Advances in Nanoparticles
Vol.2 No.2(2013), Article ID:31467,7 pages DOI:10.4236/anp.2013.22015
Synthesis and Characterization of Poly(styrene-co-acrylamide) Polymers Prior to Electrospinning
1Chemistry Department, Rhodes University, Grahamstown, South Africa
2Department of Chemistry, University of Ibadan, Ibadan, Nigeria
Email: bolaoketola@yahoo.com, n.torto@ru.ac.za
Copyright © 2013 Adebola A. Oketola, Nelson Torto. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received November 25, 2012; revised January 12, 2013; accepted January 19, 2013
Keywords: Nanofibers; Emulsion Polymerization; Electrospinning; Poly(styrene-co-acrylamide)
ABSTRACT
Electrospun nanofibers present a new and rapidly growing research area due to their pronounced micro and nano characteristics associated with high surface area to volume ratio. Poly(styrene-co-acrylamide) and polystyrene polymers were synthesized by boiling temperature soap free emulsion polymerization in aqueous medium with potassium peroxosulphate as the initiator. The resulting polymers were dissolved in dimethylformamide and teterahydrofuran (DMF: THF) (4:1) to form polymer solutions that were electrospun into fiber mats with diameters ranging from 1.84 - 2.53 µm and 5.01 µm, respectively. The fibers were characterized by Fourier transform infrared spectroscopy (FTIR) equipped with universal ATR sampling accessory (4000 - 400 cm−1). The morphology and size were examined by a scanning electron miscroscope (SEM) and the thermal properties by thermogravimetric analysis (TGA). The FTIR spectra of the poly(styrene-co-acrylamide) revealed the presence of acrylamide on the polystyrene chain. Thus, surface modification of polystyrene with acrylamide is possible in a single step polymerization reaction prior to electrospinning.
1. Introduction
Electrospinning is a technique employed for preparing fibers having diameters in the range of 10 µm - 10 nm using a high electrostatic field [1]. It is a widely employed method to produce polymer nanofiber mats with high surface area to volume ratio [2]. Electrospinning technology has attracted growing scientific interest for the preparation of fibers due to the affordability and versatility of this process and the unique properties of electrospun fibers [1,3,4]. Beside this technology, there are other methods used for fiber fabrication that include drawing [5], template synthesis [6,7], phase separation [8] and self-assembly [9,10]. However, phase separation and self-assembly techniques are time consuming in processing continuous polymer nanofibers. In drawing, only viscoelastic material that can undergo strong deformations while being cohesive enough to support the stresses developed during pulling can be made into nanofibers. Thus, the electrospinning process seems to be the only method that can be further developed for mass production of one-by-one continuous nanofibers from various polymers [11].
Electrospun nanofibers have attracted much attention in various applications in recent years. Such applications include sorbents for solid phase extraction [12], membranes [13], filtration purposes [14], chemical protective clothings [15-17], reinforcement in transparent composites [18], tissue scaffolding [19,20] and other biomedical applications [21], nanocomposites [22], sensors [23] and nanoelectronics. Electrospun nanofibers possess high specific surface area, high porosity, small fiber diameter and low weight. However, for a successful electrospinning experiment, one needs to select a polymer that has the requisite characteristics of solubility and molecular weight.
A number of methods have been used for the synthesis of different polymers [24,25]. Most of these methods have their peculiar advantages and disadvantages with high polymerization rate as a major setback. Numerous methods of surface modification or functionalization have also been studied [24,26]. Most routes of surface modification of polymers with grafted polyacrylamide chains have been based on radical polymerization, using an oxidation-reduction initiation system (with ceric ion) [27] or a living radical polymerization (LRP) by coppermediated atom transfer radical polymerization (ATRP) [28]. It was shown that the grafting of acrylamide at the surface of hydrocarbon polymers could be conveniently initiated by near-UV irradiation of a photo-initiator previously absorbed at the surface of the polymer or by thermal decomposition of photo-generated hydroperoxides in the presence of the monomer in aqueous solution.
