Journal of Agricultural Chemistry and Environment
Vol.03 No.03(2014), Article ID:48912,17 pages
10.4236/jacen.2014.33013
Comparison of Accelerated Solvent Extraction, Soxhlet and Sonication Techniques for the Extraction of Estrogens, Androgens and Progestogens from Soils
Sonya M. Havens1*, Curtis J. Hedman1,2, Jocelyn D. C. Hemming2, Mark G. Mieritz2, Martin M. Shafer1,2, James J. Schauer1
1Environmental Chemistry and Technology, University of Wisconsin―Madison, Madison, WI, USA
2Wisconsin State Laboratory of Hygiene, Environmental Health Division, Madison, WI, USA
Email: *sonyahavens@gmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 24 April 2014; revised 25 May 2014; accepted 20 June 2014
ABSTRACT
Leaching of hormones from manure amended fields to receive surface water can lead to endocrine disruption in resident fish populations. In order to determine the concentrations of hormones present in manure amended soils, and thus the potential for soils to release hormones to aquatic environments, efficient extraction methods are needed. In this study, the efficacy of three techniques (accelerated solvent extraction [ASE], Soxhlet and sonication) for the extraction of estrogens, androgens and progestogens, as well as their metabolites, from various soil types were evaluated. The stability of hormones spiked into these soils and stored for 30, 90 and 210 days at −20˚C was also investigated. Four experimental soil matrices (reagent sand, silt loam, clay and high organic) were spiked with 50 μL of 10 μg・mL−1 (in methanol; final conc. 100 ng・g−1) of a stock mix of hormones and isotopically-labeled standards (ISTDs). After equilibration, triplicate samples of the spiked soils were extracted by ASE, Soxhlet and sonication techniques and analysed, without post extraction cleanup, using HPLC-MS/MS. Sonication and ASE were effective at extracting hormones from all matrices with overall average apparent recoveries, for all 19 extracted analytes, of 71% ± 23% and 73% ± 16%, respectively. Soxhlet was significantly less efficient (p < 0.05) with overall average apparent recoveries of 58% ± 34%. Incorporation of ISTDs resulted in overall average process efficiencies of 108% ± 24%, 102% ± 24% and 180% ± 310% for ASE, Soxhlet and sonication, respectively. The hormones had variable stability in soils stored for at least 30 days, and therefore it recommended that soil samples be analysed within 30 days of sampling.
Keywords:
Hormones, Storage Stability, Pressurized Liquid Extraction, Liquid Chromatography

1. Introduction
Approximately 60,000 metric tonnes of manure are generated annually in the United States, most of which is land-applied to meet crop nutrient demands [1] . Estrogens, androgens and progestogens, as well as their metabolites, are produced by all vertebrates and are found in livestock waste [2] . Synthetic hormones (e.g., tren-bolone acetate and melengestrol acetate) that are used to enhance livestock growth and synchronize the estrous cycle have also been detected in cattle waste [3] . Alpha-zearalenol, zearalenone and zearalanone were also chosen for study since they are metabolites of the oestrogenic mycotoxin α-zearalanol that are used in some cattle implants for growth promotion and may also be produced by fungi, principally from the genus Fusarium, that grow on cereal plants (e.g., corn, barley, etc.) in agricultural environments. When applied to soils, these hormones and hormone metabolites can potentially leach into the adjacent ground and surface waters [3] -[6] and may subsequently disrupt the endocrine systems of aquatic organisms [7] -[9] . In order to determine the concentrations of hormones and hormone metabolites present in manure amended soils, robust extraction and detection methods are needed. These methods are also vital to investigate the extent to which hormones and hormone metabolites are degraded in soils, retained by soils or leached from soils to ground and surface waters.
Several techniques have been utilized to extract and analyse hormones, particularly estrogens, from solid matrices (Table 1). However, many androgens and progestogens, as well as their metabolites, may be present in

Table 1. Results of various analytical methods for the evaluation of hormones in solid matrices.
1E1 = estrone, E2 = estradiol, E3 = estriol, EE2 = ethinyl estradiol, 4A = 4-androstene-3,17-dione, AND = androsterone, P = progesterone and T = testosterone. 2Liquid chromatography-diode array detection-mass spectrometry. 3Liquid chromatography-mass spectrometry using an electrospray interface. 4Pentafluoro- propionic anhydride. 5Gas chromatography-mass spectrometry. 6Microwave assisted solvent extraction. 7N,O-Bis(trimethylsilyl) trifluoroacetamide with trimethylchlorosilane. 8N-trimethylsilyl-N-methyl trifluoroacetamide. 9Restricted access materials. 10Liquid chromatography mass selective detector equipped with an atmospheric-pressure ionization source and electrospray interface.
cattle manure amended soil [3] -[6] . Thus, a technique that can efficiently extract and analyse estrogens, androgens and progestogens, as well as their metabolites, in soils with minimal sample processing, is needed. To this end, this study evaluated the efficacy of three techniques (accelerated solvent extraction [ASE], Soxhlet and sonication) for the extraction of a large suite of synthetic and natural estrogens, androgens and progestogens, as well as their metabolites, that have been previously detected [3] [4] [18] -[21] or are likely to be present in manure amended fields (Table 2), from various soil types (e.g., sand, silt loam, clay and high organic content) in order to assess which technique was most efficient for the simultaneous extraction of this large suite of analytes. Several isotopically (deuterium-d)-labeled standards (ISTDs, Table 2) were incorporated to account for extraction inefficiency and losses that may occur during sample processing as well as matrix suppression and/or enhancement effects that may take place in the ionization chamber of the mass spectrometer during sample analysis. Proper utilization of the appropriate ISTD for each target analyte is essential to the accurate and precise measurement of these analytes [22] . Incorporation of an ISTD that does not produce a similar recovery to its target analyte will result in the over- or underestimation of the analyte concentration [22] . As such, this study assesses the applicability of several ISTDs (Table 2) to appropriately account for analyte losses during sample extraction and analysis in order to minimize the potential for over- or underestimation of analyte concentrations when incorporating these ISTDs.
The ISTDs were also used to assess the stability of these compounds after 30, 90 and 210 days of storage at −20˚C. Finally, native soils, collected from a pasture at a grass-grazing dairy farm, were analysed for estrogens, androgens and progestogens using ASE and high performance liquid chromatography with tandem mass spec- trometry (HPLC-MS/MS).
Table 2. Target analytes with corresponding unlabeled Chemical Abstracts Service (CAS) number and isotopically-labeled analogues.
This study builds upon previously published work, using a range of soil types and the inclusion of synthetic estrogens as well as synthetic and natural androgens and progestogens and their metabolites and assesses the stability of these analytes under storage conditions that are likely to occur during occurrence survey studies. Furthermore, this study evaluates the ability of ASE, sonication and Soxhlet to extract these compounds from various soil types with minimal sample processing (i.e., without drying, sieving, lyophilizing or pre-extracting soils and without the use of post extraction cleanup).
2. Experimental
2.1. Materials
The 19 hormones and hormone metabolites (target analytes; Table 2) chosen for study included natural and synthetic estrogens, androgens and progestogens that have previously been detected or could be present in cattle manure amended soil [19] . All of the analytical standards (Table 2) were of high purity (>98%) and were obtained from Sigma-Aldrich (St Loius, MO) with the exception of 17α-trenbolone, which was purchased from Hayashi Pure Chemical Inc. (Osaka, Japan). The isotopically (deuterium-d)-labeled standards (ISTDs) 17b-es- tradiol-d5, estriol-d3, testosterone-d5, nandrolone-d3, and progesterone-d9 were obtained from C/D/N Isotopes (Pointe-Claire, Quebec, Canada) and melengestrol-d3, melengestrol acetate-d3, 17β-trenbolone-d3 and α-zeara- lenol-d4 were obtained from the European Union Reference Laboratory at the National Institute for Public Health and the Environment (RIVM; Bilthoven, The Netherlands). HPLC-grade methanol, hexane and acetone were obtained from Burdick & Jackson. Sodium sulfate (Na2SO4) was purchased from Fisher Scientific and muffled at 550˚C for 16 hours prior to use.
