Eosinophils Degranulation, WHO World Health Organization Classification, Chronic Eosinophilic Leukemia Not Otherwise Specified (CEL-NOS), Fluorescent In Situ Hybridization (FISH), Tyrosine Kinase Inhibitor Imatinib, Acute Transformation into Blast Crisis

1. Introduction
Eosinophils are immune cells that are involved in many vital functions. They are formed exclusively in the bone marrow (BM) where they spend about 8 days in the process of maturation before moving into the blood vessels. They travel through the vessels for 8 to 12 hours before they finally arrive at tissues of destination. In the tissues, they remain for 1 to 2 weeks [1] .
The number of eosinophils in the blood is determined by the hemostasis between the production in the BM and the elimination by tissue migration and apoptosis [2] .
The normal upper limit of eosinophils count in the peripheral blood (PB) is 3% - 5%, with a corresponding absolute eosinophil count (AEC) of 350 - 500/ mm [3] . Normal values for BM eosinophils commonly range between 1% and 6% [3] [4] .
Eosinophilic functions are many, and involve movement to inflamed areas, trapping substances, killing cells, anti-parasitic and bactericidal activity, participating in immediate allergic reactions, and modulating inflammatory responses [5] [6] .
In their granules, eosinophils store a number of biologically active molecules, such as eosinophil peroxidase, eosinophil cationic proteins, and numerous cytokines [7] .
Upon activation, eosinophils release their mediators and influence tissue homeostasis and integrity. In addition to their functions mentioned above, eosinophils could lead to profound changes in their microenvironment, often with resultant fibrosis, thrombosis, and severe organ damage.
In patients with persistent activation, tissue specimens might show marked deposition of eosinophil granule proteins, even in the absence of massive eosinophils infiltration.
Conditions such as Loffler’s disease, eosinophilic gastroenteritis, inflammatory bowel disease, acute eosinophilic pneumonia, and eosinophilic-myalgia syndrome are currently attributed to the direct toxicity of persistently activated eosinophils and their toxic granules contents.
A careful diagnostic work-up is needed for accurate classification of hypereosinophilia (HE). Precise classification is the mainstay of appropriate therapy. In addition to regular quantitative measurements of the blood cells in the PB, smears should be screened for monocytosis, cellular dysplastic features, indicators of myelofibrosis (e.g. dacrocytes, erythroid and myeloid precursors) and blasts. BM aspiration and biopsy are mandatory for histology and cytogenetic studies.
Screening for cytogenetically fusion genes such as BCL-ABL (Breakpoint Cluster region-Abelson, or Philadelphia chromosome), and FIP1L1-PDGFRA/B should be performed at an early stage for therapy modification and prognosis purposes.
Two different screening methods have been used, namely reverse transcriptase polymerase chain reaction (RT-PCR) and fluorescence in situ hybridization (FISH) [8] [9] . The relative frequency of FIP1L1-PDGFRA fusion is markedly variable in reported series, ranging between 3% and 56% in patient with HE.
In this report, we are describing a case of a patient who suffered from severe HE with evidence of multiple organ damage due to eosinophils infiltration, particularly in the heart. With the results of molecular studies; the final diagnosis was chronic eosinophilic leukemia not otherwise specified (CEL-NOS). The out- come was dire as the case was resistant to all treatments, and eventually turned into blast crisis which led to patient’s death.
Reviewing the literature, we found no registry for a large group of such patients followed for a long period of time. With such rare diseases, case reports might be the only source of knowledge physicians could have. CEL-NOS is a rare disease, and its rate of acute transformation into acute leukemia or into blast crisis (BC) is unknown. Sharing such knowledge about the dominant clinical and hematological features would provide better understanding of the course of this disease and its prognostic risk factors. We reviewed the relevant literature and found that this case represents the first fatal CEL-NOS turning into BC.