This work describes a simple method for the synthesis of poly(styrene-co-acrylamide) polymer using a boiling temperature soap-free emulsion polymerization process which makes colloid particles of polymer dispersed in water with styrene as the monomer, acrylamide as the functional monomer and potassium peroxosulphate as an initiator. Polyacrylamide, a water soluble and hydrophilic polymer is incorporated into polystyrene chain, a hydrophobic polymer in a single chemical reaction. The introduction of hydrophilic polymer chains such as polyacrylamide onto styrene copolymers may promote better diffusion of aqueous solution through porous structure and offer numerous potential applications [27]. This method of synthesis offers several advantages such as simplified operation and experimental set-up, shorter reaction time, good repeatability, etc. [25] when compared to other conventional methods. The resulting polymer was fabricated into fiber by electrospinning at ambient temperature and humidity conditions.
2. Experimental Section
2.1. Materials
Styrene (99%) and acrylamide (99%) were purchased from Sigma Aldrich (St. Louis, USA), potassium peroxosulphate (KPS) (98%) from Saarchem (Johannesburg, South Africa) and used without further purification. Methanol, tetrahydrofuran (THF), and N, N-dimethylformamide (DMF) were purchased from Merck Chemicals, (Wadeville, South Africa) and were reagent grade. Ultrapure water from MilliQ system was used throughout.
2.2. Polymer Synthesis
Poly(styrene-co-acrylamide) (PS-AAm) and polystyrene (PS) were synthesized by a boiling temperature soap-free emulsion polymerization in a three-necked flask equipped with a reflux condenser and a mechanical stirrer immersed in silicone oil bath [25]. For the synthesis of the polymers, different monomer mole ratios and acrylamide were added to 100 ml ultrapure MilliQ water. The mixture was stirred and allowed to boil under reflux after which 0.2 g of KPS was added as the initiator. The reaction continued for 3 h, after which it was stopped. Table 1 shows the list of polymers synthesized, amount of reagents used and remark on their solubility in different solvents and solvent mixtures during washing. The synthesized polymers were dried in rotary evaporator (BÜCHI Rotavapor R-114). The polymers were washed by dissolving 1 g in 10 ml of tetrahydrofuran, stirred until complete dissolution and then precipitated with 40 ml of methanol. The precipitate was filtered, washed in methanol, and dried overnight in an oven at 70˚C.
2.3. Electrospinning
Fibers were electrospun from solutions of 5 wt% and 10 wt% polymers in a solvent mixture of DMF/THF (4:1) which was prepared by dissolving 250 mg and 500 mg, respectively of poly(styrene-co-acrylamide) in 5 ml of DMF/THF (4:1). Also, 5 wt%, 10 wt% and 20 wt% unmodified polystyrene solution was prepared by dissolv-
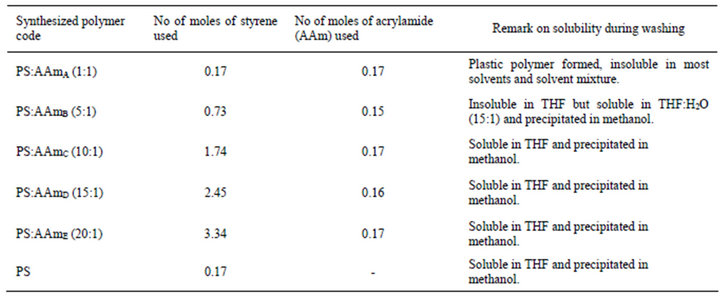
Table 1. Polymers synthesized, reagents used and solubility property.
ing 250 mg, 500 mg and 1g, respectively of PS in 5 ml of DMF/THF (4:1). The solutions were subsequently stirred for 24 h to ensure homogeneity and transferred into a 10 ml glass syringe, Poulten GmbH (Berlin Germany) equipped with a steel needle with a diameter of 0.5 mm. The needle was connected to a high-voltage supply capable of generating voltage up to 25 kV. Polymer solution was feed at 0.010 ml/h in most cases and controlled using an automatic syringe pump (New Era, NE-1000), at voltage of +15 kV and −5 kV. Fibers were collected on a copper plate pasted on an aluminum foil at the back of a round rotating disc. The collector was placed 15 cm from the tip of the needle and the solution in the tip of the needle ejected under strong electric field.