Four experimental soil matrices were used to examine the efficiencies of each of the extraction methods among various soil types (Table SI.1, Supplementary Information); 1) a reagent sand (Ottawa Sand, Fisher Scientific, Pittsburgh, PA); 2) Antigo silt loam; 3) a soil with high clay content and 4) a soil with high organic matter content. Antigo silt loam was collected from the University of Wisconsin Spooner Agricultural Research Center (Spooner, WI) and the high clay soil was collected in Platteville, WI. The high organic matter soil was a sandy loam collected from a wetland in Vilas County, WI. Prior to use, the antigo silt loam and organic matter soils were homogenized using a stainless steel commercial blender (Model 700S, Waring Commercial, Torring- ton, CT). The clay soil was placed within two ziplock bags and worked with a mallet and rolling pin until the soil was thoroughly homogenized. The reagent sand had been muffled at 550˚C for 16 hours prior to use, whereas all other experimental matrices were neither dried nor sieved. The prepared experimental soil matrices were submitted to the University of Wisconsin―Madison Soil and Plant Analysis Laboratory (Verona, WI) for evaluation of particle size, soil pH, organic matter content and CEC.
2.2. Extraction Comparison
Sub-samples of the soils (five grams of sand, silt loam and high clay and three grams of high organic matter soil) were spiked, in triplicate, with a stock solution of target analytes and ISTDs (Table 2) at 50 μL of 10 μg・mL−1 (in methanol, final conc. 100 ng・g−1, 167 ng・g−1 for high organic soil), which represents concentrations that are within the range of those used by other investigators (Table 1). In addition, a subset of these samples was spiked with ISTDs only, in triplicate, in order to quantify the background hormone concentrations in each of the soil matrices. The spiked hormones were mixed into the soils using the wooden end of sterile cotton-tipped applicators (Fisherbrand, Fisher Scientific) and allowed to equilibrate with the soils for two hours. The two-hour equilibration time was chosen to allow enough time for the analytes to partition onto the solids, but not enough time to allow for potential degradation [23] . The soils, as well as the portion of wood that came in contact with the soils during mixing, were then extracted, in triplicate, using ASE, Soxhlet and sonication with acetone-hexane (1:1, v/v) used as the solvent in all cases.
2.2.1. Accelerated Solvent Extraction
The 11 mL stainless steel extraction cells (Dionex, Sunnyvale, CA, USA) employed were sealed with stainless steel screw caps equipped with teflon O-rings. The assembled extraction cells were layered, from the bottom up, with two 19 mm muffled glass fiber filters (GF/A, Dionex Corp., Sunnyvale, CA, USA), 2.0 g of muffled Otta- wa sand, 5.0 ± 0.1 g of spiked sample (or 3.0 ± 0.1 g of high organic matter soil sample) mixed with at least 2 g Na2SO4 (up to 3 g was used for the high organic matter soil), 1.0 g of muffled Ottawa sand and topped with one 19 mm GF/A filter. After tamping down the material within the cell and affixing the cell’s top endcap, the cells were loaded onto the ASE, preheated to 120˚C and held for 5 min without solvent. The material within the cell was then subjected to two cycles of solvent exposure, wherein 6.6 mL of acetone-hexane (1:1, v/v) was pumped into the cell and the temperature and pressure were maintained at 120˚C and 1500 psi, respectively, for 5 min. The solvent was eluted with a flush of nitrogen into a 60 mL amber glass collection vial (I-CHEM, Rockwood, TN). Method blanks, consisting of three 19 mm GF/A filter discs, muffled Ottawa sand and sodium sulfate, were extracted after every five soil sample extractions to assess the potential for analyte carry over.
2.2.2. Soxhlet
The Soxhlet extractions were performed as described in USEPA Method 1698 [14] . Briefly, each spiked sample (5.0 ± 0.1 g of sand, silt loam and clay and 3.0 ± 0.1 g of high organic matter soil) was placed into a cellulose thimble (Whatman, GE Healthcare Life Sciences, Little Chalfont UK) and loaded into a glass Soxhlet extraction system (Lab Line Instruments, Inc., Melrose Park, IL). The organic samples were accidently spiked with 100 μL of 10 μg・mL−1 (final conc. 333 ng・g−1) instead of 50 μL of 10 μg・mL−1. The target analytes and ISTDs were extracted from each of the soil types by refluxing 300 mL of acetone-hexane (1:1, v/v), with the aid of boiling granules (Hengar Co., Thorofare, NJ), continuously through the sample for 16 to 24 hours at a rate of 2 - 3 cycles every 5 minutes.
2.2.3. Sonication
The spiked samples were mixed with 2.0 g of Na2SO4 and placed in silanized 100 mL glass beakers. Each soil was extracted with three cycles of ultrasonication. Each ultrasonication cycle consisted of adding 20 mL of acetone:hexane (1:1, v/v) to the sample, placing the beaker into a 3 qt. ultrasonic bath (Fisher Scientific Mechanical Ultrasonic Cleaner FS20, Fisher Scientific, Pittsburg, PA) operating at 40 kHz for 10 min. and then transferring the supernatant to a methanol rinsed 15-mL disposable glass centrifuge tube (Kimble Chase, Vineland, NJ).
2.3. Storage Stability
Five grams of sand, silt loam and high clay and 3.0 g of high organic matter soil were placed into 20 mL-sila- nized glass scintillation vials (Fisher Scientific, Pittsburgh, PA), spiked, in sextuplicate, with a stock solution of ISTDs (Table 2) at 50 μL of 10 μg・mL−1 (in methanol, final conc. 100 ng・g−1 for sand, silt loam and clay and 167 ng・g−1 for high organic soil) and then vortexed for 30 seconds. The samples were either extracted after a two hour equilibration period (t = 0) or wrapped in aluminum foil, stored at −20˚C for 30 days (t = 30), 90 days (t = 90) and 210 days (t = 210) and subsequently extracted using protocols described above for the ASE.
2.4. Native Samples
Native soil samples were collected, using a stainless steel soil corer, from a 15-acre paddock that sustained grass-grazing dairy cattle year round. To ensure that samples were representative of the entire paddock, three 20-cm deep soil cores (shallow cores) were collected along each of five transects, with an additional soil core collected from a depth of 20 - 51 cm (deep cores) from each of the five transects.
Five grams of each of the native soil samples were spiked with ISTDs at 50 μL of 10 μg・mL−1 (in methanol, final conc. 100 ng・g−1). The spiked samples were allowed to equilibrate for two hours and extracted using the ASE with the protocols described above.
2.5. Extract Concentration
All extracts were concentrated to approximately 200 μL, using a stream of nitrogen at 40˚C in a Turbovap LV Concentration Evaporator Workstation (Biotage, Uppsala, Sweden) for the ASE and sonication extracts and using a Rotary Evaporator (Buchi, Switzerland) at approximately 50˚C for the Soxhlet extracts. These extracts were then reconstituted to a final volume of 1.0 mL with methanol in 2.0 mL amber glass vials (Target LoVial, National Scientific).