2. Case Report
A 19-year-old Caucasian male presented to urgent care with four months’ history of itching, anorexia, and weight loss. He also complained of intermittent skin rash. He denied any respiratory, neurological or gastroenterology symptoms. He reports no symptoms of hay fever, sinusitis, or respiratory tract infection. He is used to be healthy otherwise with no past medical history. Family medical history is unremarkable. He smokes occasionally but does not drink alcohol. Outpatient evaluation revealed appropriate vitals with no signs of allergic rhino-con- junctivitis. His chest sounded clear. Skin exam showed urticarial rash and itching marks (Figure 1). The patient was sent home on oral antibiotics, Medrol dose pack, and antihistamines. He did not respond well, and two weeks later; he visited the urgent care once again. This time, he complained of mild abdominal pain, right leg swelling, and dyspnea on exertion. He was found to have abdominal tenderness with possible hepatosplenomegaly. There was trace edema on both legs. Blood work up was withdrawn that day and ultrasound of the lower extremities was scheduled. Labs results revealed white blood count (WBC) of 19.8 × 103/mm3, platelet count of 60 × 103/mm3, and hemoglobin of 10 gm/dl. Eosinophils were 85% of total WBC (AEC is 16.2 × 103/mm3). Deep venous thro- mbosis in the right popliteal vein was demonstrated on the ultrasound.
The patient was called and advised for immediate hospital admission. However, he did not report to the hospital till one week later. At that time, he was complaining of right sided weakness with increasing dyspnea and lethargy. Blood Pressure was stable with no fever. Respiratory rate was 28 breaths per minutes with oxygen saturation of 92% on room air. Dark skin discoloration of his left foot with weak pluses was dominant (Figure 2). Lungs exam revealed bilateral crackles. A systolic murmur at the apex was identified on heart exam. He was not told he had a murmur before, and the urgent care record was not available. Right sided muscles weakness (4/5) was appreciated. Chest X-ray showed signs of pulmonary edema and cardiomegaly (Figure 3).

Figure 1. Picture of the lower extremities upon admi- ssion. Generalized macular rash with itching marks.

Figure 2. Picture of the left foot upon admission. Dark skin discoloration involving the first four toes, consis- tent with arterial ischemia.

Figure 3. Chest X-ray upon admission. Cardiomegaly, pulmonary edema, and bilateral pleural effusion.
Patient was admitted and started on IV anticoagulation, IV diuretics, and O2 supplement. The initial impression was severe hypereosinophilic syndrome (HES). IV pulse steroids (equivalent to 1 mg/kg of prednisone), and oral Hydro- xyurea (1000 mg daily) were initiated. Labs on admission are shown in Table 1 (column one: on admission). Shortly after admission, his mental status deteriorated, and mechanical ventilation support was required. Urgent head CT showed large ischemia on the left side middle cerebral artery territory (Figure 4). The patient was not candidate for craniotomy intervention due to low platelets count. Stool ova and parasites were not detected. Transthoracic echocardiogram showed endomyocardial infiltrative process with multiple intraventricular thrombi (Fig- ure 5, Figure 6). Cardiac ejection fraction was moderately reduced at 25%. Multiple infarctions in the spleen and the kidneys were shown on abdomen CT (Figure 7). Vitamin B 12 was elevated and tryptase level was not done.
Three days following admission, BM aspirate and biopsy were done. Results showed decreased erythropoiesis and megakaryopoiesis with the following differential: Band 1.0%, Neutrophils 24%, Lymphocytes 19%, Eosinophils 55%, Basophils 1.0%, Erythroid Precursors 1.0%, Myeloid 71%, Erythroid 1.0%, and blasts cells 12% (Figure 8). No fibrosis wasidentified. There was no morphology evidence of masotcytosis or acute leukemia. Flow cytometry in the (PB) showed no atypical lymphocytes. Cytogenetic and molecular studies were obtained.
At that juncture, the patient had met all criteria for WHO-classification for CEL. Awaiting chromosomal and molecular studies, and given such aggressive presentation, tyrosine kinase inhibitor (imatinib) was initiated (200 mg initially then 400 mg daily).

Figure 4. Head CT without contrast. Parenchymal low attenuation in the left middle cerebral artery territory consistent with massive infarction (red arrow). Brain edema with right midline shift.

Figure 5. Transthoracic echocardiography, Left ventricle apex image. Se- veral masses are noted attached to the mid posterior and apicalseg- ments, involving the chordae of the mitral valve (red arrows).

Figure 6. Transthoracic echocardiography, four chamber image. Restrictive myocardium infiltrative process (red arrow).

Figure 7. Abdomen CT without contrast. Spleen infarction (red arr- ow) bilateral kidneys infarctions (yellow arrows).
No cytogenetic abnormalities were detected by conventional karyotyping. FISH testing was negative for BCR/ABL, FIP1-L1/PDGFRA, and PDGFRB fusion genes. At that point, and correlating with the most recent 2008 WHO classification for myeloid disorders [10] , the case was reclassified aschronic eosinophilic leukemia none otherwise specified (CEL-NOS).