2.4. Characterization
The morphology of the electrospun fibers was evaluated using scanning electron microscopy (TESCAN VEGA TS 5136 LM) with Scandium software. Samples for the SEM were prepared by placing the fiber on a plate, followed by vapour-deposition of a thin layer of gold. Fiber diameter was measured from an average of 20 fibers. Fourier transform infrared spectra (FT-IR) were measured with a PerkinElmer spectrum 100 FT-IR spectrometer equipped with a universal ATR sampling accessory. Thermal behaviour of the fibers was investigated using PERKIN-ELMER TGA 7 Thermogravimetric Analyzer with PERKIN-ELMER TAC 7/DX Thermal Analysis Controller. The analysis was performed with 1 - 2 mg sample in a dynamic nitrogen atmosphere with heating rate of 10˚Cmin−1 from room temperature to 650 ˚C.
3. Results and Discussion
3.1. Synthesis and Characterization
Poly(styrene-co-acrylamide) and polystyrene polymers were successfully polymerised using a boiling temperature soap free emulsion polymerization. This polymerization process provide further benefits including a reduced reaction time of less than 4 h without constant stirring and flow of nitrogen gas as in the conventional method of polymerization. It offered a practical advantage in that the acrylamide was successfully incorporated into the polystyrene chain in a single step reaction. Polymerization of styrene and acrylamide in ratio 1:1 resulted in a hard plastic polymer that was insoluble in THF and most solvents and solvent mixtures including water. As the volume of styrene in the polymerization reaction increased and the mass of acrylamide decreased, solubility of the poly(styrene-co-acrylamide) formed in THF also increased. Thus, the PS-AAm formed at ratio 10:1 (PS:AAmC), 15:1 (PS:AAmD) and 20:1 (PS:AAmE) were all soluble in THF except 5:1 (PS:AAmB) which was insoluble in THF but soluble in a solvent mixture of THF and water in the ratio (15:1). This could be attributed to the fact that polystyrene is a non-polar polymer with a glass transition temperature (Tg) around 100˚C [28] and polyacrylamide is a water soluble hydrophilic polymer.
Subsequently, the polymer was electrospun to fibers of different diameters except PS-AAmA that formed a plastic material that was insoluble in most solvents and solvent mixtures. This method of modification of styrene with acrylamide is a promising method for synthesis of poly(styrene-co-acrylamide) and other hydrophobic polymer modified with hydrophilic polymers such as acrylamide at reduced polymerization time for possible and various applications. Polystyrene has been identified as the most frequently used polymer due to its several advantages that make it suitable as hydrophobic encapsulation materials in biomedical applications. Also, it is cheap and can be functionalized [29].
3.2. FT-IR Spectra
Infrared spectra of the electrospun fibers are presented in Figure 1. The spectra of the different poly(styreneco-acrylamide) formed i.e., PS-AAmB, PS-AAmC, PSAAmD and PS-AAmE are all very similar but are rather different from the spectra of polystyrene (PS) without acrylamide modification. No remarkable changes were observed between the spectra of the four modified polymer probably because of the small concentration of acrylamide used when compared to the styrene monomer unit except PS-AAmB (5:1). In PS-AAmB spectra, there are characteristic and intense peaks found at 3200 cm−1, 1710 cm−1, 1430 cm−1 and 1024 cm−1 indicating the N-H stretching, C=O stretching and C-N stretching due to acrylamide group. In addition, the peak at 774 - 765 cm−1 band represents the out of plane bending of the weak bond N-H. These characteristics peaks were observed and prominent in the FT-IR spectra of PS-AAmB (5:1) and slightly in PS-AAmC (10:1) but totally absent in PS-AAmD (15:1), PS-AAmE (20:1) and PS spectra, respectively.