2.6. Liquid Chromatography and Mass Spectrometry
The hormone concentrations in the extracts were analysed using high-performance liquid chromatography (Agilent Technologies 1100 HPLC, Santa Clara, California) with tandem mass spectrometric detection (AB SCIEX API 4000, Framingham, Massachusetts) (HPLC-MS/MS) operating in positive Atmospheric Pressure Chemical Ionization (APCI) mode as outlined in [24] . Briefly, a sample injection volume of 15 μL was applied to a 4 μm, 4.6 × 250 mm Synergi MAX-RP column (Phenomenex, Torrance, CA) and separated with a reversed phase binary mobile phase gradient (Table SI.2, Supplementary Information) at 0.8 mL・min−1. Relevant multiple reaction monitoring (MRM) mass spectrometer settings include collision gas at 6 arbitrary units, curtain gas at 25 psig, nebulization gas at 40 psig, drying gas at 15 psig, corona discharge current of 3 volts and source temperature at 450˚C. Multiple reaction monitoring m/z values for the target analytes are listed in the Supplementary Information (Table SI.3). The instrument was calibrated with methanol dilutions of each analyte at 10, 20, 50, 100, 250, 500, 1000, 2500 and 5000 ng・mL−1. A relative response ratio, between each target analyte and its corresponding ISTD, was generated at each target analyte calibration concentration with the ISTD concentration maintained at 500 ng・mL−1. Linear or quadratic regression with 1/x weighting was used to generate calibration curves of the relative response ratios of all analytes. The calibration coefficients always exceeded 0.990. The target analyte concentration in each sample extract was calculated by normalising the relative response ratio in the sample extract to those in the calibration curve. The instrument detection limits (IDL) for all analytes monitored were less than 0.9 ng・g−1 (wet weight, Table SI.4, Supplementary Information) and were calculated by multiplying the standard deviation of ten low level standards (5.0 ng・mL−1) by the student T test variate (α = 0.01) with nine degrees of freedom. The limit of quantitation for each analyte (Table SI.4, Supplementary Information) was evaluated as three times the IDL.
2.7. Quality Controls
Negative controls analyzed with each sample group included solvent blank injections and internal standard in solvent injections. All negative controls were
2.8. Statistics
The data were analysed using the programming application R, version 2.11.1 (R Foundation, 2006). Analysis of variance (ANOVA) procedures were used to test for significant (α = 0.05) differences in the mean recoveries among the extraction treatments. The Tukey Honest Significant Difference test was then used to find the treatments that differed from each other.
3. Results & Discussion
3.1. Apparent Recovery Comparison
The apparent recoveries [25] of the extraction techniques were evaluated prior to ISTD normalisation and were calculated using the equation:
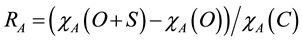
where χA(O + S) is the average peak area of analyte A in the spiked samples (n = 3), χA(O) is the average peak area of analyte A in the corresponding unspiked samples (i.e., ISTD only samples, n = 3) and χA(C) is the average peak area of analyte A in the 500 ng standards in the calibration curve on the HPLC (n = 2).
The apparent recoveries of 17β-estradiol and estriol extracted from soil with 67% clay content were low (<12%) under all of the extraction techniques employed (Figure 2(A)-(C)). Zuloaga et al. [26] reported decreasing PAH recoveries with increasing clay contents of soils extracted with ASE. While Beck et al. [15] found no correlation between clay content and estrogen recoveries at clay contents ranging from 14% to 31%, our results suggest that clay contents ≥ 67% will result in reduced recoveries of estradiol and estriol extracted with ASE, Soxhlet or sonication.
Given the low apparent recoveries of 17β-estradiol and estriol in clay samples, these analytes are excluded from the ranges and averages of apparent recoveries. When 17β-estradiol and estriol are excluded, the apparent recoveries of the remaining target analytes extracted from all soil matrices with ASE ranges from 44.8% (estriol from sand) to 99.9% (estrone from organic) with an overall average of 73% ± 16%. The overall average apparent recovery of sonication (71% ± 23%) was similar to ASE (p > 0.05). The apparent recoveries of target analytes extracted with Soxhlet were highly variable (Figure 1) and resulted in the lowest overall average apparent recovery (58% ± 34%, p < 0.05). The high variability in apparent recoveries of Soxhlet extracts was due to the high recovery of some analytes (i.e., zearalenone, zearalanone and 5α-androstane-3,17-dione) and the low recoveries of others (i.e., 17β-estradiol, estriol, 17β-trenbolone and 17α-trenbolone (Figure 2(B)).
The sonication technique resulted in the highest average apparent recoveries of the target analytes extracted from both sand and clay soils (85% ± 16% and 91% ± 11%, respectively) compared to ASE (70% ± 20% and 65% ± 12%, respectively) and Soxhlet (59% ± 34% and 51% ± 27%, respectively; p < 0.05, Figure 1(A)-(C)). The highest average apparent recovery of target analytes extracted from silt loam was achieved using ASE (81% ± 11%, p<0.05, Figure 1). Both sonication and Soxhlet were relatively ineffective (48 ± 12% and 45 ± 28%, respectively) at extracting the target analytes from silt loam (Figure 1). Sonication was particularly poor (10% ± 1% apparent recovery) at extracting estriol from silt loam (Figure 2(C)).
Sonication had the lowest apparent recovery (62% ± 19%), compared to ASE (75% ± 16%) and Soxhlet (80% ± 36%), at extracting the target analytes from the organic soil (p < 0.05, Figure 1). Soxhlet was remarkably effect- tive (>88%) at extracting 17β-estradiol and estriol from the high organic matter soil (Figure 2(B)). Though,
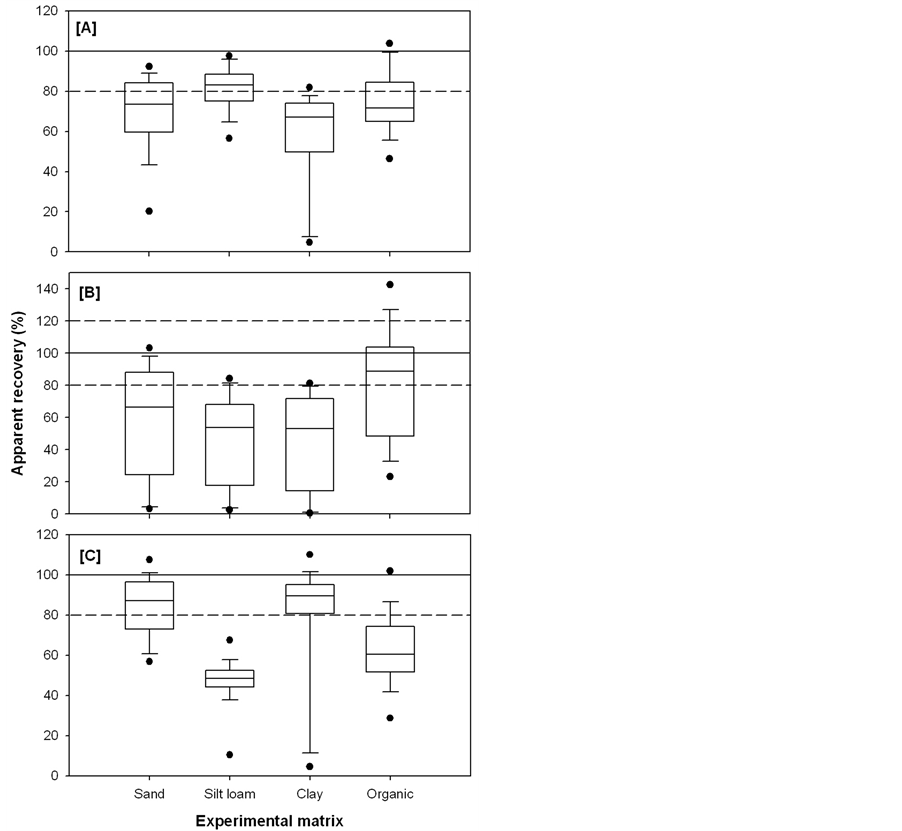
Figure 1. Ranges in the apparent recoveries (n = 57), prior to ISTD normalisation, of target analytes spiked into sand, silt loam, clay and organic soils and extracted using ASE (A); Soxhlet (B) and sonication (C).