Patient was kept on prednisone 60 mg, 1000 mg of hydroxyurea, anticoagulation and imatinib. Through the following two weeks in the hospital, he began to feel better and showed good hematological recovery with decreasing eosinophilsto 8% (Table 1: column two: labs 2 weeks on therapy). Repeated BM aspirate showed blast cells to be less than 2%.
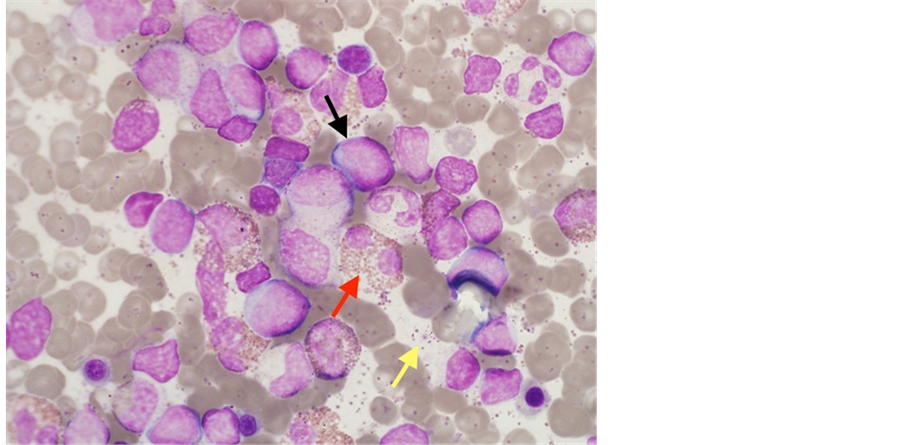
Figure 8. Bone marrow aspirate smear (Wright-Giesma stain, × 100). Mar- ked eosinophilia with mainly mature features (red arrow). An increase inblasts (black arrow). Numerous eosinophil granules from degranulated cells results in a “dirty” background (yellow arrow).

Table 1. Blood work up results on three different occasions. AEC: absolute eosinophils count.
After being on imatinib for three weeks, the patient began to deteriorate. He suffered from increasing dyspnea, low urine output, bilateral pneumonia, elevated liver function tests, and worsening heart failure. His biochemical parameters showed deranged hepatic-renal dysfunction with severe HE again. Blood labs are shown in Table 1 (column three: labs at blast crisis).
A third BM aspiration revealed blasts of 25% with dyspoieticeosinophilopoiesis. Cell differential showed: Band 11.0%, Neurtophils 55%, Lymphocytes 11%, Basophils 1.0%, Erythroid Precursors 7.0%, Myeloid 82%, Erythroid 7.0%, Promyelocyte 1.0%, Myelocytes 9%, and Metamyelocytes 5%. Blasts in PB were 17%.
Acute Myeloid Leukemia (AML) panel was done using FISH evaluation on cell culture. AML panel included the following chromosomal anomalies: RUX1/ RUNX1T1 Translocation (8, 21), PML/RARA Translocation (15, 17), CBFB (16q22) Rearrangement, MLL (11q23) Rearrangement, and PRN1/MECOM Translocation (3; 3) or inv 3. All those specified anomalies were not detected. Myelodisplastic syndrome (MDS) panel was also examined by FISH on cell culture and no chromosomal anomalies were detected. MDS panel included: (5q31) EGR1 for -5/5q-, (7q31) D7S522 for -7/7q-, 7cen and 8cen, 20q12 (D20S108) Deletion and RPN1/MECOM Translocation (3; 3) or inv (3). JAK-2V617F mutation was not detected. Since all tests were negative, transformation into acute blast crisis (BC) was confirmed.
As most patients with chronic myeloid leukemia (like CEL) eventually develop a blast phasein distinguishable from AML [11] , the patient received AML standard induction chemotherapy (cytarabine and daunorubicin). However, once AML was ruled out, chemotherapy was discontinued. Imatinib, however, was continued at a higher dose of 800 mg daily (patient received imatinib for total of 8 weeks).
Despite all of the above; the WBC continued to rise with severe HE. Eventually, the patient developed severe nosocomial pneumonia, sepsis, and with multi- organ failure. He passed away after three months of hospitalization.
3. Discussion
Eosinophilia is commonly observed in a wide range of disparate non-clonal and clonal disorders. Based on 2008 World Health Organization (WHO) classification [10] , HE is defined when PB eosinophils are higher than 500/mm3 on two different occasions 4 weeks apart. The term hypereosinophilic syndrome (HES) describes HE with evidence of organ dysfunction. Organ damage can be detected by findings such as hepatosplenomegaly, infiltrative heart failure, and pancytopenia. In our case report, the condition was initially classified as severe HES awaiting cytological and molecular studies.