3.3. Scanning Electron Microscope (SEM)
The morphology and size of the electrospun fibers produced as measured by a scanning electron microscope (SEM) are shown in Figures 2(a)-(f), respectively. 10 wt% PS polymer solution at feed rate of 0.010 ml/h resulted in the formation of beads (Figure 2(a)) while there was the formation of fiber when the polymer solution and feed rate was increased to 20 wt% and 0.020 ml/h (Figure 2(b)). These observations differ from what was reported [28] at 20% PS, where the structure con-
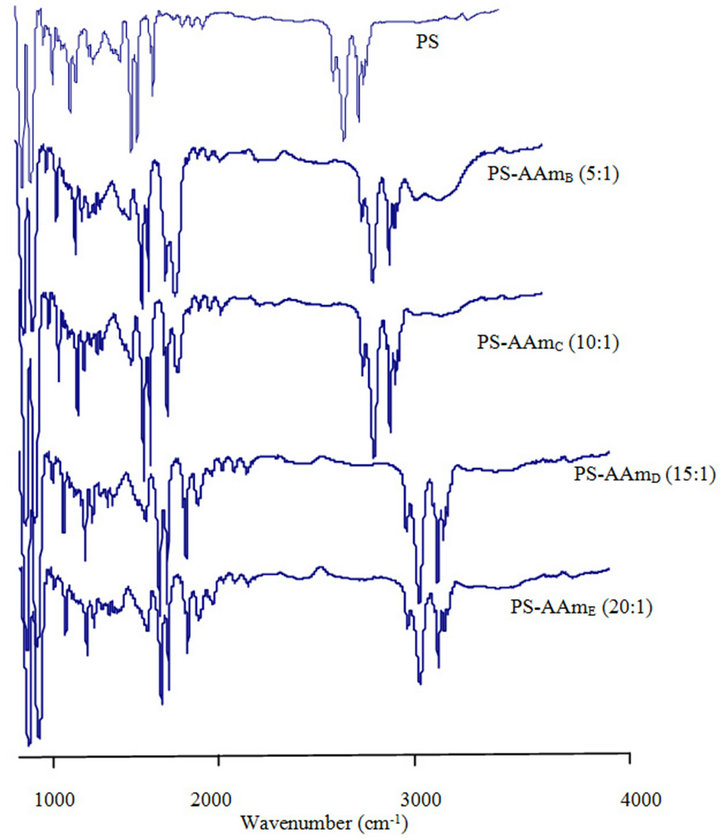
Figure 1. ATR-FTIR spectra of polystyrene (PS) and polystyrene-acrylamide (PS-AAm) of different monomer ratio.
sisted predominantly of beads with a few incipient fibers. It was reported that higher polymer concentrations (25 and 30 wt%) resulted in larger fiber/bead ratios and at 35 wt%, only fibers were obtained. There was no flow of the polymer solution from the tip of the needle at 20 wt% and 0.010 ml/h feed rate. It has been reported that formation of beads along the fibers typically occurs during the electrospinning of PS but can be avoided by the appropriate choice of parameters. It could also be prevented by increasing the polymer concentration (which resulted in larger fiber diameters) or by increasing the electrical conductivity of the spinning solution [30]. This is in line with what was obtained in this study.
Electrospun PS-AAmC and PS-AAmE resulted in the formation of fibers at 0.010 ml/h feed rate while PS-AAmB and PS-AAmD resulted in the formation of beads at the same feed rate and even when the feed rate was increased to 0.020 ml/h (Figures 2(c)-(f)). This indicated that feed rate and polymer concentration affects the morphology of electrospun fibers. The beads observed in PS-AAmB and PS-AAmD could be attributed to some of the processing conditions such as solution concentration, polymer molecular weight, solution viscosity, solution conductivities, solution surface tension, applied voltage, distance of electrode from the target substrate, electric field, solution flow rate, temperature, humidity and solvent volatility [30-32]. The beads observed have a wrinkled, raising-like surface texture that is similar to what was reported in the past [4]. Experimental observations in electrospinning confirm that for fiber formation to occur, a minimum polymer concentration is required. Below this critical value, application of voltage results in electrospraying or bead formation primarily due to a Rayleigh instability. At these low concentrations, an insufficiently deformable entangled network of polymer chains exists. As the polymer concentration increased, a mixture of beads and fiber was obtained. Further increase in concentration resulted in the formation of continuous fibers [28].
Fiber diameter is one of the most important quantities in electrospinning. Since fibers resulted from evaporation or solidification of polymer fluid jet, the fiber diameter will depend primarily on the jet size as well as the polymer content in the jet [11]. For a given molecular weight, it is an established fact that fiber diameter increases with polymer concentration [31]. The SEM image showed that very long fibers were formed having varying diameters. 10 wt% PS solution resulted in the formation of beads while increasing the concentration to 20 wt% resulted in fiber formation without beads with an average diameter of 5.01 µm. 10 wt% polymer solution of PS-AAmC (10:1) and PS-AAmE (20:1) also gave fiber with an average diameter of 1.84 µm - 2.53 µm (Figures 2(d)-(f)). 5 wt% polymer solution of both functionalized and unfuctionalized polystyrene resulted in beads formation at 0.010 ml/h feed rate. One of the most significant parameters influencing the fiber diameter is the solution viscosity. A higher viscosity resulted in a larger fiber diameter [33]. However, when a solid polymer is dissolved in a solvent, the solution viscosity is proportional to the polymer concentration. Thus, the higher the polymer concentration, the larger the resulting fiber diameter [11]. Also, a higher applied voltage ejects more fluid in a jet, resulting in a larger fiber diameter [33], which was obtained when the PS polymer concentration was increased from 10 wt% to 20 wt% in this study.