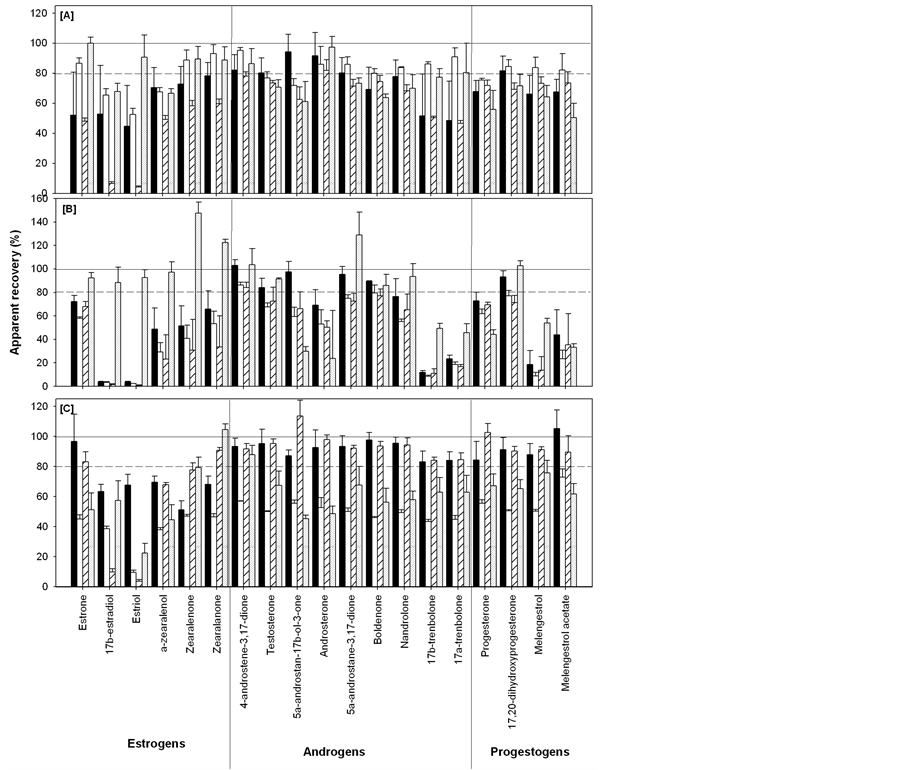
Figure 2. Apparent recoveries (average ± standard deviation, n = 3), prior to ISTD normalisation, of target analytes spiked with 50 μL of 10 μg・mL−1 (in methanol) into sand (black), silt loam (white), clay (striped) and organic soil (hatched) and extracted with ASE (A), Soxhlet (B) and sonication (C).
this may have been due to the fact that the organic samples extracted with Soxhlet were inadvertently spiked at a higher concentration (100 μL of 10 μg・mL−1) than the sand, silt loam and clay samples (50 μL of 10 μg・mL−1). The enhanced apparent recoveries in Soxhlet extracts could also have been a result of peak signal enhancement on the HPLC-MS/MS due to coextracted matrix components (i.e., humic and fulvic acids) that can increase the intensity of analyte ions [27] , particularly in soils with a high organic carbon content. Indeed, the high apparent recoveries of zearalenone (147% ± 10%), zearalanone (123% ± 3%) and 5α-androstane-3,17-dione (129% ± 20%) in organic samples extracted with Soxhlet may have been due to signal enhancement caused by coextracted matrix components.
Soxhlet was particularly ineffective at extracting 17β-estradiol, estriol, 17β-trenbolone, 17α-trenbolone and melengestrol from sand, silt loam and clay (<24%), especially compared to ASE (>50%, p < 0.05) and sonication (>43%, p < 0.05, Figure 2), excluding 17β-estradiol and estriol in clay. Androsterone extracted from organic soils with Soxhlet was only recovered in one out of three of the replicates (Figure 2(B)). The coextraction of matrix components leads to higher background noise in the chromatogram, which can result in an underestimation of analyte recovery since the baseline is set at the noise level. This high baseline noise due to the coextraction of matrix components is particularly problematic for compounds that have low peak areas on the HPLC-MS/MS. The peak areas of androsterone in the calibration curve on the HPLC-MS/MS were low (≈3.5 × 104 at 500 ng). The increased background noise caused by the coextracted matrix components “drowned out” the androsterone signal in two out of three of the samples resulting in low apparent recovery and poor precision (24% ± 41%).
The Soxhlet extraction also resulted in low apparent recoveries (<30%) of α-zearalanol in silt loam and clay and melengestrol acetate in silt loam (Figure 2(B)). This suggests that the acetone:hexane solvent exposure with heat alone (Soxhlet) may have been unable to overcome the sorption interactions between these analytes and the soil particles, resulting in poor apparent recoveries. The combination of acetone:hexane solvent exposure with increased temperature and pressure (ASE) or particle agitation (sonication) was likely more effective at disrupting the sorption interactions than heat alone (Soxhlet) and resulted in the enhanced apparent recoveries of these analytes. These low recoveries may have been a result of ion suppression due to the coextraction of matrix components that can reduce the intensity of analyte ions [27] .
3.2. Process Efficiency Comparison
The process efficiencies were calculated using the equation:
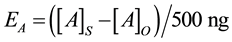
where [A]S is the average calculated concentration (i.e., normalised to the ISTD, Figure SI.1) of analyte A in the extracts of the spiked samples (n = 3), [A]O is the average calculated concentration of analyte A in their corresponding unspiked samples (n = 3, Figure SI.2) and 500 ng is the spike concentration.
The use of ISTDs as internal standards can account for analyte losses resulting from extraction inefficiency, adsorption to particles that renders the analytes unextractable, and to some extent microbial degradation. How- ever, this technique relies on adequate ISTD recovery and is only applicable to target analytes that have a similar recovery to their corresponding ISTD. If the apparent recovery of an ISTD is much lower than that of its corresponding target analyte it will lead to the overestimation of the target analyte concentration. Conversely, if the recovery of the ISTD is much higher than that of its corresponding target analyte it will lead to an underestima- tion of the target analyte concentration. Furthermore, low ISTD recovery results in high relative standard deviation of the normalised concentration through a denominator effect; small variances in the target analyte peak area result in large variances in the normalised concentration when normalising to a low ISTD recovery [22] .