HES is predominantly a disease of men (male-female ratio, 9:1), and is usually diagnosed between the ages of 20 and 50 years. In one series of 50 patients, the mean age at onset was 33 years and the mean duration of disease was 4.8 years [12] . The median survival was 9 months, and the 3-year survival was only 12%. These patients generally had advanced disease, with congestive heart failure accounting for 65% of the identified causes of death at autopsy. In addition to the development of cardiac disease, PB blasts or a WBC count more than 100 × 103/mm3 were associated with a poor prognosis.
A later report of 40 HES patients observed, a 5-year survival rate was 80%, decreasing to 42% at 15 years [13] . In this cohort, poor prognostic factors included the presence of a concurrent myeloproliferative syndrome, lack of response to corticosteroids, existence of cardiac disease, male sex, and the height of eosinophilia. It is clear that most of these risk factors for poor prognosis are found in our case.
If clonality is detected by genetic and molecular testing, then eosinophilic myeloid proliferative (Eos-MPD) disorder is being diagnosed. Among the cases of Eos-MPD, chronic eosinophilic leukemia (CEL) has its own diagnostic criteria [14] .
CELis a rare disorder with persistent hypereosinophilia, evidence of organ dysfunction, and an increased number of blood or marrow blasts (>5% and <20%). With the presence of 12 % blasts in the BM, our case was appropriately reclassified as severe CEL.
There have been many genetic abnormalities linked to CEL, the most common of which is fusion gene FILP1-L1/PDGFRA on chromosome 4. This gene is found in almost 10% of all CEL cases [15] . The resultant chimeric gene leads to production of a chimeric tyrosine kinase that is constitutively active. Persistent activation of tyrosine kinase protein causes unregulated growth and proliferation of eosinophils. The result is tissue damage and organ dysfunction. With improving in modern molecular pathogenesis, more chromosomal abnormalities have been found [15] .
Chronic eosinophilic leukemia-not otherwise specified (CEL-NOS) is also a rare disease. It is defined as Philadelphia-negative myeloproliferative neoplasm, which is due to clonal proliferation of eosinophil precursors. 2008 WHO criteria for myeloproliferative disorders, defined CEL-NOS as CEL that lacks the presence of any fusion genes abnormalities [15] . All other myeloproliferative neoplasms should also be excluded (e.g. Polycythemia Vera, Systemic Mastocytosis, and Essential Thrombosis). In this category,eosinophils in PB should be >1.5 × 103/mm3, and blasts in blood or marrow should be >5% and <20%.
In the new WHO classification, CEL-NOS is listed in the category of myeloproliferative neoplasms [15] [16] , whereas it was incorporated under the heading of myeloproliferative forms of hypereosinophilic syndromes according to the classification of the Eosinophilic Working Group [17] . Regardless of the classification used, CEL-NOS remains an extremely rare entity with variable prognosis [15] [18] . Given the above criteria, our reported case was appropriately reclassified as CEL-NOS.
Due to the rarity of the CEL-NOS, survival rate is not truly known. Mortality is related to the aggressiveness of the disease and to the burden of the tissue eosinophilic infiltration. Mortality is also related to the acute transformation into acute leukemia. In a recently reported cohort of 10 patients with CEL-NOS [19] , the median survival was 22.2 months. 5 of the 10 patients developed acute trans- formation (AT) after a median of 20 months from diagnosis of CEL-NOS. In that study, four patients developed AML-M4, one patient developed AML-M5, and one patient had T-cell lymphoblastic leukemia/lymphoma. In that study, most patients were predominantly male. Splenomegaly was the most frequent clinical manifestation.
CEL with gene abnormalities has been reported to have good response to tyrosine kinase inhibitor (imatinib) [20] . However, cases of CEL-NOS (where there are no detected gene anomalies) do not respond to tyrosine inhibitors. Some reports showed transient hematological recovery, but this response was limited, short lived, and it was thought to be drug-related myelosuppressive effect [20] .