It can be shown however, that the higher the amount of styrene in the polymer, the higher the average diameter of the fiber formed. Thus, the range of diameter of PSAAmC (10:1) was 1.84 µm - 2.31 µm while that of PSAAmE (20:1) was 2.48 µm - 2.53 µm, respectively. This is in agreement with what was obtained [1] where aligned fibers having diameters in the range of 1 - 10 µm were formed. Reference [4] also obtained fiber diameter ranging from 0.3 - 2 µm and pointed out that the fiber diameter increased with increasing polymer concentration according to a power law relationship. [32], however, also found that the fiber diameter was proportional to the cube of the polymer concentration. Another parameter which affects the fiber diameter to a remarkable extent is the applied electrical voltage. All these can be said to have contributed to the size of the fibers obtained in this study. Thus, optimization of these parameters will result in the formation of fiber mats in nano scale level.
 (a) PS 10% (w/v), Feed rate: 0.010 ml/h
(a) PS 10% (w/v), Feed rate: 0.010 ml/h
 (b) PS 20% (w/v), Feed rate: 0.02 ml/h
(b) PS 20% (w/v), Feed rate: 0.02 ml/h
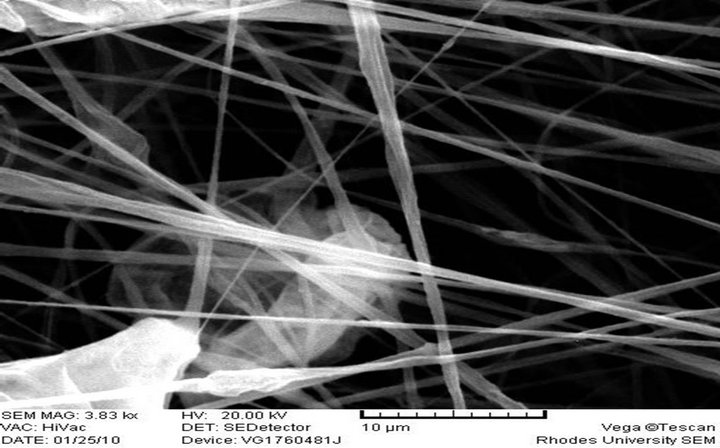 (c) PS-AAmB, Feed rate: 0.020 ml/h
(c) PS-AAmB, Feed rate: 0.020 ml/h
 (d) PS-AAmC, Feed rate: 0.010 ml/h
(d) PS-AAmC, Feed rate: 0.010 ml/h
 (e) PS-AAmD, Feed rate: 0.010 ml/h
(e) PS-AAmD, Feed rate: 0.010 ml/h
 (f) PS-AAmE, Feed rate: 0.010 ml/h
(f) PS-AAmE, Feed rate: 0.010 ml/h
Figure 2. SEM image of electrospun fibers.
 (a) PS-AAm
(a) PS-AAm
 (b) PS
(b) PS
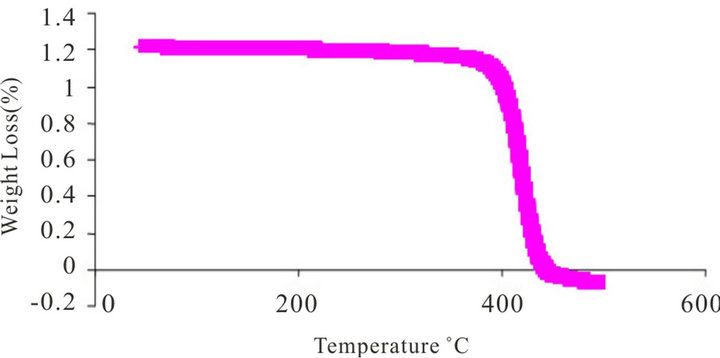
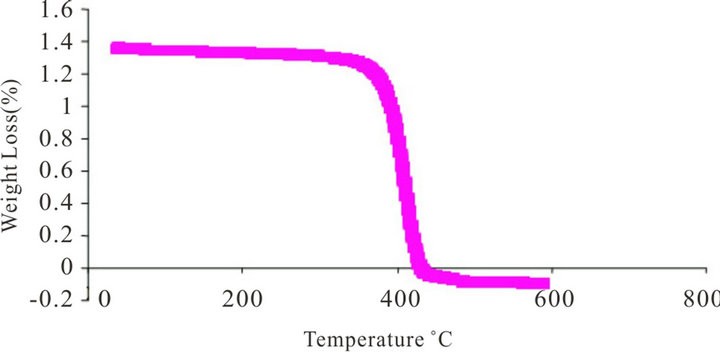
Figure 3. TGA curve of (a) polystyrene-acrylamide (PS-AAm) and (b) polystyrene (PS).