Figure 3 provides the ranges of process efficiencies in ASE, Soxhlet, and sonication extractions among each soil type. Combining ISTD normalisation with ASE, sonication and Soxhlet extractions resulted in average process efficiencies of 119% ± 95%, 109% ± 63% and 229% ± 533%, respectively for all analytes extracted from all soil types. However, the process efficiencies of estrone extracted from clay with ASE, sonication and Soxhlet were all significantly overestimated (913% ± 144%, 608% ± 48% and 3973% ± 873% respectively, Figure 4). These overestimations were due to the normalisation of the adequate apparent recoveries of estrone (48% ± 2%, 83% ± 7% and 68% ± 4% for ASE, sonication and Soxhlet, respectively, Figure 2) to the poor recovery of its corresponding ISTD, 17β-estradiol-d5 (7% ± 2%, 17% ± 3% and 2% ± 1% for ASE, sonication and Soxhlet, respectively). Thus, 17β-estradiol-d5 may not be the appropriate ISTD to account for the efficiency of estrone extraction from soil. Unfortunately, attempts to utilize estrone-d2 were unsuccessful due to its susceptibility to deuterium loss [28] . When estrone in clay extracts are omitted, the average process efficiencies were 108% ± 24%, 102% ± 24% and 180% ± 310% for ASE, sonication and Soxhlet, respectively. With the exception of Soxhlet, these recoveries are consistent with those witnessed in other studies (Table 1) and were obtained without drying, sieving or lyophilizing the soils prior to extraction, which can result is target analyte losses due to degradation, and without the use of post-extraction cleanup, which can be costly, time consuming and may also lead to target analyte losses due to inefficient recovery.
The concentrations of many of the target analytes extracted with Soxhlet were overestimated (Figure 3(B)) leading to an artificially high average process efficiency and poor precision (229% ± 533%). The process efficiencies of estrone, α-zearalanol, zearalenone and zearalanone in sand, silt loam and clay and 17,20-dihy-drox- yprogesterone in organic soil extracted with Soxhlet were overestimated (Figure 4(B)) due to the extremely low apparent recoveries of their corresponding ISTD’s. The concentrations of estriol in silt loam and clay were unable to be calculated due to the fact that estriol-d3 was not recovered in the Soxhlet extracts. The concentration of estriol extracted from sand with Soxhlet was only capable of being calculated in the one replicate, out of the three, where estriol-d3 was recovered. The peak areas of Estriol-d3 in the calibration curve (500 ng) were low
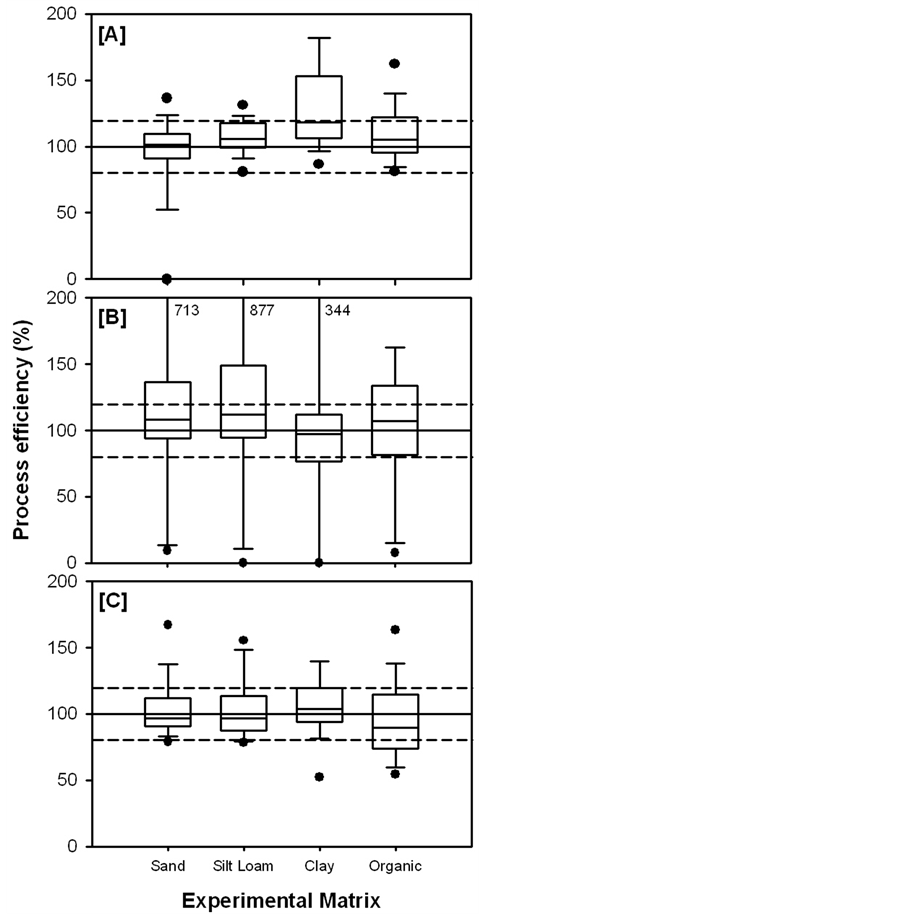
Figure 3. Ranges in the process efficiencies (n = 57) of target analytes spiked into sand, silt loam, clay and organic soil and extracted using ASE (A); Soxhlet (B) and sonication (C).
(≈5.5 × 104). Thus, even small losses of estriol during extraction can result in not only poor recovery, but also high variability as well due to the denominator effect [22] . Estriol-d3 was sufficiently recovered from organic soil, but this was likely due to the inadvertent double spiking of organic soils extracted with Soxhlet.
Both ASE and sonication resulted in adequate process efficiencies for the extraction of estriol from all soil types except clay (Figure 4). While the apparent recoveries of estriol were low in some instances (Figure 2), the use of estriol-d3 accounted for the low apparent recoveries that may have been done to extraction inefficiency or matrix induced signal suppression (Figure 4). These estriol results are an improvement to those obtained in the study conducted by Peng et al. [12] (45% ± 10% and 27% ± 3% using Soxhlet and sonication, respectively) due to the use of an ISTD with a similar apparent recovery to estriol (i.e., estriol-d3). The lower process efficiency of estriol from sonication in the Peng et al. [12] study (i.e., 27% ± 3%) was due to the use of an ISTD (i.e., 17β-estradiol acetate) that had an apparent recovery (i.e., 107% - 110%) that differed considerably from that of estriol.