In the described case, the patient received 400 mg daily imatinib within three days of admission with no delay. While waiting for the fusion gene FIP1L-1/ PDGFR/A result, it is recommended by the 2015-WHO recommendation to initiate imatinib therapy [21] . In cases of clonal HE, a trial of imatinib treatment is also recommended even in patients with no genes anomalies if there is evidence of organ dysfunction. There might be some cryptic molecular abnormalities that might respond to therapy but they are yet to be detected by future improvement in molecular testing methods. In our case the patient did respond initially to imatinib, however, the response lasted only for 2 - 3 weeks. Upon transforming into BC the dose was increased to 800 mg daily. Unfortunately, there was no good response, neither was there a response to AML standard induction chemotherapy (no maintenance therapy was given as AML was ruled out).
Most patients with chronic myeloid leukemia-a myeloproliferative disorder- eventually develop a blast phase indistinguishable from AML [12] . However, extensive cytogenetic testingin our patient failed to demonstrate any evidence for such transformation.
Blast crisis (BC) is the major remaining challenge in the management of chronic myeloid leukemia, and CEL is not an exception [22] . However, BC is usually associated with genetic abnormities.
Based on the recent World Health Organization, blast crisis is defined if BM blastsare more than 20% in analogy to the definition of acute myeloid leukemia (AML) [10] . In the reported case, there was a sudden increase in PB blasts (17%) and BM blasts (25%). With negative AML and MDS panels, the diagnosis of BC transformation was made. It is the first report that describes fatal BC transformation of CEL-NOS in the literature.
Several features have been associated with an unfavorable prognosis during BC, such as clonal evolution, more than 50% blast cells, high platelet counts, short duration of the chronic phase, unsatisfactory response to initial therapy during the chronic phase, and extra-medullary disease [22] .
4. Conclusion
We are reporting a first documented fatal case of severe CEL-NOS that transformed into blast crisis in a 19-year-old Caucasian male. The disease was aggressive since the presentation. Risk factors for such transformation would be: young age, cardiac involvement, high eosinophils count, negative molecular abnormalities, early transformation into blast crisis, and blasts count more than 25% in the BM. In order to keep the leukemia in its chronic phase, patients with similar features should be treated with tyrosine kinase inhibitor as early as possible.
Cite this paper
Al-Sanouri, B., Maslamani, Y. and Al-Sanouri, I. (2016) A Fatal Case of Chronic Eosinophilic Leukemia Not Otherwise Specified (CEL-NOS) in a 19-Year-Old Male with Acute Transformation into Blast Crisis. Case Reports in Clinical Medicine, 5, 528-540. http://dx.doi.org/10.4236/crcm.2016.512067
References
- 1. Kato, M., Kephart, G.M., Talley, N.J., Wagner, J.M., Sarr, M.G., Bonno, M., et al. (1998) Eosinophil Infiltration and Degranulation in Normal Human Tissue. Anatomical Record, 252, 418-425.
https://doi.org/10.1002/(SICI)1097-0185(199811)252:3<418::AID-AR10>3.0.CO;2-1 - 2. Ackerman, S.J. and Bochner, B.S. (2007) Mechanisms of Eosinophilia in the Pathogenesis of Hypereosinophilic Disorders. Immunology and Allergy Clinics of North America, 27, 357-375.
https://doi.org/10.1016/j.iac.2007.07.004 - 3. Brigden, M. and Graydon, C. (1997) Eosinophilia Detected by Automated Blood Cell Counting in Ambulatory North American Outpatients. Incidence and Clinical Significance. Archives of Pathology & Laboratory Medicine, 121, 963-967.
- 4. Hogan, S.P., Rosenberg, H.F., Moqbel, R., Phipps, S., Foster, P.S., Lacy, P., et al. (2008) Eosinophils: Biological Properties and Role in Health and Disease. Clinical & Experimental Allergy, 38, 709-750.
https://doi.org/10.1111/j.1365-2222.2008.02958.x - 5. Rothenberg, M. and Hogan, S. (2006) The Eosinophil. Annual Review of Immunology, 24, 147-174.
https://doi.org/10.1146/annurev.immunol.24.021605.090720 - 6. Shi, H. (2004) Eosinophils Function as Antigen-Presenting Cells. Journal of Leukocyte Biology, 76, 520-537.
https://doi.org/10.1189/jlb.0404228 - 7. Gleich, G.J. (2000) Mechanisms of Eosinophil-Associated Inflammation. Journal of Allergy and Clinical Immunology, 105, 651-663.
https://doi.org/10.1067/mai.2000.105712 - 8. Langer-Safer, P.R., Levine, M. and Ward, D.C. (1982) Immunological Method for Mapping Genes on Drosophila Polytene Chromosomes. Proceedings of the National Academy of Sciences of the United States of America, 79, 4381-4385.