3.4. Thermogravimetry
Polystyrene is an amorphous polymer with a glass transition temperature (Tg) around 100˚C. The weight loss on heating the electrospun fibers was measured using thermogravimetric analysis. The thermal behaviour of the fibers was examined up to 650˚C. In all the TG curves, rapid weight loss was observed around 356˚C, and no further weight loss above 437˚C in PS and PS-AAm, respectively (Figures 3(a) and (b)). It was observed that the thermal stability of PS-AAm is slightly higher than that of pure PS without the amide modification. The observed weight loss was due to the degradation of the fiber mats which are mainly organic.
4. Conclusion
The aim of this study was to describe a simple boiling temperature soap-free emulsion polymerization for the synthesis of poly(styrene-co-acrylamide) at reduced polymerization time. FT-IR characteristic of the polymer showed that there was acrylamide group on the styrene. Thus, acrylamide was successfully incorporated into the polystyrene chain prior to electrospinning. Electrospun fibers of different diameters were fabricated from the styrene and acrylamide of different mole ratios to give poly (styrene-co-acrylamide), some of which resulted in bead formation. Many parameters can influence the transformation of polymer solutions into fibers through electrospinning. These parameters include: 1) the solution properties such as viscosity, elasticity, conductivity, and surface tension; 2) governing variables such as hydrostatic pressure in the capillary tube, electric potential at the capillary tip, and the gap (distance between the tip and the collecting screen); and 3) ambient parameters such as solution temperature, humidity, and air velocity in the electrospinning chamber. Variation and optimization of these parameters will results in fiber of different nanometer sizes that can have diverse and various applications including drug delivery, electrical and optical application etc. The poly(styrene-co-acrylamide) fiber formed is anticipated for diverse application such as Solid Phase Extraction (SPE) materials, filters, textile etc.
5. Acknowledgements
AAO is most grateful to the Organization for the Prohibition of Chemical Weapons (OPCW) for the financial support (L/ICA/ICB/149824/09). The contribution of Mr. Godfred Darko and Mr. Samuel Chigome are highly appreciated.
REFERENCES
- B. Sundaray, V. Subramanian, T. S. Natarajan, R. Z. Xiang, C. C. Chang and W. S. Fann, “Electrospinning of Continuous Aligned Polymer Fibers,” Applied Physics Letters, Vol. 84, No. 7, 2004, pp. 1222-1224. doi:10.1063/1.1647685
- C. Zhang, Q. Liu, N. Zhan, Q. Yang, Y. Song, L. Sun, H. Wang and Y. Li, “A Novel Approach to Prepare Silver Chloride Nanoparticles Grown on the Surface of Nanofiber via Electrospinning Combined with Gas-Solid Reaction,” Colloid and Surface A: Physicochemical and Engineering Aspects, Vol. 353, No. 1, 2010, pp. 64-68. doi:10.1016/j.colsurfa.2009.10.022