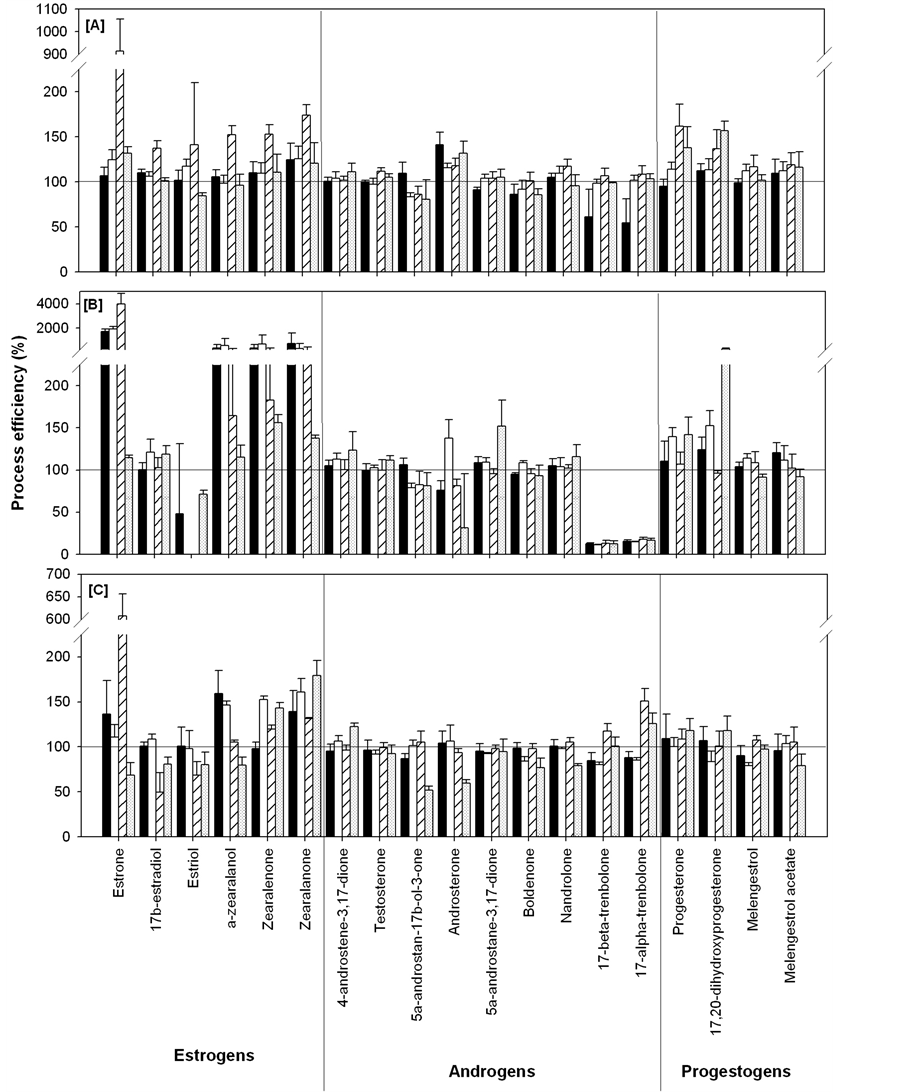
Figure 4. Process efficiencies (average ± standard deviation, n = 3) of target analytes spiked with 50 μL of 10 μg・mL−1 (in methanol) into sand (black), silt loam (white), clay (striped) and organic soil (hatched) and extracted with ASE (A); Soxhlet (B) and sonication (C).
The employment of post extraction cleanup may improve the Soxhlet extraction recoveries by removing matrix components (e.g., humic and fulvic acids) that may cause interference, either signal enhancement or suppression, on the HPLC-MS. Indeed, other investigators [14] [12] have obtained adequate recoveries in Soxhlet extracts filtered through silica or alumina-florisil, respectively (Table 1). Post extraction cleanup reduces the potential for coextracted matrix components to cause signal enhancement or suppression on the HPLC-MS/MS. However, this additional processing step is costly and time consuming and can result in target analyte losses, which can be problematic for the measurement of analytes at low concentrations; particularly hormones, which can induce endocrinological effects on fish at extremely low concentrations [7] -[9] [29] [30] . The incorporation of ISTDs should account for any matrix suppression and/or enhancement effects, providing that the target analyte and its corresponding ISTD undergo the same matrix enhancement/suppression. The overestimations of analyte concentrations in this study were primarily due to the normalisation of adequate analyte recoveries to the low recoveries of their corresponding ISTD. Thus, it is essential to employ an ISTD that has a similar apparent recovery to the target analyte in order to achieve an accurate and precise concentration.
3.3. Storage Stability
The storage stability of the target analytes in the various soil types was assessed by comparing the recovery of the ISTDs in samples stored for 30 (t = 30), 90 (t = 90) and 210 days (t = 210) at −20˚C with the recovery of samples that had been extracted two hours (t = 0) after spiking. Figure 5 shows the average apparent recoveries and 95% confidence intervals for each of the eight ISTDs spiked into the t = 0, t = 30, t = 90 and t = 210 samples. All of the ISTDs spiked into sand were statistically stable for at least 210 days of storage. However, there was a notable decrease in the average apparent recoveries of α-zearalenol-d4 in the t = 210 day samples that was not considered statistically significant due to the large variance of apparent recoveries ranging from 4% to 54%. The recoveries of 17β-estradiol-d5, estriol-d3, α-zearalenol-d4 and testosterone-d5 spiked into antigo silt loam significantly decreased within 30 days storage (Figure 5). While there was a decrease in nandrolone-d5 and progeste- rone-d9 apparent recoveries within 30 days of storage, the decrease was not statistically significant until 90 and 210 days of storage, respectively, and were still within acceptable levels of recovery (73% ± 2% and 71% ± 8%, respectively) at 210 days of storage. Therefore, silt loam soils spiked with ISTDs prior to storage should account for any nandrolone and progesterone losses that can occur during storage.
Similar to what was observed with the analyte apparent recovery, the recovery of estriol-d4 and 17β-estradiol-
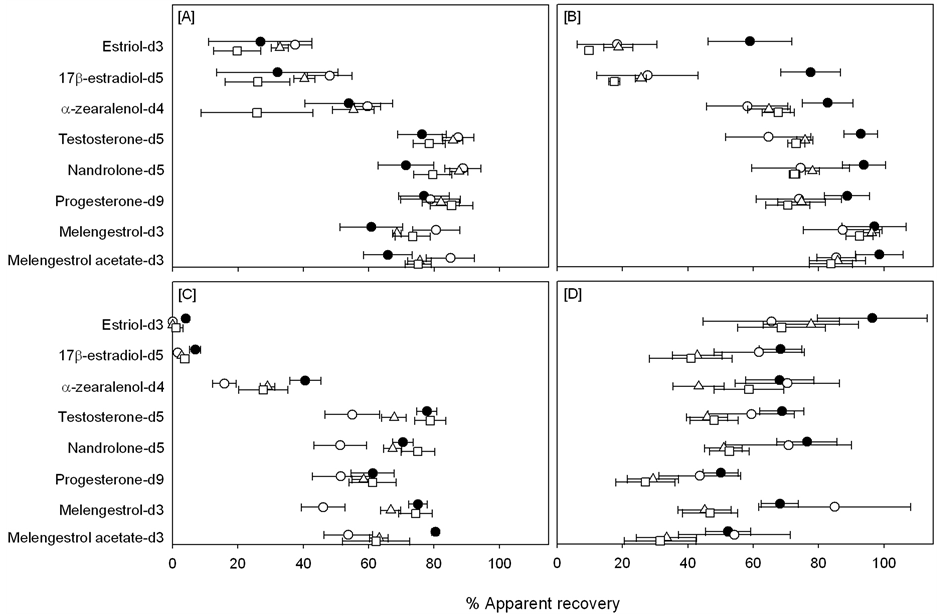
Figure 5. Comparison of the apparent recoveries (average ± 95% confidence interval, n = 6) of isotopically-labeled standards spiked into sand (A), silt loam (B), clay (C) and organic soils (D) and extracted with ASE immediately (black circle), after 30 (white circle), 90 (white triangle) or 210 (white square) days of storage at −20˚C.
d5 spiked into clay soil and immediately (after 2 hours) extracted were low (4% ± 1% and 7% ± 2%, respect- tively). After 30 days of storage the recoveries of estriol-d4 and 17b-estradiol-d5 were less than 1% ± 3% and 3.7% ± 0.2%, respectively, however, given the low initial apparent recoveries, the storage stability of these com- pounds could not be properly assessed. The apparent recoveries of melengestrol acetate-d3 and α-zearalenol-d4 spiked into clay-rich soil significantly decreased within 30 days of storage. A significant decrease in the apparent recovery of testosterone-d5, nandrolone-d5 and melengestrol-d3 extracted from organic soils was witnessed within 90 days of storage.
Given these variable results in storage stability, soil samples should be processes within 30 days of sample collection in order to avoid possible analyte losses during sample storage.
3.4. Native Soils
The native soil samples collected from the 15-acre paddock were best characterised as clay loam soils (34% sand, 37% silt and 29% clay). The overall average apparent recovery of all eight ISTDs spiked into the 22 native soil samples and extracted with ASE was fair (77% ± 13%) revealing ASE to be a reasonably efficient method for extracting estrogens, androgens and progestogens from clay loam soils.