https://doi.org/10.1073/pnas.79.14.4381 - 9. Amann, R. and Fuchs, B.M. (2008) Single-Cell Identification in Microbial Communities by Improved Fluorescence in Situ Hybridization Techniques. Nature Reviews Microbiology, 6, 339-348.
https://doi.org/10.1038/nrmicro1888 - 10. Bain, B.J., Gilliland, D.G., Horny, H.-P., et al. (2008) Chronic Eosinophilic Leukaemia, Not Otherwise Specified. In: Swerdlow, S., Harris, N.L., Stein, H., Jaffe, E.S., Theile, J. and Vardiman, J.W., Eds., World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, IARC Press, Lyon, 51-53.
- 11. Hehlmann, R. (2012) How I Treat CML Blast Crisis. Blood, 120, 737-747.
https://doi.org/10.1182/blood-2012-03-380147 - 12. Fauci, A.S., Harley, J.B., Roberts, W.C., Ferrans, V.J., Gralnick, H.R. and Bjornson, B.H. (1982) The Idiopathic Hypereosinophilic Syndrome. Clinical, Pathophysiologic, and Therapeutic Considerations. Annals of Internal Medicine, 97, 78-92.
https://doi.org/10.7326/0003-4819-97-1-78 - 13. Lefebvre, C., Bletry, O., Degoulet, P., et al. (1989) Prognostic Factors of Hypereosinophilic Syndrome. Study of 40 Cases. Annals of Internal Medicine, 140, 253-257.
- 14. Bain, B.J. (2003) Cytogenetic and Molecular Genetic Aspects of Eosinophilic Leukaemias. British Journal of Haematology, 122, 173-179.
https://doi.org/10.1046/j.1365-2141.2003.04458.x - 15. Bain, B.J., Gilliland, D.G., Vardiman, J.W. and Horny, H.P. (2008) Chronic Eosinophilic Leukemia, Not Otherwise Specified. In: Swierdlow, S., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. and Vardiman, J.W., Eds., World Health Organization Classification of Tumours: Tumours of Haematopoietic and Lymphoid Tissue, International Agency for Research on Cancer, Lyon, 68-73.
- 16. Swierdlow, S., Campo, E., Harris, N.L., et al. (2008) World Health Organization Classification of Tumours: Tumours of Haematopoietic and Lymphoid Tissue. International Agency for Research on Cancer, Lyon.
- 17. Simon, H.U., Rothenberg, M.E., Bochner, B., et al. (2010) Refining the Definition of Hypereosinophilic Syndrome. Journal of Allergy and Clinical Immunology, 126, 45-49.
https://doi.org/10.1016/j.jaci.2010.03.042 - 18. Crane, M.M., Chang, C.M., Kobayashi, M.G. and Weller, P.F. (2010) Incidence of Myeloproliferative Hypereo-Sinophilic Syndrome in the United States and an Estimate of All Hypereosinophilic Syndrome Incidence. Journal of Allergy and Clinical Immunology, 126, 179-181.
https://doi.org/10.1016/j.jaci.2010.03.035 - 19. Helbig, G., Soja, A., Bartkowska-Chrobok, A. and Kyrcz-Krzemien, S. (2012) Chronic Eosinophilic Leukemia-Not Otherwise Specified Has a Poor Prognosis with Unresponsiveness to Conventional Treatment and High Risk of Acute Transformation. American Journal of Hematology, 87, 643-645.
https://doi.org/10.1002/ajh.23193 - 20. Coutré, S. and Gotlib, J. (2004) Targeted Treatment of Hy-pereosinophilic Syndromes and Chronic Eosin-Ophilicleukemias with Imatinib Mesylate. Seminars in Cancer Biology, 14, 23-31.
https://doi.org/10.1016/j.semcancer.2003.11.004 - 21. Gotlib, J. (2015) World Health Organization-Defined Eosinophilic Dis-orders: 2015 Update on Diagnosis, Risk Stratification, and Management. American Journal of Hematology, 90, 1077-1089.
http://onlinelibrary.wiley.com/doi/10.1002/ajh.24196/full#ajh24196-fig-0001 https://doi.org/10.1002/ajh.24196 - 22. Wadhwa, J., Szydlo, R.M., Apperley, J.F., et al. (2002) Factors Affecting Duration of Survival after Onset of Blastic Transformation of Chronic Myeloid Leukemia. Blood, 99, 2304-2309.
https://doi.org/10.1182/blood.V99.7.2304