- S. Chigome and N. Torto, “A Review of Opportunities for Electrospun Nanofibers in Analytical Chemistry,” Analytica Chimica Acta, Vol. 706, No. 1, 2011, pp. 25-36. doi:10.1016/j.aca.2011.08.021
- J. M. Deitzel, J. Kleinmeyer, D. Harris and N. C. B. Tan, “The Effect of Processing Variables on the Morphology of Electrospun Nanofibers and Textiles,” Polymer, Vol. 42, No. 1, 2001, pp. 261-272. doi:10.1016/S0032-3861(00)00250-0
- T. Ondarcuhu and C. Joachim, “Drawing a Single Nanofibre over Hundreds of Microns,” Europhysics Letters, Vol. 42, No. 2, 1998, pp. 215-220. doi:10.1209/epl/i1998-00233-9
- L. Feng, S. Li, H. Li, J. Zhai, Y. Song, L. Jiang and D. Zhu, “Super-Hydrophobic Surface of Aligned Polyacrylonitrile Nanofibers,” Angewandte Chemie International Edition, Vol. 41, No. 7, 2002, pp. 1221-1223. doi:10.1002/1521-3773(20020402)41:7<1221::AID-ANIE1221>3.0.CO;2-G
- C. R. Martin, “Membrane-Based Synthesis of Nanomaterials,” Chemistry of Materials, Vol. 8, No. 8, 1996, pp. 1739-1746. doi:10.1021/cm960166s
- P. X. Ma and R. Zhang, “Synthetic Nano-Scale Fibrous Extracellular Matrix,” Journal of Biomedical Materials Research, Vol. 46, No. 1, 1999, pp. 60-72. doi:10.1002/(SICI)1097-4636(199907)46:1<60::AID-JBM7>3.0.CO;2-H
- G. J. Liu, J. F. Ding, L. J. Qiao, A. Guo, B. P. Dymov and J. T. Gleeson,” Polystyrene-block-poly(2-cinnamoylethyl methacrylate) Nanofibers—Preparation, Characterization, and Liquid Crystalline Properties,” Chemistry—A European Journal, Vol. 5, No. 9, 1999, pp. 2740-2749. doi:10.1002/(SICI)1521-3765(19990903)5:9<2740::AID-CHEM2740>3.0.CO;2-V
- G. M. Whitesides and B. Grzybowski, “Self-Assembly at All Scales,” Science, Vol. 295, No. 5564, 2002, pp. 2418- 2421. doi:10.1126/science.1070821
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, “A Review on Polymer Nanofibers by Electrospinning and Their Applications in Nanocomposites,” Composites Science and Technology, Vol. 63, No. 15, 2003, pp. 2223-2253. doi:10.1016/S0266-3538(03)00178-7
- Y. Zhang, X. Kang, L. Chen, C. Pao, Y. Yao and Z. Z. Gu, “Fiber-Packed SPE Tips Based on Electrospun Fibers” Analytical and Bioanalytical Chemistry, Vol. 391, No. 6, 2008, pp. 2189-2197. doi:10.1007/s00216-008-2145-2
- H. Q. Liu and Y. L. Hsieh, “Ultrafine Fibrous Cellulose Membranes from Electrospinning of Cellulose Acetate,” Journal of Polymer Science Part B: Polymer Physics, Vol. 40, No. 18, 2002, pp. 2119-2129. doi:10.1002/polb.10261
- P. P. Tsaia, H. Schreuder-Gibson and P. Gibson, “Different Electrostatic Methods for Making Electrets Filters,” Journal of Electrostatics, Vol. 54, No. 3-4, 2002, pp. 333-341. doi:10.1016/S0304-3886(01)00160-7
- P. W. Gibson, H. L. Schreuder-Gibson and D. Rivin, “Transport Properties of Porous Membranes Based on Electrospun Nanofibers,” Colloids and Surfaces A: Physicochemical and Engineering Aspects, Vol. 187-188, 2001, pp. 469-481. doi:10.1016/S0927-7757(01)00616-1
- H. L. Schreuder-Gibson, P. Gibson, K. Senecal, M. Sennett, J. Walker, W. Yeomans, et al., “Protective Textile Materials Based on Electrospun Nanofibers,” Journal of Advanced Materials, Vol. 32, No. 2, 2002, pp. 44-55.