We evaluated the presence of all of the target analytes in the native soils and occasionally detected some of the hormones. The only analyte detected in the shallow cores was 5α-androstane-3,17-dione (8 out of 17 samples) with concentrations ranging from 0.9 to 8.6 ng・g−1. Progesterone was detected in two of the shallow cores, however, at concentrations (0.8 and 0.9 ng・g−1) near the instrument detection limit. Both 5α-androstane-3, 17-dione (three out of 5 samples) and zearalenone (two out of samples) were detected in the deep cores samples at concentrations of 3.4 to 4.2 ng・g−1 and 2.5 and 3.6 ng・g−1, respectively.
4. Conclusion
With the exception of estrone from clay soils, the ASE and sonication techniques were efficient at extracting a large suite of estrogens, androgens and progestogens, as well as their metabolites, from varying soils types. Coupling these techniques with the incorporation of ISTDs provided accurate and reliable detection of hormones at environmentally relevant concentrations without the need for soil pretreatment (drying, sieving or lyophilizing) or post-extraction cleanup. Choosing the proper ISTD to account for target analyte losses and matrix affects is vital to achieve accurate target analyte concentrations since differences in their apparent recoveries can lead to under- or overestimations of target analytes. Finally, to avoid possible losses during sample storage, it is recommended that soil samples be processed within 30 days of sample collected.
Acknowledgements
The present study was funded by the United States Environmental Protection Agency’s National Center for Environmental Research Science To Achieve Results (STAR) program under the grant number R833421 and the Wisconsin Department of Natural Resources Bureau of Drinking and Groundwater. Special thanks to Steven Quinlan and Yajaira Pluess for their help with soil sample collection and extractions. We’re grateful to the University of Wisconsin Spooner Agricultural Research Station for providing the Antigo silt loam soil as well as the University of Wisconsin-Discovery Farms and the United States Geological Survey for their assistance with the collection of the native soil samples.
References
- United States Environmental Protection Agency (2000) Proposed Regulations to Address Water Pollution from Concentrated Animal Feeding Operations. Report No. EPA 833-F-00-016, Office of Water, Washington DC.
- Zheng, W., Yates, S.R. and Bradford, S.A. (2008) Analysis of Steroid Hormones in a Typical Dairy Waste Disposal System. Environmental Science & Technology, 42, 530-535. http://dx.doi.org/10.1021/es071896b
- Schiffer, B., Daxenberger, A., Meyer, K. and Meyer, H.H.D. (2001) The Fate of Trenbolone Acetate and Melengestrol Acetate after Application as Growth Promoters in Cattle: Environmental Studies. Environmental Health Perspectives, 109, 1145-1151. http://dx.doi.org/10.1289/ehp.011091145
- Kolodziej, E.P. and Sedlak, D.L. (2007) Rangeland Grazing as a Source of Steroid Hormones to Surface Waters. Environmental Science & Technology, 41, 3514-3520. http://dx.doi.org/10.1021/es063050y
- Lange, I.G., Daxenberger, A., Schiffer, B., Witters, H., Ibarreta, D. and Meyer, H.H.D. (2002) Sex Hormones Originating from Different Livestock Production Systems: Fate and Potential Disrupting Activity in the Environment. 4th International Symposium on Hormone and Veterinary Drug Residue Analysis, Antwerp, 4-7 June 2002, 27-37.
- Shore, L.S., Reichmann, O., Shemesh, M., Wenzel, A. and Litaor, M. (2004) Washout of Accumulated Testosterone in a Watershed. Science of the Total Environment, 332, 193-202. http://dx.doi.org/10.1016/j.scitotenv.2004.04.009
- Ankley, G.T., Jensen, K.M., Makynen, E.A., Kahl, M.D., Korte, J.J., Hornung, M.W., Henry, T.R., Denny, J.S., Leino, R.L., Wilson, V.S., Cardon, M.C., Hartig, P.C. and Gray, L.E. (2003) Effects of the Androgenic Growth Promoter 17-Beta-Trenbolone on Fecundity and Reproductive Endocrinology of the Fathead Minnow. Environmental Toxicology and Chemistry, 22, 1350-1360. http://dx.doi.org/10.1002/etc.5620220623
- Jensen, K.M., Makynen, E.A., Kahl, M.D. and Ankley, G.T. (2006) Effects of the Feedlot Contaminant 17 Alpha- Trenholone on Reproductive Endocrinology of the Fathead Minnow. Environmental Science & Technology, 40, 3112- 3117. http://dx.doi.org/10.1021/es052174s
- Orlando, E.F., Kolok, A.S., Binzcik, G.A., Gates, J.L., Horton, M.K., Lambright, C.S., Gray, L.E., Soto, A.M. and Guillette, L.J. (2004) Endocrine-Disrupting Effects of Cattle Feedlot Effluent on an Aquatic Sentinel Species, the Fathead Minnow. Environmental Health Perspectives, 112, 353-358. http://dx.doi.org/10.1289/ehp.6591
- de Alda, M.J.L., Gil, A., Paz, E. and Barcelo, D. (2002) Occurrence and Analysis of Estrogens and Progestogens in River Sediments by Liquid Chromatography-Electrospray-Mass Spectrometry. Analyst, 127, 1299-1304. http://dx.doi.org/10.1039/b207658f
- Cespedes, R., Petrovic, M., Raldua, D., Saura, U., Pina, B., Lacorte, S., Viana, P. and Barcelo, D. (2004) Integrated Procedure for Determination of Endocrine-Disrupting Activity in Surface Waters and Sediments by Use of the Biological Technique Recombinant Yeast Assay and Chemical Analysis by LC-ESI-MS. Analytical and Bioanalytical Chemistry, 378, 697-708. http://dx.doi.org/10.1007/s00216-003-2303-5
- Peng, X.Z., Wang, Z.D., Yang, C., Chen, F.R. and Mai, B.X. (2006) Simultaneous Determination of Endocrine-Dis- rupting Phenols and Steroid Estrogens in Sediment by Gas Chromatography-Mass Spectrometry. Journal of Chromatography A, 1116, 51-56. http://dx.doi.org/10.1016/j.chroma.2006.03.017
- Labadie, P. and Hill, E.M. (2007) Analysis of Estrogens in River Sediments by Liquid Chromatography-Electrospray Ionisation Mass Spectrometry: Comparison of Tandem Mass Spectrometry and Time-of-Flight Mass Spectrometry. Journal of Chromatography A, 1141, 174-181. http://dx.doi.org/10.1016/j.chroma.2006.12.045
- United States Environmental Protection Agency (2007) Method 1698: Steroids and Hormones in Water, Soil, Sediment and Biosolids by HRGC/HRMS. Report No. 821-R-08-003, Office of Water, Washington DC, 69.