- L. Chen, L. Bromberg, T. Hatton and G. Rutledge, “Catalytic Hydrolysis of p-Nitrophenyl Acetate by Electrospun Polyacrylamidoxime Nanofibers,” Polymer, Vol. 48, No. 16, 2007, pp. 4675-4682. doi:10.1016/j.polymer.2007.05.084
- S. Chand, “Review: Carbon Fibers for Composites,” Journal of Materials Science, Vol. 35, No. 6, 2000, pp. 1303- 1313. doi:10.1023/A:1004780301489
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, “Electrospinning of Collagen Nanofibers,” Biomacromolecules, Vol. 3, No. 2, 2002, pp. 232-238. doi:10.1021/bm015533u
- V. N. Morozov, T. Y. Morozova and N. R. Kallenbach, “Atomic Force Microscopy of Structures Produced by Electrospraying Polymer Solutions,” International Journal of Mass Spectrometry, Vol. 178, No. 3, 1998, pp. 143- 159. doi:10.1016/S1387-3806(98)14083-6
- M. M. Hohman, M. Shin, G. Rutledge and M. P. Brenner, “Electrospinning and Electrically Forced Jets. II. Applications,” Physics of Fluids, Vol. 13, No. 8, 2001, pp. 2221-2236. doi:10.1063/1.1384013
- P. S. Nair, T. Radhakrishnan, N. Revaprasadu, C. G. C. E. van Sittert, V. Djokovic and A. S. Luyt, “Characterisation of Polystyrene Filled with HgS Nanoparticles,” Materials Letters, Vol. 58, No. 3-4, 2004, pp. 361-364. doi:10.1016/S0167-577X(03)00501-9
- Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hellwig and M. Steinhart, “Nanostructured Fibers via Electrospinning,” Advanced Materials, Vol. 13, No. 1, 2001, pp. 70- 72. doi:10.1002/1521-4095(200101)13:1<70::AID-ADMA70>3.3.CO;2-8
- J. M. Jin, J. M. Lee, M. H. Ha, K. Lee and S. Choe, “Highly Cross-Linked Poly(glycidyl methacrylate-co-divinylbenzene) Particles by Precipitation Polymerization,” Polymer, Vol. 48, No. 11, 2007, pp. 3107-3115. doi:10.1016/j.polymer.2007.03.068
- Z. Z. Gu, H. Chen, S. Zhang, L. Sun, Z. Xie and Y. Ge, “Rapid Synthesis of Monodisperse Polymer Spheres for Self-Assembled Photonic Crystals,” Colloids and Surfaces A: Physicochemical and Engineering Aspects, Vol. 302, No. 1-3, 2007, pp. 312-319. doi:10.1016/j.colsurfa.2007.02.064
- J. Zhao and G. Geuskens, “Surface Modification of Polymers VI. Thermal and Radiochemical Grafting of Acrylamide on Polyethylene and Polystyrene,” European Polymer Journal, Vol. 35, No. 12, 1999, pp. 2115-2123. doi:10.1016/S0014-3057(99)00026-9
- L. da Cunha, F. M. B. Coutinho, V. G. Teixeira, E. F. O. de Jesus and A. S. Gomes, “Surface Modification of Styrene-Divinylbenzene Copolymers by Polyacrylamide Grafting via Gamma Irradiation,” Polymer Bulletin, Vol. 61, No. 3, 2008, pp. 319-330. doi:10.1007/s00289-008-0962-2
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, “Role of Chain Entanglements on Fiber Formation during Electrospinning of Polymer Solutions: Good Solvent, NonSpecific Polymer-Polymer Interaction Limit,” Polymer, Vol. 46, No. 10, 2005, pp. 3372-3384. doi:10.1016/j.polymer.2005.03.011
- J. Huang, H. Pen, Z. Xu and C. Yi, “Magnetic Fe3O4/ Poly(styrene-co-acrylamide) Composite Nanoparticles Prepared by Microwave-Assisted Emulsion Polymerization,” Reactive and functional Polymers, Vol. 68, No .1, 2008, pp. 332-339. doi:10.1016/j.reactfunctpolym.2007.08.002
- Greiner and J. H. Wendorff, “Electrospinning: A Fascinating Method for the Preparation of Ultrathin Fibers,” Angewandte Chemie, Vol. 16, No. 46, 2007, pp. 5670- 5703. doi:10.1002/anie.200604646
- J. Doshi and D. H. Reneker, “Electrospinning Process and Applications of Electrospun Fibers,” Journal of Electrostatics, Vol. 35, No. 2-3, 1995, pp. 151-160. doi:10.1016/0304-3886(95)00041-8
- M. M. Demir, I. Yilgor, E. Yilgor and B. Erman, “Electrospinning of Polyurethane Fibers,” Polymer, Vol. 43, No. 11, 2002, pp. 3303-3309. doi:10.1016/S0032-3861(02)00136-2
- H. Fong, I. Chun and D. H. Reneker, “Beaded Nanofibers Formed during Electrospinning,” Polymer, Vol. 40, No. 16, 1999, pp. 4585-4592. doi:10.1016/S0032-3861(99)00068-3