- Beck, J., Totsche, K.U. and Kogel-Knabner, I. (2008) A Rapid and Efficient Determination of Natural Estrogens in Soils by Pressurised Liquid Extraction and Gas Chromatography-Mass Spectrometry. Chemosphere, 71, 954-960. http://dx.doi.org/10.1016/j.chemosphere.2007.11.062
- Petrovic, M., Tavazzi, S. and Barcelo, D.A. (2002) Column-Switching System with Restricted Access Pre-Column Packing for an Integrated Sample Cleanup and Liquid Chromatographic-Mass Spectrometric Analysis of Alkylphenolic Compounds and Steroid Sex Hormones in Sediment. Journal of Chromatography A, 971, 37-45. http://dx.doi.org/10.1016/S0021-9673(02)01026-9
- Hajkova, K., Pulkrabova, J., Schurek, J., Hajslova, J., Poustka, J., Napravnikova, M. and Kocourek, V. (2007) Novel Approaches to the Analysis of Steroid Estrogens in River Sediments. Analytical and Bioanalytical Chemistry, 387, 1351-1363. http://dx.doi.org/10.1007/s00216-006-1026-9
- Velicu, M. and Suri, R. (2009) Presence of Steroid Hormones and Antibiotics in Surface Water of Agricultural, Suburban and Mixed-Use Areas. Environmental Monitoring and Assessment, 154, 349-359. http://dx.doi.org/10.1007/s10661-008-0402-7
- Shore, L.S. and Pruden, A. (2009) Hormones and Pharmaceuticals Generated by Concentrated Animal Feeding Operations. Springer, New York, NY.
- Kolok, A.S., Snow, D.D., Kohno, S., Sellin, M.K. and Guillette, L.J. (2007) Occurrence and Biological Effect of Exogenous Steroids in the Elkhorn River, Nebraska, USA. Science of the Total Environment, 388, 104-115. http://dx.doi.org/10.1016/j.scitotenv.2007.08.001
- Adejumo, T.O., Hettwer, U. and Karlovsky, P. (2007) Survey of Maize from South-Western Nigeria for Zearalenone, Alpha- and Beta-Zearalenols, Fumonisin B-1 and Enniatins Produced by Fusarium Species. Food Additives and Contaminants, 24, 993-1000. http://dx.doi.org/10.1080/02652030701317285
- Havens, S.M., Hedman, C.J., Hemming, J.D.C., Mieritz, M.G., Shafer, M.M. and Schauer, J.J. (2010) Stability, Preservation, and Quantification of Hormones and Estrogenic and Androgenic Activities in Surface Water Runoff. Environmental Toxicology and Chemistry, 29, 2481-2490. http://dx.doi.org/10.1002/etc.307
- Cabrera, A., Papiernik, S.K., Koskinen, W.C. and Rice, P.J. (2012) Sorption and Dissipation of Aged Metalochlor Residues in Eroded and Rehabilitated Soils. Pest Management Science, 68, 1272-1277. http://dx.doi.org/10.1002/ps.3294
- Vanderford, B.J., Pearson, R.A., Rexing, D.J. and Snyder, S.A. (2003) Analysis of Endocrine Disruptors, Pharmaceuticals, and Personal Care Products in Water Using Liquid Chromatography/Tandem Mass Spectrometry. Analytical Chemistry, 75, 6265-6274. http://dx.doi.org/10.1021/ac034210g
- Burns, D.T., Danzer, K. and Townshend, A. (2002) Use of the Terms “Recovery” and “Apparent Recovery” in Analytical Procedures. Pure and Applied Chemistry, 74, 2201-2205. http://dx.doi.org/10.1351/pac200274112201
- Zuloaga, O., Fitzpatrick, L.J., Etxebarria, N. and Dean, J.R. (2000) Influence of Solvent and Soil Type on the Pressurised Fluid Extraction of PAHs. Journal of Environmental Monitoring, 2, 634-638. http://dx.doi.org/10.1039/b006178f
- Matuszewski, B.K., Constanzer, M.L. and Chavez-Eng, C.M. (2003) Strategies for the Assessment of Matric Effect in Quantitative Bioanalytical Methods Based on HPLC-MS/MS. Analytical Chemistry, 75, 3019-3030. http://dx.doi.org/10.1021/ac020361s
- Foreman, W.T., ReVello, R.C. and Gray, J.L. (2010) Deuterium Exchange Complicated Isotope Dilution Methods for Steroid Hormones. SETAC North America, 31 Annual Meeting, Portland.
- Palace, V.P., Evans, R.E., Wautier, K., Baron, C., Vandenbyllardt, L., Vandersteen, W. and Kidd, K. (2002) Induction of Vitellogenin and Histological Effects in Wild Fathead Minnows from a Lake Experimentally Treated with the Synthetic Estrogen, Ethynylestradiol. Water Quality Research Journal of Canada, 37, 637-650.
- DeQuattro, Z.A., Peissig, E.J., Antkiewics, D.S., Lundgren, E.J., Hedman, C.J., Hemming, J.D.C. and Barry, T.P. (2012) Effects of Progesterone on Reproduction and Embryonic Development in the Fathead Minnow (Pimephales promelas). Environmental Toxicology and Chemistry, 31, 851-856. http://dx.doi.org/10.1002/etc.1754
Supplemental Information
Table SI.1. Physical and chemical characteristics of the experimental matrices.
Table SI.2. Binary mobile phase gradient program (A = 0.1% formic acid, B = methanol).
Table SI.3. MRM m/z values and residence time for the target analytes and isotopically-labeled analogues (*denotes compounds for which APCI molecular ion generation involves the loss of H20). Note: DP = declustering potential; CE = collision energy; CXP = collision cell exit potential.
Table SI.4. Instrumental detection limit (IDL) and limits of quantitation (LOQ) for each target analyte analyzed on the HPLC-MS/MS.
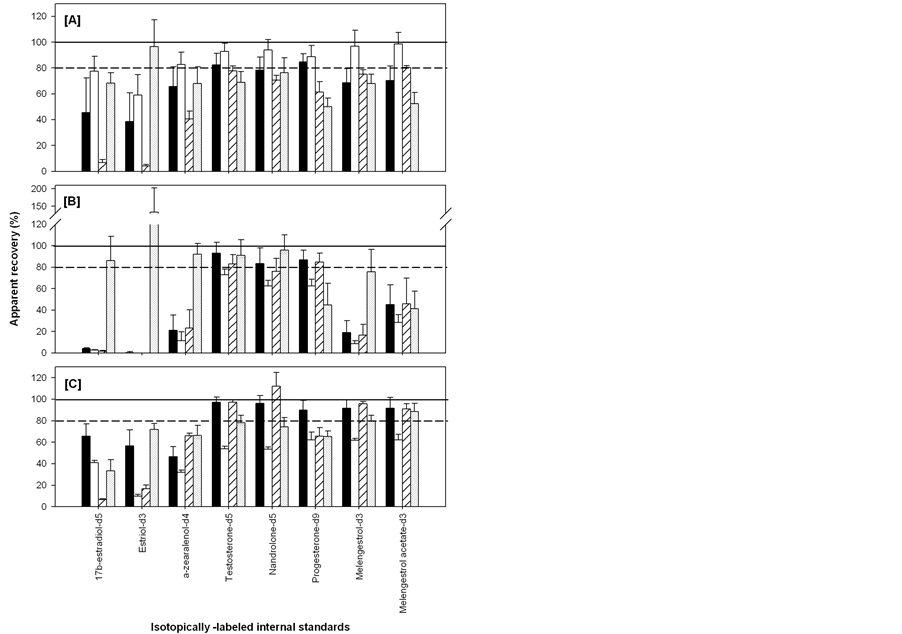
Figure SI.1. Apparent recoveries (average ± standard deviation, n = 3) of isotopically-labeled standards spiked with 50 μL of 10 μg・mL−1 (in methanol) into sand (black), silt loam (white), clay (striped) and organic (hatched) soil and extracted with ASE (A); Soxhlet (B) and sonication (C).
Figure SI.2. Background concentrations in sand (black), silt loam (white), clay (striped) and organic (hatched) soil samples extracted with ASE (A); Soxhlet (B) and sonication (C).
NOTES

*Corresponding author.